
Tumor microenvironment and immunology of cholangiocarcinoma
 0
0Abstract
Cholangiocarcinoma (CCA), an aggressive tumor originating from both intra- and extra-hepatic biliary cells, represents an unmet need in liver oncology, as treatment remains largely unsatisfactory. A typical feature of CCA is the presence of a complex tumor microenvironment (TME) composed of neoplastic cells, a rich inflammatory infiltrate, and cancer-associated fibroblasts and desmoplastic matrix that makes it extremely chemoresistant to traditional chemotherapeutic drugs. In this review, we describe the cell populations within the TME, in particular those involved in the innate and adaptive immune response and how they interact with tumor cells and with matrix proteins. The TME is crucial for CCA to mount an immune escape response and is the battlefield where molecularly targeted therapies and immune therapy, particularly in combination, may actually prove their therapeutic value.
Keywords
INTRODUCTION
Cholangiocarcinoma (CCA) is a highly malignant cancer that can develop from different segments of the biliary tree. The anatomical classification does not include gallbladder and ampullary cancers and distinguishes among intrahepatic (iCCA) that develops inside the liver up to the secondary bile ducts
CCA incidence shows a strong geographical variation, ranging from 0.4:100,000 inhabitants in Canada to 85:100,000 in the northeast of Thailand[3]. Furthermore, in the United States, distinct ethnic groups show different incidences with higher rates among Asians and Hispanics (2.8:100,000 and 3.3:100,000, respectively) and lower ones in Caucasians and African Americans (1.4:100,000 and 1.7:100,000)[4]. The most significant known risk factor for the development of CCA in East Asia is parasitic infestations of Opisthorchis viverrini or Clonorchis sinensis. After their encystation in the biliary network, these parasites cause chronic irritation, leading to neoplasm development[5]. In Western countries, the most prominent risk factor for CCA development is the primary sclerosing cholangitis (PSC) with an odds ratio (OR) of 164 (CI: 73.3-369, P < 0.001)[6]. PSC is an inflammatory disease affecting both the intra- and extra-hepatic biliary tract, causing inflammation of the biliary epithelium, periductal fibrosis, and biliary stenosis[5]. Other additional risk factors, particularly for iCCA, are HBV- and HCV-related cirrhosis, choledochal cysts, cholelithiasis, type 2 diabetes mellitus, obesity, non-alcoholic fatty liver disease, smoking, and hypertension[7,8]. It is worth noting that all these conditions are associated with liver inflammation.
Genetically, CCA is a heterogeneous tumor. CCAs originating from large ducts show a high mutation frequency of oncogenes and of tumor suppressor genes, such as Kirsten rat sarcoma virus (15%-30%) and tumor protein P53 (TP53) (10%-40%) They may also harbor mutations of BRAF, BRCA1 associated protein 1 (BAP1), phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA), guanine nucleotide binding protein GNAS, AT-rich interaction domain 1A (ARID1A), SMAD family member 4 (SMAD4), phosphatase and tensin homolog (PTEN), mouse double minute 2 homolog (MDM2), epidermal growth factor receptor (EGFR), and Erb-B2 receptor tyrosine kinase 2 (ERBB2), among others. Microsatellite instability is another prognostically and therapeutically relevant marker for CCA, as it has been shown that tumors with deficiency of mismatch DNA repair mechanisms (e.g., those associated with liver fluke infestation) are significantly more sensitive to immune checkpoint blockade[9]. In contrast, small duct type CCA exhibits a mass-forming growth pattern and exhibits isocitrate dehydrogenase 1/2 mutations (10%-30%) and fibroblast growth factor receptor 2 (FGFR2) fusions (10%-25%), among others[10,11].
REACTIVE TUMOR STROMA
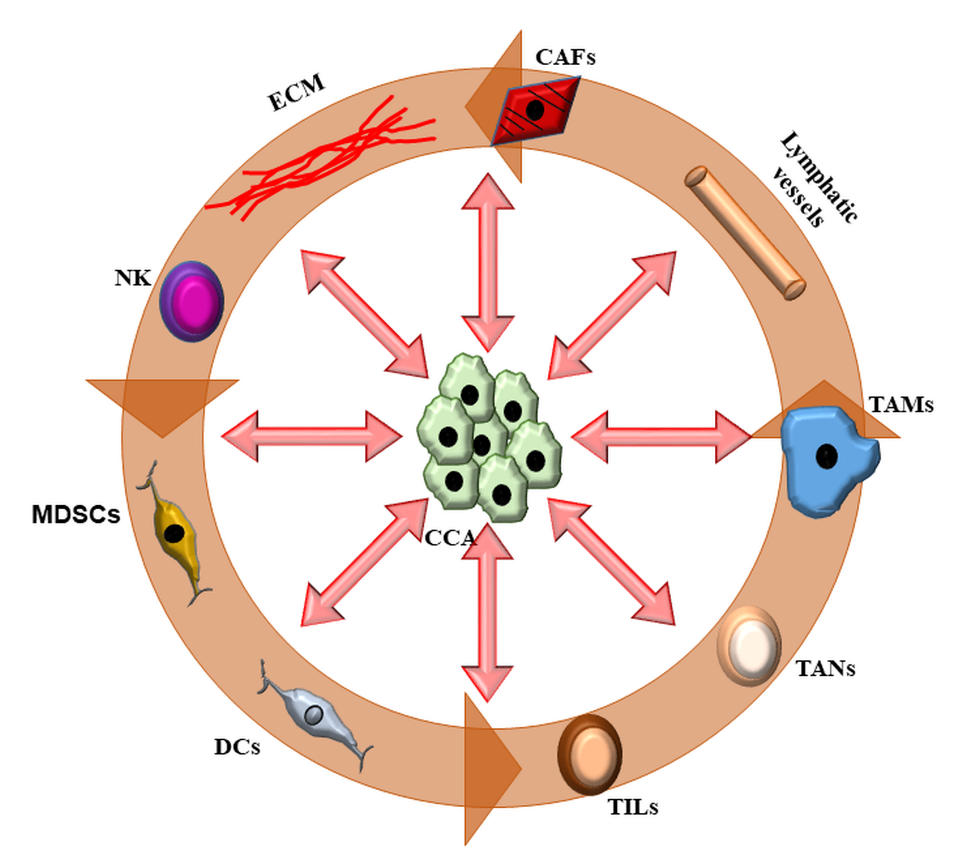
Similar to other cancers, such as pancreatic or breast adenocarcinoma, CCA is characterized by an intense desmoplastic reaction [tumor reactive stroma (TRS)] supported by a rich cellular microenvironment and by modifications of the matrix composition. This tumor microenvironment (TME)[2] has a structural component, the extracellular matrix (ECM), and a cellular component with a plethora of infiltrating cells. The matrix of the TME is significantly different from the normal one, in both quantity and quality. The TRS within the tumor is in fact continuously modified by the interaction between neoplastic and infiltrating cells. The cellular component of TME is variably composed of neoplastic epithelial cells, endothelial cells of the blood and lymphatic vessels, cancer-associated fibroblasts (CAFs), and cells of the innate [tumor-associated macrophages (TAM), tumor-associated neutrophils (TAN), dendritic cells (DC), natural killer (NK), and myeloid-derived suppressor cells (MDSC)] and adaptive immunity [tumor-infiltrating lymphocytes (TIL)][12,13] [Figure 1]. The structural component provides a dense and rigid scaffolding, which confers the characteristic desmoplasia to the tumor. This is composed of numerous and specific extracellular matrix proteins (see below). It is believed that the stroma does not have a simple passive function, but it actively participates in the intense communication between cells in the microenvironment and supports these interactions [Figure 2] and could be the target of therapeutic interventions [Table 1][2].
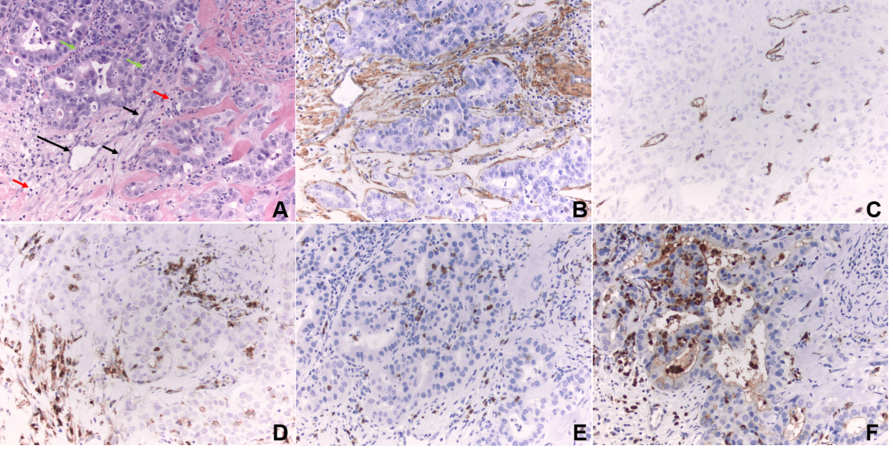
Figure 1. Tumor microenvironment. (A) Intrahepatic cholangiocarcinoma (hematoxylin and eosin stain, magnification 200×): black arrow, cancer-associated fibroblasts; green arrow, neutrophils; red arrow, tumor-infiltrating lymphocytes; long black arrow, microvessel. (B) Cancer-associated fibroblasts in the desmoplastic stroma are immunoreactive for α-smooth muscle actin, a biomarker of myofibroblast differentiation (immunohistochemistry, 200×). (C) Microvessels are highlighted by CD34 immunostain. (D) Tumor-infiltrating CD4 positive lymphocytes are highlighted by CD4 immunostain. (E) Tumor-infiltrating CD8 positive lymphocytes are highlighted by CD8 immunostain. (F) Tumor-associated neutrophils are highlighted by myeloperoxidase immunostain. (B-F) Brown stain indicates positive (mmunohistochemistry, magnification 200×).
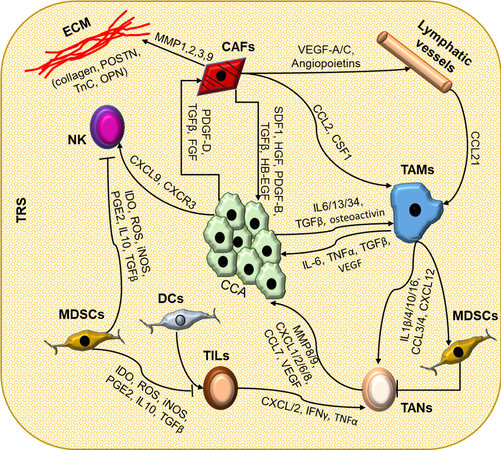
Figure 2. The complex interactions among the cell types composing TRS in CCA. In cholangiocarcinoma, neoplastic cells (CCA) are at the center of a complex interplay with a number of cell types that infiltrate the tumor microenvironment (TME) and a strongly modified extracellular matrix (ECM). Inflammatory cells and cancer-associated fibroblasts (CAFs), residing in close vicinity to CCA, can influence each other through the secretion of soluble mediators. CCA cells are able to recruit CAFs by secreting platelet-derived growth factor (PDGF)-D, transforming growth factor (TGF) β, and fibroblast growth factor (FGF); natural killer (NK) cells via the C-X-C motif ligand (CXCL) 9 - C-X-C motif chemokine receptor (CXCR) 3 axis; and cancer-associated macrophages (TAMs) through IL-6, IL-13, IL-34, TGFβ, and osteoactivin. In turn, these cells exert a trophic effect on neoplastic cells by secretion of soluble mediators. CAFs are also actively involved in the recruitment of lymphatic vessels and in the modifications of the ECM by secreting metalloproteinases (MMPs)-1, -2, -3, and -9, collagens, and other structural proteins such as osteopontin (OPN), tenascin C (TnC), and periostin (POSTN). The TME is also the site of an intense modulation of the innate and adaptive immune responses. Immune cells recruited into the tumor reactive stroma (TRS) influence each other in a difficult balance between immune surveillance and immune tolerance. Dendritic cells (DCs) stimulate the activation of tumor-infiltrating lymphocytes (TILs), while tumor-associated macrophages (TAMs) actively recruit tumor-associated neutrophils (TANs). Conversely, myeloid-derived suppressor cells (MDSC) inhibit the activity of immune cells, such as TILs, TANs, and NKs. The ability of each cancer to respond to immune therapy depends on the balance between these factors and the adaptation of the TME. Association of different targets (immune checkpoints and/or signaling targets) may be a pathway to therapeutic success.
Clinical trials targeting ligands and receptors shred by different cell type of the TME
| Ligands/receptors | Drugs | Clinical trial |
| VEGFRs, PDGFRs, c-MET | Lenvatinib | NCT03895970 NCT04211168 NCT04550624 |
| TT-00420 Pazopanib Bevacizumab | NCT04742959 NCT01855724 NCT00350753 NCT00426829 NCT00356889 NCT01007552 NCT00410956 NCT04164069 | |
| Apatinib | NCT03251443 NCT04454905 | |
| Tivozanib | NCT04645160 NCT05000294 | |
| FGFRs | Pemigatinib | NCT02924376 NCT03656536 |
| Erdafitinib E7090 Futibatinib BGJ398 | NCT02699606 NCT04238715 NCT02052778 NCT03773302 NCT02150967 | |
| RLY-4008 Derazantinib INCB062079 | NCT04526106 NCT01752920 NCT03144661 | |
| EGFR | A166 Varlitinib Regorafenib KSP/QRH dimer Erlotinib | NCT03602079 NCT02609958 NCT02053376 NCT04304781 NCT00955149 |
| Panitumumab | NCT00397384 NCT00033462 NCT01320254 | |
| TGFβ | M7824 | NCT04708067 NCT03833661 NCT04066491 |
| CSF1 | SNDX-6352 | NCT04301778 |
Matrix
The matrix in the normal liver is usually limited to the portal space and the space of Disse. The persistence of a chronic inflammatory stimulus induces a process of pathologic repair that, losing the fine regulation and self-limitation, leads to scarring. During the process of cholangiocarcinogenesis, there is also an aberrant deposition of both structural and non-structural ECM components, which creates a thick and stiff layer of ECM proteins around the neoplastic bile ducts. Aberrant deposition of ECM components is considered a pathological hallmark of cholangiocarcinomas, and it is believed to be responsible for the pronounced aggressiveness of CCA and its low response to current therapies[14,15]. CCA cells secrete a wide range of proteolytic enzymes, such as metalloproteinase (MMP)-2 and -9, that dismantle the laminin-rich basal membrane, allowing the tumoral cells to invade the peritumoral matrix reaching lymphatic and blood vessels and thus to dive into the blood and lymphatic stream and disseminate to distant metastatic loci. This mechanism is further facilitated by the interaction of CCA cells with other cell types that that are recruited into the TME and primed to express a prosecretory phenotype able to further modify the surrounding ECM[16,17]. In CCA, there is indeed an abnormal deposition of several matricellular proteins, including periostin (POSTN), tenascin C (TnC), and osteopontin (OPN). These proteins are associated with an increase in tumor size and lymphatic metastasis and reduced overall survival[3,12]. These non-structural ECM proteins have an important role during embryonic development but in adult life are only expressed during tissue remodeling and wound repair[18-20]. POSTN can act as both a promoter and a suppressor of cancer cell invasiveness, interacting with other ECM proteins, such as collagen types I and V, fibronectin, TnC, and heparin, and contribute to the activation of pathways of tissue remodeling, fibrogenesis, cell motility, angiogenesis, tumor invasiveness, and metastasis. POSTN and TnC cooperate to promote metastasis through the activation of the Wnt and Notch signaling. POSTN is also capable of recruiting TAM, making this matrix protein a potential curative target for drug development[21]. TnC binds many ECM proteins, including fibronectin, POSTN, collagen, fibrillin-2, and proteoglycans, probably playing a structural role and defining the stiffness of the microenvironment. In iCCA, TnC is selectively expressed by the tumor invasion front, and its expression correlates with adverse outcomes[22]. OPN is a glycosylated phosphoprotein produced by many cell types that is normally involved in bone remodeling, immune-regulation, inflammation, and vascularization. The role of OPN in CCA has been covered recently[23,24]. OPN is in fact an important regulator of the repair response based on the progenitor liver cells that produce it and with an autocrine loop stimulates their proliferation and migration, which eventually leads to the ductular reaction. It also regulates the interaction between these cells and stromal cells; for example, through the mediation with transforming growth factor (TGF) β, it allows the activation of the fibroblasts that produce the other proteins, as well as the migration of macrophages[23,24]. For further information on the tumor matrix, see the work of Fabris et al.[25].
Cancer-associated fibroblasts
CAFs, the most represented cell type in the TME, are cells of mesenchymal origin that lay embedded into the tumoral ECM and have a prominent role in the production of ECM components and the degradation of the native ECM[12,26]. CAFs are constitutively activated fibroblasts and express α-smooth muscle actin (α-SMA), cluster of differentiation (CD) 10, and S100 calcium binding protein A4 (S100A4). The origin of these cells is debated, as it has been proposed that CAFs may derive from resident portal fibroblasts, from hepatic stellate, from bone marrow-derived mesenchymal cells, and/or epithelial/tumor cells, via EMT[15] and are recruited around neoplastic biliary epithelia by the secretion of platelet derived growth factor (PDGF)-D[27,28]. Independently from the histogenesis, CAFs actively influence tumor progression thanks to a complex cross talk with the other components of the TME[29]. Once recruited, CAFs can stimulate tumor growth by secreting factors such as hepatocyte growth factor, TGFβ, PDGF-B, heparin-binding epidermal growth factor, and stromal cell-derived factor (SDF)-1[12,30,31]. Notably, SDF-1 is only weakly expressed by fibroblasts in the peritumoral area, but it is highly expressed and secreted by CAFs. Strong evidence for a role of CAFs in promoting CCA aggressiveness was demonstrated in a study in which a syngeneic rat model of CCA was treated with navitoclax (a small BH3-mimetic compound) to induce selective CAF depletion, thus suppressing tumor growth and improving host survival[26]. The relationship between CAF and other components of the TME, such as inflammatory cells and vessels, is very complex, and it is mediated by a series of growth factors [vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF)], TGFβ, cytokines, and chemokines [monocyte chemoattractant protein (MCP-1) and C-X-C motif ligand (CXCL) 12 and 14] and MMPs that promote tumor growth and spread, modifying the matrix, attracting the precursors of vascular and lymphatic vascular cells, and favoring an immunosuppressive microenvironment[15].
Endothelial cells
An important but still little studied element in the TME of CCA is its lymphatic vascular bed. Lymphatic metastatic spread occurs early during the course of CCA progression and often precludes curative surgical approaches. Notably, lymphatic endothelial cells are more represented in the TRS than blood endothelial cells and are localized in close proximity to CAFs[15]. Their involvement in the progression and metastatic spread of CCA seems to be due to the ability of both neoplastic and stromal cells to secrete lymphangiogenic growth factors (including VEGF-C, VEGF-D, and angiopoietins)[32,33]. Lymphatic vessels have large fenestrations that make them more permeable to the passage of immune cells and they secrete many chemokines [e.g., C-C motif ligand (CCL) 21] that promote the intravasation of macrophages and other inflammatory cells[32,34]. It has been shown that increased lymphatic density is associated with a worse prognosis and reduced disease-free and overall survival[35,36].
Innate immune cells
Tumor-associated macrophages
Two macrophage populations coexist in the TME of CCA: the liver resident macrophages (or Kupffer cells), and TAMs. On the contrary to the non-tumoral tissue, populated more by the M1 (or classically activated) subtype, which exert proinflammatory effects and defend the organism from invasion of pathogens, TAMs are mostly of the M2 (or alternatively activated) type and derive from circulating CD14+CD16+ monocytes that are usually involved in tissue repair and remodeling, angiogenesis, and matrix deposition[37]. TAMs play an active role in suppressing T cell activation and proliferation, in the promotion of angiogenesis, in the induction of tissue remodeling, and in stimulating apoptosis of M1 macrophages, which, however, contrast the neoplastic cells[38,39]. Whereas CAFs are spread over the entire tumor mass, TAMs are mainly located at the tumor invasive front, putatively recruited by neoplastic cells. CCA cells in fact secrete interleukins (IL) -6, -13, and -34, TGFβ, and osteoactivin, all molecules able to recruit monocytes and stimulate their M2 transdifferentiation[40,41]. Conversely, CAFs support TAM recruitment to the tumoral microenvironment by secreting CCL2 and colony stimulating factor 1 (CSF1)[42]. Many other secreted chemotactic mediators such as the cytokines IL-1β, IL-4, IL-10, and IL-16, CCL3, CCL4, and CXCL12 play a supportive role[16,43].
TAMs can act as a trophic cell population for neoplastic cells by secreting IL6, tumor necrosis factor (TNF) α, TGFβ, and VEGF and activating cyclooxygenase-2 and WNT/β-catenin signaling[44-47]. TAM-secreted VEGF can also stimulate neoangiogenesis, thus contributing to CCA metastasis[48]. Moreover, TAMs are the main source of metalloproteases, in particular MMP-9, which, by degrading the matrix, favors metastasis[39]. Finally, TAMs can attract immunosuppressive cells, such as TANs and MDSCs, through the secretion of different soluble mediators (IL-4, IL-8, IL-10, CCL2, CCL17, and CCL22) to generate an immunosuppressive environment that favors the malignant behavior of CCA[49].
Neutral killer cells
NK cells are a subpopulation of CD3-CD56+ lymphatic cells characterized by their ability to kill tumor- or virus-infected cells. Although NK cells are also known for their activity in recognizing and killing cancer cells, few studies have been performed in CCA. NKs can carry out their cytotoxicity through two pathways, one antigen-nonspecific, exploiting the release of enzymes such as perforin, proteases, and granzymes, and a direct one, through the activation of the Fas cell surface death receptor ligand (FasL)/TNF-related apoptosis-inducing ligand signal pathway[50]. The responsiveness of NK cells to the Fas/FasL pathway also has a drawback; in fact, a recent study demonstrated that, in vitro, iCCA cells express high levels of Fas and FasL, which induce apoptosis of NK cells, as an immune escape mechanism[51]. Conversely, the overexpression in CCA tumor cells of CXCL9, a ligand of C-X-C motif chemokine receptor (CXCR) 3, induces the recruitment of NK cells[52] that infiltrate the tumor and positively correlated with postoperative overall survival in a cohort of 70 patients[53]. Similarly, using a xenograft model in which iCCA-derived HuCCT-1 cells were xenotransplanted into non-immunocompetent NCr athymic nude mice, infusion of NK cells (SMT01) induced significant inhibition of tumor growth[54]. Furthermore, in vitro treatment of HuCCT-1 and NK co-cultures with cetuximab, an EGFR inhibitor, demonstrated a significant increase in NK cytolytic activity against tumor cells[55]. These data suggest a potential use of NK in the treatment of CCA. NK cells are characterized by the expression of natural killer group 2 member D (NKG2D) receptor, a receptor whose polymorphisms are linked to the susceptibility to cancer development[56]. A study on 82 patients with eCCA who underwent surgical resection showed that overexpression of the NKG2D receptor on NK cells and its ligands in the cancer cells correlated with a better patient prognosis[57].
Tumor-associated neutrophils
Despite their importance in the immune response[58], there are very few studies on the involvement of neutrophils in the pathogenesis of CCA. Similar to TAMs, TANs are divided into two subcategories, N1, which is endowed with antitumor function, and N2, which has protumoral activity[59]. Circulating neutrophils are recruited to the tumor site by cells of the TME, such as CAFs [that secrete granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), VEGF, and IL-1β], TAMs (IL-6 and IL-8), T lymphocytes (CXCL1, CXCL2, interferon γ, and TNFα)[60]. The neoplastic cells themselves secrete CXCL5, which induces neutrophils recruitment by activating the phosphatidylinositol 3-kinases/Akt and ERK1/2 pathways[61]. Once they reach the tumor site, TANs secrete a vast set of factors potentially involved in the biology of CCA, as mentioned above (such as MMP-8, MMP-9, CXCL1, CXCL2, CXCL6, CXCL8, CCL7, and VEGF)[16,58]. It has been shown that, in both iCCA and eCCA, the accumulation of TANs leads to a worse overall and disease-free survival of tumoral patients[62-64]. Unfortunately, only a few studies have evaluated the impact of TANs on CCA and they are mostly observational.
Dendritic cells
DCs are antigen-presenting cells and are usually found in small numbers in healthy tissues. It should be noted that the TME often has fewer DCs than the surrounding healthy tissue[41]. Similar to TANs, the studies regarding DCs in CCA are also rather sporadic. From a topographical point of view, mature DCs tend to accumulate on the tumor invasion front, while immature DCs are found in the tumor bulk[65]. A more recent study also demonstrated in a cohort of 350 patients with iCCAs that accumulation of DCs in the peritumoral tissue, but not in the CCA, is associated with a worse outcome[66]. A strong interaction between DCs and T cells has also been demonstrated; in fact, the inhibition of IL-10 and TGFβ receptors and the overexpression of cAMP-dependent protein kinase type I-alpha regulatory subunit (PRKAR1A) on DCs stimulates the antitumor activity of T cells against CCA[67,68]. Finally, a recent work with four different mouse models of iCCA showed that anti-CD40/PD-1 treatments, accompanied by gemcitabine/cisplatin, is able to activate the DC compartment and decrease tumor burden, an effect dramatically reduced by DC depletion[69].
Myeloid-derived suppressor cells
A family of immune cells only recently studied in CCA is that of the MDSCs[70]. MDSCs are a large group of myeloid-derived cells whose number expands in diseases such as cancer or chronic inflammation and can exert an immunosuppressive effect[70,71]. MDSCs inhibit the action of cytotoxic T cells and NK cells by producing indoleamine 2,3-dioxygenase, reactive oxygen species (ROS), inducible nitric oxide synthase, prostaglandin E2, arginase, and immunomodulatory cytokines such as IL-10 and TGFβ[70]. A study performed on a very small cohort of 17 patients with CCA showed an increase in circulating MDSCs compared to controls[72], an observation confirmed in a more recent study[73]. Zhang et al.[74], using an Mdr2-/- mouse in which iCCA was induced by hydrodynamic tail injection of plasmids known to favor its development (NICD + AKT and YAP + AKT), showed that accumulation of MDSCs favored tumor progression. Notably, gut sterilization was able to reduce MDSC recruitment. This observation demonstrates that hepatic recruitment of MDSCs can be modulated by the gut microbiota. Furthermore, using a different mouse model of iCCA [LSL-KrasG12D; Trp53Flox/Flox; Alb-Cre (KPPC) mouse], it was shown that neoplastic cells can recruit MDSCs via GM-CSF and that administering a blocking monoclonal antibody halts the recruitment of myeloid cells and decreases the growth and spread of the tumor[73]. In addition, the use of antibodies targeting a specific ApoE MDSC subset, coupled with TAM depletion, can increase the antitumoral effect of immune checkpoint blockade monotherapy[75].
Adaptive immunity
TILs are T cells that accumulate within the tumor stroma and counteract tumor development in an antigen-specific manner. The TIL population is composed by different cell types and includes CD4+ T cells (T helper or Th lymphocytes), CD4+CD25+ regulatory T cells (Tregs), CD8+ T cells (cytotoxic T lymphocytes), and CD20+ B lymphocytes. In general, in biliary tract tumors, the cells that mount the adaptive response tend to decrease during the process that goes from dysplasia to frank tumor and are also more numerous in eCCA than in iCCA[76]. The different components of TILs accumulate in specific areas of the tumor. While CD20+ B cells are present throughout the tumor, CD4+ cells accumulate in the peritumor area and CD8+ T cells on the tumor front[62,76,77]. Several studies showed that an enrichment in CD4+, CD8+, and CD20+ cells correlates with a better overall survival and lower recurrence rates in patients with both iCCA and eCCA[63,76,78-81].
The adaptive immune response is also finely tuned by a set of stimulatory or inhibitory molecules expressed on the membrane of T cells called immune checkpoints that mostly function to avoid autoimmune reactions against self-cells. The downside of this mechanism is that it can be used by cancer cells to avoid being recognized by immune surveillance, a mechanism known as immune escape[13,82]. The main stimulatory molecules belonging to immune checkpoints include CD27, CD28, CD40, CD137, CD278, OX40, and glucocorticoid-induced TNF receptor, while among the inhibitory ones the best known and studied are cytotoxic T lymphocyte antigen-4 (CTLA-4), programmed death-1 (PD-1) and its ligand PD-L1, lymphocyte activation gene-3, T-cell immunoglobulin, and mucin protein-3[83]. Pharmacological blockage of inhibitory checkpoint molecules is currently exploited for the development of anti-tumor drugs, and there are several anti PD-1, PD-L1, and CTLA-4 molecules approved for use or currently in clinical trials for the treatment of several solid cancers, including CCA[84-88]. Data regarding the predictive value of PD-1 and PD-L1 expression on patient outcome are discordant and conflicting. One recent meta-analysis of 11 studies with more than 1000 patients showed that the expression of PD-L1 by tumor cells does not correlate with a worse overall survival of the patients even after stratifying by type of CCA[89], but another meta-analysis provided opposite results[90]. As for PD-1, recent work has shown that, in iCCA, the increase in CD68+ macrophages and CD8+ T lymphocytes expressing PD-1 correlates with a worse postoperative survival[91]. Furthermore, in eCCA, high PD-1 expression appears to correlate with increased lymphatic metastases and lower patient survival[92]. Only one study evaluated the role of CTLA-4 expression as a prognostic indicator in CCA, demonstrating that, in eCCA, a high CTLA-4 H-score predicts better overall and disease-free survival[93].
Immunosuppressive tumor microenvironment
It is becoming clear that TME can generate an immunosuppressive environment and confer to the tumor cell a survival advantage by inducing tumor immune evasion. This is likely one of the main mechanisms responsible for the still disappointing results of immunotherapy in CCA[3,12]. For example, secretion of CCL2 by cells populating the TME, such as CAFs and tumor cells, leads to the enrichment of Tregs and of MDSCs. MDSCs in turn secrete ROS and other immunomodulatory compounds able to repress the activity of cytotoxic T cells (in particular CD8+ T cells) and NK cells. CAFs within the TRS of CCA secrete CXCL12, which may prevent migration of T cells[94].
The effects of Tregs and their contribution to the pathogenesis of CCA, as well as their relevance as a prognostic index of CCA, are still debated. Data from two cohorts of patient with eCCA indicate the presence of Tregs as an indicator of poor outcome[63], while other papers identify their over-representation as a positive prognostic factor[76]. From a biological point of view, Tregs secrete immunosuppressive mediators, such as IL10 and TGFβ, and further depress the antitumoral activity of CD8 T and NK cells[95,96]. A subset of Tregs, forkhead box P3 (FoxP3)+CD25+, bind IL-2, reducing the IL-2-mediated activation of the immune milieu[95]. The accumulation of FoxP3-positive Tregs is a distinctive trait of CCAs, showing worst outcome and greater tendency to lymphocyte metastasis[63,66]. Moreover, the accumulation of FoxP3+ Tregs is accompanied by an increase in expression of CTLA-4, also an indicator of poor outcome[97].
IMMUNOTHERAPEUTIC STRATEGIES TO TREAT CCA
To date, only pembrolizumab (an anti-PD-1 monoclonal antibody) is an approved treatment in CCA. Several other drugs and methods that exploit an immunotherapeutic approach are under evaluation. These studies are summarized in Table 2.
Clinical trials involving use of immune checkpoint inhibitors
| Clinical trial | Phase | Drugs | Target | Status | Results |
| NCT01876511 | 2 | Pembrolizumab | PD-1 | Completed | 1 patient in remission, 3 patients stable |
| NCT02628067 | 2 | Pembrolizumab | PD-1 | Recruiting | ORR: 5.8% |
| NCT02829918 | 2 | Nivolumab | PD-1 | Active, not recruiting | Failed |
| NCT02834013 | 2 | Nivolumab Ipilimumab | PD-1 CTLA-4 | Recruiting | ORR: 18% PFS: 2 months OS: 12 months |
| NCT02923934 | 2 | Nivolumab Ipilimumab | PD-1 CTLA-4 | Active, not recruiting | ORR: 23% OS: 5.7 months |
| NCT04634058 | 2 | anti-CTLA-4 abs anti-PD-1L abs | CTLA-4 PD-1L | Not yet recruiting | No data available |
| NCT04550624 | 2 | Pembrolizumab Lenvatinib Mesylate | PD-1 VEGFR1/2/3 | Recruiting | ORR: 25% DCR: 78% |
| NCT04550624 | 2 | Pembrolizumab Lenvatinib Mesylate | PD-1 VEGFR1/2/3 | Recruiting | No data available |
| NCT02443324 | 1a/1b | Pembrolizumab Ramucirumab | PD-1 VEGFR2 | Active, not recruiting | ORR: 4% Disease stabilization: 35% |
| NCT01853618 | 1/2 | Tremelimumab TACE | CTLA-4 | Completed | Partial response: 13% Disease stabilization: 31% |
| NCT01869166 | 1/2 | CART-EGFR | EGFR | Unknown | Partial response: 29% Disease stabilization: 57% |
| NCT01935843 | 1/2 | CART-HER-2 | HER-2 | Unknown | Partial response: 1 patients Disease stabilization: 5 patients |
Immune checkpoint inhibitors
Although the theoretical premises for the use of immune checkpoint inhibitors for the treatment of CCA are solid, the expectations were not fulfilled. The first clinical trials gave encouraging results, especially in patients with microsatellite instability due to mismatch repair deficiency. In these patients, the use of pembrolizumab, a humanized anti-PD-1 antibody (NCT01876511), gave a good response, with one patient in remission and three with stable disease[98]. Moreover, the analysis of five single-arm open-label clinical trials (KEYNOTE-012, -016, -028, -158, and -164) demonstrated an overall response rate (ORR) of 78%. However, these results were not confirmed in a larger study on 104 patients, KEYNOTE-158 (NCT02628067), in which the ORR was 5.8%. A clinical trial using nivolumab, an anti-PD-1 antibody (NCT02829918), failed to show favorable results[99]. CTLA-4 inhibitors are little studied in CCA and mostly in combined treatments in the hope to increase their efficacy. Some clinical trials have studied the combination of treatment with nivolumab and ipilimumab, an anti-CTLA-4 antibody (NCT02834013 and NCT02923934), in advanced solid tumors. Treatment of 39 patients showed an ORR of 23% and a disease control rate (DCR) of 44%, but with an overall survival (OS) of only 5.7 months[100]. The use of anti-CTLA-4 antibodies with anti-PD-1L antibodies is currently being evaluated in a phase II clinical trial (NCT04634058) whose results are not yet available. Pembrolizumab was also used in combination with levatinib, a tyrosine kinase inhibitor active on VEGFR1, VEGFR2, and VEGFR3, giving an ORR of 25% and a DCR of 78%[101]. Currently, a clinical trial for advanced CCA is in the recruitment phase (NCT04550624). Another phase I study, KEYNOTE-098 (NCT02443324), used pembrolizumab in combination with ramucirumab, a monoclonal antibody against VEGFR2, but the response rate was 4% with disease stabilization in 35% of cases[102]. Tremelinumab, an anti CTLA-4 inhibitor, is being evaluated in a phase I clinical trial in combination with radiofrequency ablation (NCT01853618). Among 20 patients, 13% showed a partial response and 31% a stabilization of the disease. A recent study divided iCCA into four subtypes based on the differences of cell components of the TME (immune desert, immunogenic, myeloid, and mesenchymal). This stratification was proposed to better allocate patients to a more correct therapeutic intervention. In particular, the authors suggested treating with immune checkpoints inhibitor only the patients belonging to the immunogenic subtype, to maximize the potential anti-tumorigenic effects of the compounds[103].
Other immune strategies
These are mostly in the experimental or preclinical stage. The rationale behind the study of cancer vaccines is to identify proteins specifically expressed by cancer cells that could be recognized and destroyed by the immune cells. Few studies, usually composed of small cohorts, have been conducted in CCA. Identifying tumor-related proteins that can be specifically targeted by the vaccine is a daunting task. Studies conducted using Wilms’ tumor 1 (WT1) and Mucin-1 (MUC-1), proteins expressed by 70%-80% of iCCAs, as targets gave unsatisfactory results[104,105]. Similar disappointing results were obtained by co-treatment with gemcitabine and WT1 vaccine in four CCA patients[106].
Another strategy to manipulate the immune system and train it to search and destroy tumoral cells is the use of adoptive cell therapies (ACTs), such as the autologous infusion of TILs or engineered T cells expressing with chimeric antigen receptor (CAR) or T cell receptors. The use of these therapeutic approaches in CCA are limited to case reports or small clinical trials, however the preliminary results are promising[107-109]. More recently, small phase I clinical trials have used CART technology. In one study, 14 patients with CCA were treated with infusions of EGFR-targeting CART cells (NCT01869166)[110], while in a second clinical trial (NCT01935843) patients were infused with specific CARTs for human epidermal growth factor receptor 2[111]. Both trials resulted in an increase in disease-free survival with respect to patients treated with chemotherapy drug only. Several recent studies used new and improved (fourth-generation) CARTs that target proteins highly expressed by CCA, such as CD133[112], MUC-1[113], and integrin αVβ6[114]; these have been shown to be extremely efficient both in the expansion of the CART population and in the lysis of different CCA cell lines, in vitro.
CONCLUSION
Despite the increased interest of the scientific community, treatment of cholangiocarcinomas remains an unmet need. Currently, a therapeutic option that has shown good efficacy is liver transplantation, which, only in very recent years, is being proposed for patients with iCCA and pCCA without distant metastases and with an early disease[115]. Aside from this option, there are no other truly effective treatment options. The genetic and phenotypic diversity that characterizes this family of rare cancers has negatively affected the progress in this field. An important strategy to overcome this impasse is to better understand the mechanisms that mediate the crosstalk between tumor cells, the variety of TME cells described above, and the components of the ECM. In recent years, several studies have aimed at classifying CCA on the basis of not only of their immunohistochemical and anatomopathological phenotype but also their genetics, signal pathways, and actionable targets. A seminal work by Sia et al.[116] classifies iCCAs into proliferation class and inflammation class, based on specific signaling pathways and mutations. Notably, proliferation class shows a worse outcome with respect to the inflammatory one. Using a similar approach, mixed hepatocellular CCA (HCC/CCA)[117] and eCCA[118] have also been subcategorized. Using a molecular approach, eCCAs were classified in metabolic, mesenchymal, proliferative, and immune classes, all characterized in terms of mutations and modulation of different actionable targets. Furthermore, the development of single-cell RNA sequencing now allows analysis of the discrete cell populations in the TRS, including CAFs[119] and immune response cells[120], and to study in more detail the crosstalk between these cell milieus[121]. Finally, a recent study used nano liquid chromatography coupled to matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF/TOF) analysis to study the composition of the matrix (or matrisome) in CCA[122]. Using this approach, these authors demonstrated that the aberrant deposition of collagen type III alpha 1 chain directly stimulates the migration of neoplastic cells[122]. Such studies will not only lead to a better understanding of the mechanisms of development and growth of the CCA but also open new avenues for a better allocation of patients to the most appropriate treatments. For example, when planning combination treatment with immune checkpoint inhibitors and other molecularly targeted drugs, a personalized approach based on genetic mutations and signaling pathways deregulated in specific CCA subclasses may confer a therapeutic advantage.
DECLARATIONS
Authors’ contributionsMade substantial contributions to conception and writing of the manuscript: Cadamuro M, Fabris L, Zhang X, Strazzabosco M
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis project was supported in part by the Yale Liver Center award NIH P30 DK034989 Clinical -Translational core.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Razumilava N, Gores GJ. Classification, diagnosis, and management of cholangiocarcinoma. Clin Gastroenterol Hepatol 2013;11:13-21.e1; quiz e3.
3. Banales JM, Marin JJG, Lamarca A, et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol 2020;17:557-88.
4. McLean L, Patel T. Racial and ethnic variations in the epidemiology of intrahepatic cholangiocarcinoma in the United States. Liver Int 2006;26:1047-53.
6. Choi J, Ghoz HM, Peeraphatdit T, et al. Aspirin use and the risk of cholangiocarcinoma. Hepatology 2016;64:785-96.
7. Zhang H, Zhu B, Zhang H, Liang J, Zeng W. HBV Infection status and the risk of cholangiocarcinoma in Asia: a meta-analysis. Biomed Res Int 2016;2016:3417976.
8. Clements O, Eliahoo J, Kim JU, Taylor-Robinson SD, Khan SA. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma: a systematic review and meta-analysis. J Hepatol 2020;72:95-103.
9. Limpaiboon T, Krissadarak K, Sripa B, et al. Microsatellite alterations in liver fluke related cholangiocarcinoma are associated with poor prognosis. Cancer Letters 2002;181:215-22.
10. Moeini A, Sia D, Bardeesy N, Mazzaferro V, Llovet JM. Molecular pathogenesis and targeted therapies for intrahepatic cholangiocarcinoma. Clin Cancer Res 2016;22:291-300.
11. Boerner T, Drill E, Pak LM, et al. Genetic determinants of outcome in intrahepatic cholangiocarcinoma. Hepatology 2021;74:1429-44.
12. Fabris L, Perugorria MJ, Mertens J, et al. The tumour microenvironment and immune milieu of cholangiocarcinoma. Liver Int 2019;39 Suppl 1:63-78.
13. Rimassa L, Personeni N, Aghemo A, Lleo A. The immune milieu of cholangiocarcinoma: from molecular pathogenesis to precision medicine. J Autoimmun 2019;100:17-26.
14. Lee JI, Campbell JS. Role of desmoplasia in cholangiocarcinoma and hepatocellular carcinoma. J Hepatol 2014;61:432-4.
15. Cadamuro M, Stecca T, Brivio S, et al. The deleterious interplay between tumor epithelia and stroma in cholangiocarcinoma. Biochim Biophys Acta Mol Basis Dis 2018;1864:1435-43.
16. Høgdall D, Lewinska M, Andersen JB. Desmoplastic tumor microenvironment and immunotherapy in cholangiocarcinoma. Trends Cancer 2018;4:239-55.
17. Sirica AE, Strazzabosco M, Cadamuro M. Intrahepatic cholangiocarcinoma: morpho-molecular pathology, tumor reactive microenvironment, and malignant progression. Adv Cancer Res 2021;149:321-87.
18. Conway SJ, Izuhara K, Kudo Y, et al. The role of periostin in tissue remodeling across health and disease. Cell Mol Life Sci 2014;71:1279-88.
19. Terada T, Kitamura Y, Nakanuma Y. Normal and abnormal development of the human intrahepatic biliary system: a review. Tohoku J Exp Med 1997;181:19-32.
20. Brown LF, Berse B, Van de Water L, et al. Expression and distribution of osteopontin in human tissues: widespread association with luminal epithelial surfaces. Mol Biol Cell 1992;3:1169-80.
21. Zeng J, Liu Z, Sun S, et al. Tumor-associated macrophages recruited by periostin in intrahepatic cholangiocarcinoma stem cells. Oncol Lett 2018;15:8681-6.
22. Aishima S, Taguchi K, Terashi T, Matsuura S, Shimada M, Tsuneyoshi M. Tenascin expression at the invasive front is associated with poor prognosis in intrahepatic cholangiocarcinoma. Mod Pathol 2003;16:1019-27.
23. Sulpice L, Rayar M, Desille M, et al. Molecular profiling of stroma identifies osteopontin as an independent predictor of poor prognosis in intrahepatic cholangiocarcinoma. Hepatology 2013;58:1992-2000.
24. Liu Y, Cao L, Chen R, et al. Osteopontin promotes hepatic progenitor cell expansion and tumorigenicity via activation of β-catenin in mice. Stem Cells 2015;33:3569-80.
25. Fabris L, Cadamuro M, Cagnin S, Strazzabosco M, Gores GJ. Liver matrix in benign and malignant biliary tract disease. Semin Liver Dis 2020;40:282-97.
26. Mertens JC, Fingas CD, Christensen JD, et al. Therapeutic effects of deleting cancer-associated fibroblasts in cholangiocarcinoma. Cancer Res 2013;73:897-907.
27. Cadamuro M, Nardo G, Indraccolo S, et al. Platelet-derived growth factor-D and Rho GTPases regulate recruitment of cancer-associated fibroblasts in cholangiocarcinoma. Hepatology 2013;58:1042-53.
28. Vaquero J, Aoudjehane L, Fouassier L. Cancer-associated fibroblasts in cholangiocarcinoma. Curr Opin Gastroenterol 2020;36:63-9.
29. Brivio S, Cadamuro M, Strazzabosco M, Fabris L. Tumor reactive stroma in cholangiocarcinoma: the fuel behind cancer aggressiveness. World J Hepatol 2017;9:455-68.
30. Kuperwasser C, Chavarria T, Wu M, et al. Reconstruction of functionally normal and malignant human breast tissues in mice. Proc Natl Acad Sci U S A 2004;101:4966-71.
31. Gascard P, Tlsty TD. Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev 2016;30:1002-19.
32. Stacker SA, Williams SP, Karnezis T, Shayan R, Fox SB, Achen MG. Lymphangiogenesis and lymphatic vessel remodelling in cancer. Nat Rev Cancer 2014;14:159-72.
33. Cadamuro M, Brivio S, Mertens J, et al. Platelet-derived growth factor-D enables liver myofibroblasts to promote tumor lymphangiogenesis in cholangiocarcinoma. J Hepatol 2019;70:700-9.
34. Kim H, Kataru RP, Koh GY. Inflammation-associated lymphangiogenesis: a double-edged sword? J Clin Invest 2014;124:936-42.
35. Thelen A, Scholz A, Benckert C, et al. Microvessel density correlates with lymph node metastases and prognosis in hilar cholangiocarcinoma. J Gastroenterol 2008;43:959-66.
36. Thelen A, Scholz A, Benckert C, et al. Tumor-associated lymphangiogenesis correlates with lymph node metastases and prognosis in hilar cholangiocarcinoma. Ann Surg Oncol 2008;15:791-9.
37. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol 2017;14:399-416.
38. Duluc D, Delneste Y, Tan F, et al. Tumor-associated leukemia inhibitory factor and IL-6 skew monocyte differentiation into tumor-associated macrophage-like cells. Blood 2007;110:4319-30.
39. Rőszer T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflamm 2015;2015:816460.
40. Hasita H, Komohara Y, Okabe H, et al. Significance of alternatively activated macrophages in patients with intrahepatic cholangiocarcinoma. Cancer Sci 2010;101:1913-9.
41. Paillet J, Kroemer G, Pol JG. Immune contexture of cholangiocarcinoma. Curr Opin Gastroenterol 2020;36:70-6.
42. Dalen FJ, van Stevendaal MHME, Fennemann FL, Verdoes M, Ilina O. Molecular repolarisation of tumour-associated macrophages. Molecules 2018;24:9.
43. Pathria P, Louis TL, Varner JA. Targeting tumor-associated macrophages in cancer. Trends Immunol 2019;40:310-27.
44. Loilome W, Bungkanjana P, Techasen A, et al. Activated macrophages promote Wnt/β-catenin signaling in cholangiocarcinoma cells. Tumour Biol 2014;35:5357-67.
45. Boulter L, Guest RV, Kendall TJ, et al. WNT signaling drives cholangiocarcinoma growth and can be pharmacologically inhibited. J Clin Invest 2015;125:1269-85.
46. Henze AT, Mazzone M. The impact of hypoxia on tumor-associated macrophages. J Clin Invest 2016;126:3672-9.
47. Liu S, Jiang J, Huang L, et al. iNOS is associated with tumorigenicity as an independent prognosticator in human intrahepatic cholangiocarcinoma. Cancer Manag Res 2019;11:8005-22.
48. Thanee M, Loilome W, Techasen A, et al. Quantitative changes in tumor-associated M2 macrophages characterize cholangiocarcinoma and their association with metastasis. Asian Pac J Cancer Prev 2015;16:3043-50.
49. Zhou M, Wang C, Lu S, et al. Tumor-associated macrophages in cholangiocarcinoma: complex interplay and potential therapeutic target. EBioMedicine 2021;67:103375.
50. Chiossone L, Dumas PY, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol 2018;18:671-88.
51. Carnevale G, Carpino G, Cardinale V, et al. Activation of Fas/FasL pathway and the role of c-FLIP in primary culture of human cholangiocarcinoma cells. Sci Rep 2017;7:14419.
52. Wendel M, Galani IE, Suri-Payer E, Cerwenka A. Natural killer cell accumulation in tumors is dependent on IFN-gamma and CXCR3 ligands. Cancer Res 2008;68:8437-45.
53. Fukuda Y, Asaoka T, Eguchi H, et al. Endogenous CXCL9 affects prognosis by regulating tumor-infiltrating natural killer cells in intrahepatic cholangiocarcinoma. Cancer Sci 2020;111:323-33.
54. Jung IH, Kim DH, Yoo DK, et al. In vivo study of natural killer (NK) cell cytotoxicity against cholangiocarcinoma in a nude mouse model. In Vivo 2018;32:771-81.
55. Morisaki T, Umebayashi M, Kiyota A, et al. Combining cetuximab with killer lymphocytes synergistically inhibits human cholangiocarcinoma cells in vitro. Anticancer Res 2012;32:2249-56.
56. Melum E, Karlsen TH, Schrumpf E, et al. Cholangiocarcinoma in primary sclerosing cholangitis is associated with NKG2D polymorphisms. Hepatology 2008;47:90-6.
57. Tsukagoshi M, Wada S, Yokobori T, et al. Overexpression of natural killer group 2 member D ligands predicts favorable prognosis in cholangiocarcinoma. Cancer Sci 2016;107:116-22.
58. Fridlender ZG, Albelda SM. Tumor-associated neutrophils: friend or foe? Carcinogenesis 2012;33:949-55.
59. Masucci MT, Minopoli M, Carriero MV. Tumor associated neutrophils. Their role in tumorigenesis, metastasis, prognosis and therapy. Front Oncol 2019;9:1146.
60. Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res 2014;2014:149185.
61. Zhou SL, Dai Z, Zhou ZJ, et al. CXCL5 contributes to tumor metastasis and recurrence of intrahepatic cholangiocarcinoma by recruiting infiltrative intratumoral neutrophils. Carcinogenesis 2014;35:597-605.
62. Gu FM, Gao Q, Shi GM, et al. Intratumoral IL-17+ cells and neutrophils show strong prognostic significance in intrahepatic cholangiocarcinoma. Ann Surg Oncol 2012;19:2506-14.
63. Kitano Y, Okabe H, Yamashita YI, et al. Tumour-infiltrating inflammatory and immune cells in patients with extrahepatic cholangiocarcinoma. Br J Cancer 2018;118:171-80.
64. Zhou Z, Wang P, Sun R, et al. Tumor-associated neutrophils and macrophages interaction contributes to intrahepatic cholangiocarcinoma progression by activating STAT3. J Immunother Cancer 2021;9:e001946.
65. Takagi S, Miyagawa S, Ichikawa E, et al. Dendritic cells, T-cell infiltration, and Grp94 expression in cholangiocellular carcinoma. Hum Pathol 2004;35:881-6.
66. Hu ZQ, Zhou ZJ, Luo CB, et al. Peritumoral plasmacytoid dendritic cells predict a poor prognosis for intrahepatic cholangiocarcinoma after curative resection. Cancer Cell Int 2020;20:582.
67. Panya A, Thepmalee C, Sawasdee N, et al. Cytotoxic activity of effector T cells against cholangiocarcinoma is enhanced by self-differentiated monocyte-derived dendritic cells. Cancer Immunol Immunother 2018;67:1579-88.
68. Thepmalee C, Panya A, Junking M, Chieochansin T, Yenchitsomanus PT. Inhibition of IL-10 and TGF-β receptors on dendritic cells enhances activation of effector T-cells to kill cholangiocarcinoma cells. Hum Vaccin Immunother 2018;14:1423-31.
69. Diggs LP, Ruf B, Ma C, et al. CD40-mediated immune cell activation enhances response to anti-PD-1 in murine intrahepatic cholangiocarcinoma. J Hepatol 2021;74:1145-54.
70. Ma C, Zhang Q, Greten TF. MDSCs in liver cancer: a critical tumor-promoting player and a potential therapeutic target. Cell Immunol 2021;361:104295.
71. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol 2018;19:108-19.
72. Xu X, Hu J, Wang M, et al. Circulating myeloid-derived suppressor cells in patients with pancreatic cancer. Hepatobiliary Pancreat Dis Int 2016;15:099-105.
73. Ruffolo LI, Jackson KM, Kuhlers PC, et al. GM-CSF drives myelopoiesis, recruitment and polarisation of tumour-associated macrophages in cholangiocarcinoma and systemic blockade facilitates antitumour immunity. Gut 2021; doi: 10.1136/gutjnl-2021-324109.
74. Zhang Q, Ma C, Duan Y, et al. Gut microbiome directs hepatocytes to recruit MDSCs and promote cholangiocarcinoma. Cancer Discov 2021;11:1248-67.
75. Loeuillard E, Yang J, Buckarma E, et al. Targeting tumor-associated macrophages and granulocytic myeloid-derived suppressor cells augments PD-1 blockade in cholangiocarcinoma. J Clin Invest 2020;130:5380-96.
76. Goeppert B, Frauenschuh L, Zucknick M, et al. Prognostic impact of tumour-infiltrating immune cells on biliary tract cancer. Br J Cancer 2013;109:2665-74.
77. Kasper HU, Drebber U, Stippel DL, Dienes HP, Gillessen A. Liver tumor infiltrating lymphocytes: comparison of hepatocellular and cholangiolar carcinoma. World J Gastroenterol 2009;15:5053-7.
78. Vigano L, Soldani C, Franceschini B, et al. Tumor-infiltrating lymphocytes and macrophages in intrahepatic cholangiocellular carcinoma. Impact on prognosis after complete surgery. J Gastrointest Surg 2019;23:2216-24.
79. Yugawa K, Itoh S, Yoshizumi T, et al. Prognostic impact of tumor microvessels in intrahepatic cholangiocarcinoma: association with tumor-infiltrating lymphocytes. Mod Pathol 2021;34:798-807.
80. Huang YH, Zhang CZ, Huang QS, et al. Clinicopathologic features, tumor immune microenvironment and genomic landscape of Epstein-Barr virus-associated intrahepatic cholangiocarcinoma. J Hepatol 2021;74:838-49.
81. Miyazaki K, Morine Y, Imura S, et al. Preoperative lymphocyte/C-reactive protein ratio and its correlation with CD8+ tumor-infiltrating lymphocytes as a predictor of prognosis after resection of intrahepatic cholangiocarcinoma. Surg Today 2021;51:1985-95.
82. Xu F, Jin T, Zhu Y, Dai C. Immune checkpoint therapy in liver cancer. J Exp Clin Cancer Res 2018;37:110.
84. Gok Yavuz B, Hasanov E, Lee SS, et al. Current landscape and future directions of biomarkers for immunotherapy in hepatocellular carcinoma. J Hepatocell Carcinoma 2021;8:1195-207.
85. Shek D, Akhuba L, Carlino MS, et al. Immune-checkpoint inhibitors for metastatic colorectal cancer: a systematic review of clinical outcomes. Cancers (Basel) 2021;13:4345.
86. Xiong W, Zhao Y, Du H, Guo X. Current status of immune checkpoint inhibitor immunotherapy for lung cancer. Front Oncol 2021;11:704336.
87. Jalalvand M, Darbeheshti F, Rezaei N. Immune checkpoint inhibitors: review of the existing evidence and challenges in breast cancer. Immunotherapy 2021;13:587-603.
88. Zeng FL, Chen JF. Application of immune checkpoint inhibitors in the treatment of cholangiocarcinoma. Technol Cancer Res Treat 2021;20:15330338211039952.
89. Xu G, Sun L, Li Y, et al. The clinicopathological and prognostic value of PD-L1 expression in cholangiocarcinoma: a meta-analysis. Front Oncol 2019;9:897.
90. Xie Q, Wang L, Zheng S. Prognostic and clinicopathological significance of PD-L1 in patients with cholangiocarcinoma: a meta-analysis. Dis Markers 2020;2020:1817931.
91. Tian L, Ma J, Ma L, et al. PD-1/PD-L1 expression profiles within intrahepatic cholangiocarcinoma predict clinical outcome. World J Surg Oncol 2020;18:303.
92. Ma K, Wei X, Dong D, Wu Y, Geng Q, Li E. PD-L1 and PD-1 expression correlate with prognosis in extrahepatic cholangiocarcinoma. Oncol Lett 2017;14:250-6.
93. Lim YJ, Koh J, Kim K, et al. Clinical implications of cytotoxic T lymphocyte antigen-4 expression on tumor cells and tumor-infiltrating lymphocytes in extrahepatic bile duct cancer patients undergoing surgery plus adjuvant chemoradiotherapy. Target Oncol 2017;12:211-8.
94. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015;348:74-80.
95. Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of treg-mediated T cell suppression. Front Immunol 2012;3:51.
96. Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol 2012;12:239-52.
97. Ghidini M, Cascione L, Carotenuto P, et al. Characterisation of the immune-related transcriptome in resected biliary tract cancers. Eur J Cancer 2017;86:158-65.
98. Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409-13.
99. Kim RD, Chung V, Alese OB, et al. A phase 2 multi-institutional study of nivolumab for patients with advanced refractory biliary tract cancer. JAMA Oncol 2020;6:888-94.
100. Klein O, Kee D, Nagrial A, et al. Evaluation of combination nivolumab and ipilimumab immunotherapy in patients with advanced biliary tract cancers: subgroup analysis of a phase 2 nonrandomized clinical trial. JAMA Oncol 2020;6:1405-9.
101. Lin J, Yang X, Long J, et al. Pembrolizumab combined with lenvatinib as non-first-line therapy in patients with refractory biliary tract carcinoma. Hepatobiliary Surg Nutr 2020;9:414-24.
102. Arkenau HT, Martin-Liberal J, Calvo E, et al. Ramucirumab plus pembrolizumab in patients with previously treated advanced or metastatic biliary tract cancer: nonrandomized, open-label, phase I trial (JVDF). Oncologist 2018;23:1407-e136.
103. Job S, Rapoud D, Dos Santos A, et al. Identification of four immune subtypes characterized by distinct composition and functions of tumor microenvironment in intrahepatic cholangiocarcinoma. Hepatology 2020;72:965-81.
104. Yamamoto K, Ueno T, Kawaoka T, et al. MUC1 peptide vaccination in patients with advanced pancreas or biliary tract cancer. Anticancer Res 2005;25:3575-9.
105. Lepisto AJ, Moser AJ, Zeh H, et al. A phase I/II study of a MUC1 peptide pulsed autologous dendritic cell vaccine as adjuvant therapy in patients with resected pancreatic and biliary tumors. Cancer Ther 2008;6:955-64.
106. Kaida M, Morita-Hoshi Y, Soeda A, et al. Phase 1 trial of Wilms tumor 1 (WT1) peptide vaccine and gemcitabine combination therapy in patients with advanced pancreatic or biliary tract cancer. J Immunother 2011;34:92-9.
107. Higuchi R, Yamamoto M, Hatori T, Shimizu K, Imai K, Takasaki K. Intrahepatic cholangiocarcinoma with lymph node metastasis successfully treated by immunotherapy with CD3-activated T cells and dendritic cells after surgery: report of a case. Surg Today 2006;36:559-62.
108. Tran E, Turcotte S, Gros A, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014;344:641-5.
109. Feng KC, Guo YL, Liu Y, et al. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J Hematol Oncol 2017;10:4.
110. Guo Y, Feng K, Liu Y, et al. Phase I study of chimeric antigen receptor-modified T cells in patients with EGFR-positive advanced biliary tract cancers. Clin Cancer Res 2018;24:1277-86.
111. Feng K, Liu Y, Guo Y, et al. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell 2018;9:838-47.
112. Sangsuwannukul T, Supimon K, Sujjitjoon J, et al. Anti-tumour effect of the fourth-generation chimeric antigen receptor T cells targeting CD133 against cholangiocarcinoma cells. Int Immunopharmacol 2020;89:107069.
113. Supimon K, Sangsuwannukul T, Sujjitjoon J, et al. Anti-mucin 1 chimeric antigen receptor T cells for adoptive T cell therapy of cholangiocarcinoma. Sci Rep 2021;11:6276.
114. Phanthaphol N, Somboonpatarakun C, Suwanchiwasiri K, et al. Chimeric antigen receptor T cells targeting integrin αvβ6 expressed on cholangiocarcinoma cells. Front Oncol 2021;11:657868.
115. Cillo U, Fondevila C, Donadon M, et al. Surgery for cholangiocarcinoma. Liver Int 2019;39 Suppl 1:143-55.
116. Sia D, Hoshida Y, Villanueva A, et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 2013;144:829-40.
117. Moeini A, Sia D, Zhang Z, et al. Mixed hepatocellular cholangiocarcinoma tumors: Cholangiolocellular carcinoma is a distinct molecular entity. J Hepatol 2017;66:952-61.
118. Montal R, Sia D, Montironi C, et al. Molecular classification and therapeutic targets in extrahepatic cholangiocarcinoma. J Hepatol 2020;73:315-27.
119. Affo S, Nair A, Brundu F, et al. Promotion of cholangiocarcinoma growth by diverse cancer-associated fibroblast subpopulations. Cancer Cell 2021;39:866-882.e11.
120. Su M, Qiao KY, Xie XL, et al. Development of a prognostic signature based on single-cell RNA sequencing data of immune cells in intrahepatic cholangiocarcinoma. Front Genet 2020;11:615680.
121. Zhang M, Yang H, Wan L, et al. Single-cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J Hepatol 2020;73:1118-30.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Cadamuro M, Fabris L, Zhang X, Strazzabosco M. Tumor microenvironment and immunology of cholangiocarcinoma. Hepatoma Res 2022;8:11. http://dx.doi.org/10.20517/2394-5079.2021.140
AMA Style
Cadamuro M, Fabris L, Zhang X, Strazzabosco M. Tumor microenvironment and immunology of cholangiocarcinoma. Hepatoma Research. 2022; 8: 11. http://dx.doi.org/10.20517/2394-5079.2021.140
Chicago/Turabian Style
Cadamuro, Massimiliano, Luca Fabris, Xuchen Zhang, Mario Strazzabosco. 2022. "Tumor microenvironment and immunology of cholangiocarcinoma" Hepatoma Research. 8: 11. http://dx.doi.org/10.20517/2394-5079.2021.140
ACS Style
Cadamuro, M.; Fabris L.; Zhang X.; Strazzabosco M. Tumor microenvironment and immunology of cholangiocarcinoma. Hepatoma. Res. 2022, 8, 11. http://dx.doi.org/10.20517/2394-5079.2021.140
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 25 clicks
Cite This Article 25 clicks

 Like This Article 18
likes
Like This Article 18
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.