
Role of CD4+ T-cells in the pathology of non-alcoholic fatty liver disease and related diseases

Abstract
Non-alcoholic fatty liver disease (NAFLD), which is considered a liver phenotype of metabolic diseases, is becoming a major cause of chronic liver disease. Multiple factors influence and interact with each other in a complex manner to form this pathological condition. As evidenced by low-grade chronic inflammation in obesity, which is a basic pathological feature of NAFLD, immune cell infiltration can occur in various organs, and immune cell infiltration into the liver plays an important role in the development of steatohepatitis. In recent years, an increasing number of reports indicate the involvement of innate immunity and adaptive immunity in the pathogenesis of NAFLD. CD4+ T-cells, which serve as an essential and complex element of the immune system and major regulators of host health and disease, are differentiated into functional T helper 1 (Th1), Th2, Th9, Th17, Th22, T follicular helper, and regulatory T-cells upon antigen stimulation in a special cytokine environment. In NAFLD patients, various pathological conditions such as obesity, diabetes, dyslipidemia, and adipose tissue inflammation coexist. Hence, T-cells can be affected by each of these pathological conditions. This review covers and discusses the reports on NAFLD and its associated pathologies as well as their effects on CD4+ T-cells.
Keywords
INTRODUCTION
The number of patients with non-alcoholic fatty liver disease (NAFLD) has increased along with an increase in the obese population, exhibiting an approximate frequency of 25% worldwide[1]. NAFLD is associated with metabolic syndrome[2,3] as well as diseases associated with lifestyle such as cardiovascular disease[4], type 2 diabetes (T2DM)[5], and dyslipidemia[6]. It is a major cause of chronic liver disease and becoming an increasingly important cause of hepatocellular carcinoma (HCC)[7]. Furthermore, it is becoming a common indication for liver transplantation in the United States[8]. It also has a large impact on social costs, which are expressed as medical expenses[9,10]. Therefore, NAFLD is considered an important public health disease.
NAFLD includes various phases such as simple steatosis, steatohepatitis, liver fibrosis, cirrhosis, and HCC[11]. Multiple factors such as oxidative stress, adipokine, lipid peroxidation, insulin resistance, diet, intestinal bacteria, and genetic factors associate with each other, resulting in the pathology of the disease[12]. The liver is the central organ of glucose/protein/lipid metabolism that maintains homeostasis in the body by sensing biological stress, inflammation, overnutrition, organ-derived humoral factors, and dynamically changing gene expression[13]. In contrast, approximately 15% of the cells that compose the liver are immune cells[14]. The liver also plays an important role as an immune organ owing to the anatomical characteristics that require immediate reaction to foreign antigens derived from the portal blood flow[15-17]. As such, hepatocyte injury and the influence of immune cells in the surrounding microenvironment must be considered in liver disease.
In general, innate immune responses dominate the initial response to liver injury; however, adaptive immune responses play a vital role in the persistence of inflammation for chronic liver injury[18]. Liver inflammation is considered to increase the recruitment of lymphocytes to the liver, and the type and distribution of these infiltrating cells determine the nature of inflammation[19].
Recently, there have been several reports on the involvement of adaptive immunity in NAFLD pathology[20,21]. Antigens from oxidative stress trigger the adaptive immune response[22]. Additionally, the in vivo cluster of differentiation for positive (CD4+) T-cell depletion reduces inflammation and fibrosis in the liver of immunocompromised mice transplanted with human immune cells that have been fed a high-fat, high-calorie diet[23]. These studies provide evidence that CD4+ T-cells play an important role in the pathology of NAFLD.
A review discussing the relationship between obesity and T2DM in NAFLD[20] and a review discussing the role of T-cells and B-cells in inflammation and fibrosis in NAFLD[21] have previously been published. However, the regulatory mechanism of T-cells with effector function is complex, and there is an incomplete understanding of the significance and role of T-cells in the pathophysiology of NAFLD. This review discusses the dynamics of CD4+ T-cells with effector function in the peripheral blood and liver tissue and of their related cytokines in NAFLD patients and mouse models. It also summarizes the association between factors related to metabolic syndrome that underlie NAFLD and CD4+ T-cells. This review also discusses changes in CD4+ T-cells associated with therapeutic intervention for NAFLD, the effects of immunometabolism on immune cells in NAFLD, and the association between HCC and T-cells. Finally, we explore the role of CD4+ T-cells in the pathophysiology of NAFLD along with some future issues.
EVALUATING THE DEGREE OF IMMUNE CELL INFILTRATION IS ESSENTIAL FOR UNDERSTANDING THE PATHOLOGY OF NAFLD
Pathological features such as steatosis, lobular and portal inflammation, and hepatocellular ballooning are characteristic of non-alcoholic steatohepatitis (NASH)[24]. In the Matteoni classification[25], Brunt classification[26], and NAFLD activity score (NAS)[27], which are known as the methods for the classification of pathological conditions or disease states based on the pathological findings, the degree of infiltration by inflammatory cells is an important component of the type, grading, and scoring. These scoring systems demonstrate that hepatic inflammation with infiltration of various immune cell subsets is essential for progression from fatty liver to NASH[28] and that the immune cells play an important role in the pathology of NAFLD[29].
HEPATOCYTES ARE ANATOMICALLY EASIER TO CONTACT WITH IMMUNE CELLS AND ARE INVOLVED IN IMMUNOMODULATION
The liver receives an abundance of blood flow. Approximately 30% of the whole blood passes through the liver every minute[30], carrying approximately 108 peripheral blood lymphocytes through the liver in 24 h[31]. In addition, nearly 1010 lymphocytes, such as T-cells, B-cells, and natural kicker (NK) cells, reside in the liver, which weighs 1.5 kg[14,32]. The cells are distributed not only in the vessels but also in the liver parenchyma.
Circulating T-cells pass through sinusoids in the liver and interact with Kupffer cells, liver sinusoidal endothelial cells, resident dendritic cells, and hepatocytes. The diameter of sinusoids (6-15 μm) is smaller than that of lymphocytes (7-12 μm)[14]. Therefore, when the lymphocytes pass through the sinusoid, the blood flow becomes slower, increasing the contact time between the lymphocytes and the antigen-presenting cells (APCs). Similarly, a fenestrated endothelium and the lack of basement membrane facilitate extravasation of lymphocytes, and the unique access of lymphocytes to hepatocytes directly or indirectly primes them[33]. Thus, the anatomical location of the liver makes communication with immune cells convenient.
Antigen recognition in the liver causes T-cells to undergo activation, suppression, immune escape leading to differentiation into a regulatory phenotype, or apoptosis[33]. In addition, the liver induces the apoptosis of activated T-cells in a non-antigen-specific manner to regulate the immune response[34].
THE ORIGIN OF LYMPHOCYTES IN THE LIVER
Liver damage increases lymphocyte infiltration into the liver, and the type, distribution, activation, and function of these infiltrating cells in the liver determine the nature of inflammation[19,35]. The chemokine system is a factor that plays an important role in regulating the continuous influx of lymphocytes into the liver[18,36].
As lymphocytes are generally activated by direct contact with the antigen, they migrate to secondary lymphoid organs in which antigens are present. Then, they are activated by antigen presentation from APCs and migrate to target organs[37,38]. The continuous recirculation and homing of lymphocytes between specific sites also occurs between the liver and lymph nodes[39]. In the pathology of NAFLD, the mesenteric lymph node[40,41] and mesenteric adipose tissue[42] are considered potential sources of lymphocytes in the liver. The upregulation of C-C motif chemokine ligand 5 (CCL5) in the liver and C-C motif chemokine receptor 3 (CCR3) (the receptor of CCL5) in mesenteric lymph node cells have been associated with increased lymphocyte infiltration into the liver[41]. B-cells involved in the early inflammation of mesenteric adipose tissue also migrate to the liver and contribute to hepatocyte inflammation[42].
There have also been reports that naïve T-cells may be activated in the liver[33]. This is because various types of cells capable of presenting antigens exist in the liver, and their unique structure enables direct contact with circulating T-cells[43]. This indicates that circulating T-cells may elicit an immune response without the intervention of secondary lymphoid organs, suggesting that circulating T-cells may directly infiltrate the liver and modify the pathology of chronic liver injury.
The exact route by which lymphocytes enter the liver is not yet well understood. Therefore, clarifying the source of lymphocytes that are found in the liver may provide a means to modify the immunokinetics in the liver and prevent disease progression without interfering with the mobilization of physiological lymphocytes[44].
THE HOMEOSTATIC MECHANISM OF IMMUNE CELLS MAY BE DISRUPTED IN LIVER INJURY
The liver plays a role in maintaining immune homeostasis. Changes in liver architecture as a result of acute or chronic inflammatory conditions lead to a remarkable change in the organization and localization of lymphocyte populations[45]. Therefore, the mechanism of homeostasis maintenance by immune cells might be disrupted during liver injury. The relationship between the presence of lymphocytes in liver tissue and liver disease has been known for a long time, and there are reports related to autoimmune hepatitis (AIH)[46], primary biliary cholangitis (PBC)[47], viral hepatitis[48], and hepatocarcinogenesis[49].
NAFLD HAS AN ENVIRONMENT THAT AFFECTS THE IMMUNE SYSTEM
NAFLD sometimes coexists with AIH[50] or PBC[51]. Approximately 30% and 5%, of NASH patients[52] are positive for the anti-nuclear antibody (ANA) and anti-mitochondrial antibody (AMA), respectively, and the frequencies of both are higher than in the general population[53]. The relationship between the presence of these antibodies and the pathogenesis of NAFLD is controversial[54]. However, this tendency does not simply demonstrate the comorbidity of AIH or PBC in NAFLD patients, but it also indicates that the environment that drives NAFLD may influence the immune response of the host, including autoimmunity.
Furthermore, in obesity resulting from excessive intake of nutrients, chronic low-grade inflammation primarily occurs in the adipose tissue[55], as well as systemically[56,57]. Chronic inflammation is involved in the development of metabolic disease[58] and insulin resistance[59]. In obesity, factors such as chronic inflammation[60,61], changes in intestinal flora[62], insulin resistance[63], arteriosclerosis[64], and dyslipidemia[65] affect T-cells.
RELATIONSHIP BETWEEN GUT MICROBIOTA AND IMMUNE SYSTEM
The changes in the microbial flora, especially gut microbiota, highly co-evolving with the immune system[66], are considered to be associated with various diseases. In NAFLD patients, there are qualitative and quantitative abnormalities of gut microbiota and intestinal hyperpermeability[67]. Endotoxemia, the result of these changes, is exacerbated as the disease progresses in NAFLD[68]. Leptin-induced upregulation of CD14 in Kupffer cells enhances responsivity to endotoxin in fatty liver[69]. Furthermore, endotoxin is considered to be deeply involved in the establishment of steatohepatitis by activating Kupffer cells via Toll-like receptor 4 (TLR4) and producing tumor necrosis factor alpha (TNF-α) and reactive oxygen species (ROS)[12]. Thus, TLR-mediated endotoxin is involved in innate immune system activation in NAFLD.
TLRs are thought to act primarily on innate immunity and contribute to the maturation of APCs and the production of inflammatory cytokines[70]. These effects may secondarily modify adaptive immunity. However, recent studies have shown the direct role of TLR signaling pathways in adaptive immunity. TRL4 signaling in effector CD4+ T-cells regulates T-cell receptor (TCR) activation by inhibiting MAPK phosphatase 3 (MKP-3) induction-mediated activation of ERK1/2 and suppresses interferon-gamma
The expression of the TLR4 in naïve T-cells is reported to be significantly higher in NASH patients than that in healthy individuals and non-alcoholic fatty liver (NAFL) patients[74]. Therefore, in the context of NAFLD, endotoxin may modify the pathophysiology of NAFLD by exerting direct effects on T-cells as well as secondary effects mediated by innate immunity.
EFFECT OF OXIDATIVE STRESS ON CD4+ T-CELL
It is well known that oxidative stress is involved in the pathophysiology of NAFLD by promoting hepatocellular death, inflammation, fibrosis, and carcinogenesis[75]. The highly reactive aldehydes generated during lipid peroxidation modify self-molecules and form antigenic adducts, known as oxidation specific epitopes (OSEs)[76,77]. Therefore, hepatocyte oxidative stress may be an important trigger for both humoral and cellular immune responses in the liver by forming OSEs[21,22,77].
Fatty acids involved in oxidative stress can also affect T-cells in a dose-dependent manner[78-80]. These direct effects of fatty acids on T-cells can be stimulatory or lipotoxic, depending on the ability of the T-cells to avoid the toxic effects of fatty acids[81]. Therefore, similar to the oxidative stress response in hepatocytes, lipid peroxides can affect cell membranes, proteins, and deoxyribonucleic acid, affecting T-cell function.
T-CELLS ARE DIVIDED INTO VARIOUS SUBSETS AND HAVE DIFFERENT EFFECTOR FUNCTIONS
The immune system of mammals is roughly classified into adaptive immunity (specific) and innate immunity (non-specific). The former is believed to have been established early during the emergence of jawed vertebrates, and the origin of the latter might be even older[82]. Recent advances in understanding immune mechanisms at molecular and cellular levels have led to unclear boundaries between adaptive and innate immunity[83]. However, as part of the innate immunity, pattern recognition receptors (PRRs) recognize pathogen-associated molecular patterns (PAMPs) on foreign invaders. The innate immunity is characterized by a rapid reaction after foreign body recognition[84], while adaptive immunity is based on antigen-specific recognition and is characterized by the presence of immune memory.
T-cells, which express TCR, an antigen receptor, after differentiation and maturation of progenitor cells in bone marrow via selection in the thymus, play a central role in adaptive immunity[85]. The TCR recognizes peptide antigens presented by major histocompatibility complex (MHC) molecules on APCs[86]. MHC molecules are divided into Class I and Class II molecules. Several CD8+ T-cells and CD4+ T-cells demonstrate a binding affinity for each MHC molecule[87].
CD4+ T-cells are essential and complex elements of the immune system and are the key regulators of host health and diseases[88,89]. T-cells are differentiated into highly functional effector T-cells in response to foreign antigen stimulation in a special cytokine environment[90,91] and regulate immune response via the secretion of specific cytokines[92]. The effector T-cells are classified based on the cytokine production pattern[93], cell surface antigen[93,94], transcription factor[88], and intracellular metabolism[95,96]. Each subtype of T-cells exhibits different functions.
THE CHARACTERISTICS OF PERIPHERAL CD4+ T-CELL IN NAFLD PATIENTS
The dynamics of CD4+ T-cells in obesity are different from healthy subjects
NAFLD is associated with systemic metabolic disorders, as reflected by the novel term metabolic (dysfunction) associated fatty liver disease (MAFLD)[97-100]. Therefore, given that various pathological conditions such as T2DM, dyslipidemia, and adipose tissue inflammation are associated with NAFLD, peripheral T-cells can be affected by each of these conditions. For example, there are several reports on the dynamics of CD4+ T-cells in obesity associated with these pathologies. In obese patients, the total number of CD4+ T-cells in peripheral blood was significantly higher than that in the lean control group[101,102] and exhibited a significant positive correlation with fasting insulin levels in addition to serum interleukin-7
In contrast, with respect to the hypothesis that the immune mechanism may be impaired in obesity[107], the levels of CD4+ T-cells decreased in genetically obese Zucker rats[108]. Furthermore, the frequency of CD4+ T-cells and the blastogenic response in obese patients were lower than that in the lean control group, and both of these factors were recovered with weight loss[109].
The dynamics of CD4+ T-cells in obesity associated with various pathological conditions are still controversial. Therefore, it can be conjectured that understanding the dynamics of CD4+ T-cells in NAFLD pathological conditions is also not easy.
Peripheral CD4+ T-cells in NAFLD patients may be increased
The dynamics of peripheral CD4+ T-cells and CD8+ T-cells in NAFLD patients are summarized in Table 1. Few reports have compared the peripheral T-cell dynamics of healthy subjects and NAFLD patients. Compared to the healthy subjects, the frequency of peripheral CD4+ T-cells in NAFLD patients tended to increase[28] or was significantly increased[110-112], but the frequency of CD8+ T-cells remained unchanged[28,110,111] or was significantly decreased[112]. These results are consistent with many previous reports of increased frequency of peripheral CD4+ T-cells in obese conditions. However, it has been reported that the frequencies of peripheral CD4+ T-cells and CD8+ T-cells are not related to the characteristic histological features of NASH[110]. In C57BL/6 mice that were fed a high-fat diet (HFD) for 12 weeks, the frequencies of peripheral CD4+ T-cells and CD8+ T-cells were similar to that in the controls[41].
Dynamics of peripheral CD4+ T-cells and CD8+ T-cells in NAFLD patients
| CD4+ T-cells | CD8+ T-cells | |
| Inzaugarat et al.[110] | The CD4+ T-cell frequency in PBMCs significantly increases compared to HC (36.3% in 10 NASH vs. 29.8 % in 10 HC) | There is no change in the CD8+ T-cell frequency in PBMCs compared to HC (16.7% in 10 NASH vs. 13.6% in 10 HC) |
| Maricic et al.[111] | The CD4+ T-cell frequency in PBMCs significantly increases compared to HC (67.6% in 18 NASH vs. 59.6% in 19 HC) | There is no change in the CD8+ T-cell frequency in PBMCs compared to HC (data not shown) |
| Seike et al.[28] | The CD4+ T-cell frequency in CD3+ T-cell tends to increase compared to HC (66.0% in 40 NAFLD vs. 60.8% in 5 HC) | There is no change in the CD8+ T-cell frequency in CD3+ T-cell compared to HC (22.0% in 40 NAFLD vs. 26.1% 5 HC) |
| Diedrich et al.[112] | The CD4+ T-cell frequency in CD3+ T-cell significantly increases compared to HC (67.9% in 27 NAFLD vs. 59.9% 26 HC) | The CD4+ T-cell frequency in CD3+ T-cell significantly decreases compared to HC (26.12 % in 27 NAFLD vs. 32.47% in 26 HC) |
THE CHARACTERISTICS OF INTRAHEPATIC CD4+ T-CELL IN NAFLD PATIENTS
Lymphocytes tend to infiltrate the liver during NAFLD conditions
Several studies show that the homing of circulating lymphocytes to the liver may enhance liver inflammation[113]. This is supported by the results demonstrating that the genes upregulated in NASH patients encoding for chemokines and chemokine receptors are involved in leukocyte recruitment[114], including the couples C-X-C motif chemokine ligand 8 (CXCL8)/C-X-C motif chemokine receptor 1 (CXCR1), CXCL1,3/CXCR2, and CCL3-5/CCR5 and the chemokines CXCL9-11 and CCL2[115]. This suggests that lymphocytes tend to infiltrate the liver during NAFLD conditions.
The intrahepatic CD4+ T-cells in NAFLD patients may be increased
Similar to the study in peripheral blood, few reports have been published examining the intrahepatic CD4+ T-cell and CD8+ T-cell levels in NAFLD patients. The frequency of CD4+ T-cells in all cells in the liver tissue significantly increased compared to that in healthy individuals[116]. In contrast, the frequencies of CD4+ T-cells in CD3+ T-cells in the liver tissue did not change compared to that in the healthy subjects, and the frequencies of CD8+ T-cells in CD3+ T-cells tended to decrease slightly[112]. Characteristically, CD4+ T-cells and CD8+ T-cells significantly infiltrate the portal tract but not the lobules[29], and, under NASH conditions, the levels of both cells increased upon the progression of fibrosis[28,29].
The dynamics of the intrahepatic CD4+ T-cell in the NASH mouse model are controversial
In the animal model, the frequency of CD4+ T-cells in the liver tissue was significantly increased in C57BL/6 mice that were fed an HFD for 12 weeks compared to that in the controls. However, no difference was observed in the frequency of CD8+ T-cells. The increase in CD4+ T-cells that caused liver damage was attributed to the propensity of the migration of gut-derived lymphocytes to the liver, which was associated with the upregulation of CCL5 in the liver and its receptor CCR3 in lymphocytes[41]. In C57BL/6J mice that were fed an HFD for 16 weeks, the frequency of CD4+ T-cells in CD3+ T-cells in the liver tissue increased significantly compared to that in the controls. This observation may be related to the activation of CD4+ T-cells by intrahepatic B-cells that produce IL-6, TNF-α, and immunoglobulin G2a (IgG2a)[117]. In C57BL/6 mice that were fed an HFD for 16 weeks, the frequencies of CD4+ T-cells and CD8+ T-cells in CD45+ cells in the liver tissue significantly increased compared to those in the control, and CD4+ T-cells were activated (evaluated by CD69 and OX40). OX40 was reported to be an important regulator in the NAFLD context, as the genetic deletion of OX40 reduced the frequency of CD4+ T-cells in the liver tissue and inhibited activation[118].
In C57BL/6 mice that were fed a methionine- and choline-deficient (MCD) diet for 8 weeks, the frequency of CD4+ T-cells increased significantly over the 8 weeks in hepatic mononuclear cells of the liver tissue compared to that in the controls. From the second week onward, intrahepatic CD4+ T-cells were activated (evaluated by CD25) with the progression in the pathological conditions[119]. Furthermore, in C57BL/6 mice that were fed an MCD diet for 8 weeks, the frequencies of CD4+ T-cells and CD8+ T-cells in CD45+ cells that respond to oxidative stress-derived antigens increased in the liver tissue over time, and CD3+ T-cells were activated (evaluated by CD69). CD4+ T-cell infiltration in liver tissues was further increased upon immunization by oxidative stress-derived antigens[22]. These results show that the frequencies of activated CD4+ T-cells increased in the liver tissue in various NASH mouse models.
In contrast, recent reports demonstrated a decrease in the frequency of CD4+ T-cells in the liver tissue in several NASH mouse models. In C57BL/6J mice that were fed an HFD for 12 weeks, the frequency of CD4+ T-cells in the liver tissue was significantly lower than that of the controls but was recovered by the administration of antibiotics or Lactobacillus[40]. C57BL/6 mice that were fed a western diet (WD) for 24 weeks showed a selective loss of the total number of CD4+ T-cells in the liver tissue[120]. Furthermore, the frequency of CD4+ T-cells in the liver tissue was reduced in MYC-ON/OFF mice that were fed an MCD diet for 4 and 8 weeks, MYC-ON mice that were fed a choline-deficient and amino-acid-defined (CDAA) diet for 16 weeks, C57BL/6 mice that were fed a CDAA diet for 16 weeks, C57BL/6 mice that were fed an HFD or a low-fat diet (LFD) for 24 weeks, and 12-week-old ob/ob mice, which could be considered as a selective loss of CD4+ T-cells owing to the oxidative damage of fatty acids and was involved in the development of HCC[121].
Since changes in CD4+ T-cells are the sum of the changes in various types of CD4+ T-cells, it is not easy to evaluate them all together. Even if there is no change in CD4+ T-cells, the ratio of the cell fractions that compose them may be different. To elucidate the dynamics of the various effector T-cells that are described below, such as which cell fraction the activated CD4+ T-cells will differentiate into and what functions they will acquire or lose, may be important in determining the involvement of CD4+ T-cell in the NAFLD pathology.
DYNAMICS OF CD4+ EFFECTOR T-CELLS IN NAFLD
As shown in Figure 1, CD4+ T-cells are divided into subsets such as T helper 1 (Th1), Th2, Th9, Th17, Th22, regulatory T-cell (Treg), and T follicular helper (Tfh), and each subset has its own function. The dynamics of Th1, Th2, Th17, and Treg subsets in peripheral blood and liver in NAFLD patients and healthy controls are summarized in Table 2[28,74,110,112,116,122-126].
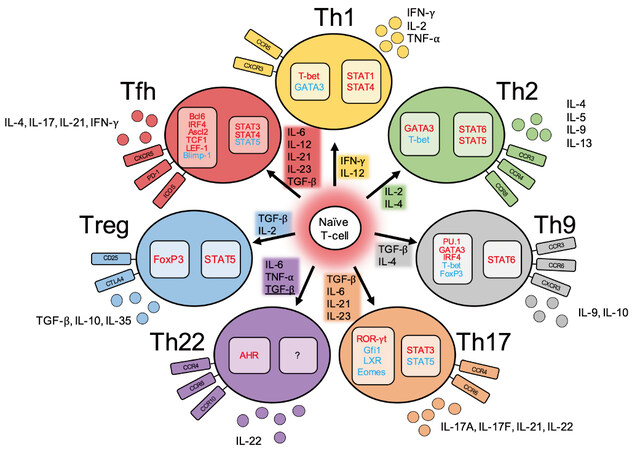
Figure 1. Summary of the types, characteristics, and functions of CD4+ effector T-cells. Each effector T-cell develops from naïve T-cells under stimulation by several cytokines. Transforming growth factor-beta (TGF-β) suppresses Th22 differentiation. Transcription factors and signal transducer and activator of transcription (STAT) molecules required for differentiation are listed, with those in red involved in positive regulation and those in blue involved in negative regulation. Th: T helper; Treg: regulatory T-cell; Tfh: T follicular helper; T-bet: T-box expressed in T-cells; GATA-3: GATA binding protein 3; IL: interleukin; IFN-γ: interferon-gamma; TNF-α: tumor necrosis factor-alpha; CCR: C-C motif chemokine receptor; CXCR: C-X-C motif chemokine receptor; IRF4: IFN regulatory factor 4; FoxP3: forkhead box P3; ROR-γt: retinoic acid-related orphan receptor gamma-t; Gfi1: growth factor independent 1; LXR: liver X receptor; Eomes: eomesodermin; AHR: aryl hydrocarbon receptor; PD-1: programmed cell death-1; ICOS: inducible costimulatory; Ascl2: achaetescute homolog 2; TCF-1: T-cell factor 1; LEF-1: lymphoid enhancer-binding factor-1; Blimp-1: B lymphocyte-induced maturation protein-1.
Dynamics of peripheral and liver effector T-cells in NAFLD patients
| Ref. | Patients population | Blood | Liver | ||||||
| Th1 | Th2 | Th17 | Treg | Th1 | Th2 | Th17 | Treg | ||
| Inzaugarat et al.[110] | 10 NASH, 10 HC | ↑a | → | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. |
| Ferreyra Solari et al.[122] | 6 pediatric NASH, 5 aged matched HC | ↑ | → | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. |
| Rau et al.[123] | 51 NAFL, 30 NASH, 43 HC | ↑b | ↑b | → | ↓c | →d | →d | ↑d | →e |
| Söderberg et al.[124] | 12 non-NASH (NAS 0-2), 33 NASH (NAS3-6) | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | ↑f |
| Tang et al.[125] | 14 NASH, 4 HC | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | ↑ | N.D. |
| Seike et al.[28] | 40 NAFLD, 5 HC | → | ↑ | ↑ | → | N.D. | N.D. | N.D. | N.D. |
| Diedrich et al.[112] | 27 NAFLD blood, 26 HC, 15 NAFLD liver, 3 HL | → | ↑ | → | → | N.D. | N.D. | N.D. | N.D. |
| Alegre et al.[126] | 9 NASH, 11 HC | ↑ | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. |
| Wang et al.[74] | 25 NASH, 25 HC | ↑ | N.D. | ↑ | N.D. | N.D. | N.D. | N.D. | N.D. |
| Inzaugarat et al.[116] | 9 NAFLD, 9 HC | N.D. | N.D. | N.D. | N.D. | ↑g | N.D. | N.D. | N.D. |
TH1 CELLS
Basic characteristics of Th1 cells
Th1 cells are characterized by the production of IFN-γ, IL-2, and TNF-α[127] and by the expression of CCR5[128] and CXCR3[129]. They develop from naïve T-cells under stimulation by IFN-γ and IL-12[92]. In the differentiation of Th1, an important transcription factor is T-box expressed in T-cells (T-bet)[130], and important signal transducer and activator of transcription (STAT) molecules are STAT4 and STAT1, activated by IL-12[131] and IFN-γ[132], respectively. GATA binding protein 3 (GATA3) suppresses the Th1 programs through the repression of IL-12 signaling[133]. Th1 cells induce macrophage activation primarily via the production of IFN-γ and are involved in intracellular pathogen elimination, antiviral response, and antitumor response [Figure 1]. IFN-γ plays a crucial role in generating efficient innate and adaptive immune responses[134]. Studies have found an association of Th1 cells with organ-specific autoimmune diseases such as autoimmune type 1 diabetes[135], rheumatoid arthritis[136], multiple sclerosis[137], and Crohn’s disease[138]. In terms of liver diseases, Th1 cells are associated with AIH[139], PBC[140], and alcohol-related cirrhosis[141].
Th1 cells are altered in many pathological conditions associated with NAFLD patients
Compared to a lean control subject, there is an increase in the peripheral Th1 cells in metabolically healthy obese subjects[142]. In addition, peripheral Th1 cells increase in T2DM patients compared to lean control subjects[142,143]. Under obese conditions, Th1 cells infiltrate the adipose tissue, and the frequency of Th1 cells in adipose tissue has a significant positive correlation with the levels of high-sensitivity c-reactive protein (hs-CRP) in plasma[144].
Th1 cells increase in adipose tissue and skeletal muscle in obese mice
In C57BL/6 mice that were fed an HFD for 14-18 weeks, the absolute number of Th1 cells per gram of fat was found to significantly increase compared to that in the controls[145]. In C57BL/6 mice that were fed an HFD for 10 weeks, the frequency of Th1 cells in visceral adipose tissue (VAT) was significantly increased compared to that in the control[146]. In C57BL/6 mice that were fed an HFD for 12 weeks, the frequency of Th1 cells in the perigonadal adipose tissue and skeletal muscle was significantly increased compared to that in the control[147]. In C57BL/6J mice that were fed an HFD for 21 weeks, the expression level of IFN-γ messenger ribonucleic acid (mRNA) in adipose tissue and the production of IFN-γ in T-cells from adipose tissue were significantly higher than in the lean controls[148]. In addition, T-bet-deficient mice showed increased insulin sensitivity despite increased VAT mass[149]. These results suggest that Th1 cells are increased in the adipose tissue and skeletal muscle in the obese mouse model; this may affect glucose metabolism via IFN-γ.
IFN-γ may be involved in insulin resistance via adipose tissue inflammation
Lymphocytes infiltrating the adipose tissue stimulate preadipocytes via IFN-γ to release monocyte chemoattractant protein-1 (MCP-1). This is important for monocyte recruitment and may promote monocyte infiltration, which plays an important role in adipose tissue inflammation and the development of insulin resistance[150]. Studies have also demonstrated that IFN-γ-induced adipose tissue inflammation and oxidative stress are associated with endothelial dysfunction in T2DM; this plays an important role in the pathogenesis of cardiovascular disorders[151].
Global deletion of IFN-γ in mice has been observed to improve glucose intolerance, hepatic insulin resistance, and weight loss associated with negative energy balance[152]. In addition, IFN-γ−/− mice fed an HFD showed a decrease in the adipocyte size, improvement in insulin sensitivity, and an M2-shift in adipose tissue macrophage phenotype and cytokine expression compared with obese wild-type control mice[153].
Th1 cells increase in the mesenteric lymph node, small intestine, and colon of obese mice
In C57BL/6J mice fed an HFD for 12 weeks, the frequency of Th1 cells in the mesenteric lymph nodes increased compared to that in the control[40,41]. In C57BL/6J mice that were fed an HFD for 30 days[154], 10 weeks[146], and 12-16 weeks[155], the frequency of Th1 cells increased in the small intestinal mucosa[146,154,155]. For C57BL/6J mice that were fed an HFD for 12-16 weeks, there was an increased frequency of Th1 cells in the colon[155].
Beta7null mice experiencing hypoplasia of the gut lymphoid system showed an improvement in HFD-induced insulin resistance and a reduction in the number of intestinal Th1 cells[155]. IFN-γ may also have direct pathological effects on the disruption of barrier function[155,156]. Therefore, IFN-γ may affect metabolic function by altering intestinal permeability.
IFN-γ may be involved in liver inflammation and fibrosis in NAFLD
In NAFLD patients, IFN-γ levels were found to increase in peripheral blood[157]. In C57BL/6 mice that were fed an HFD[158] or a choline deficiency diet (CDD)[159], studies have observed the harmful role of IFN-γ associated with the initiation or maintenance of proinflammatory activation in the development of steatohepatitis. In addition, hepatitis and fibrosis in methionine and choline-deficient HFD mouse models were alleviated by IFN-γ deficiency[134]. These results indicate that IFN-γ has a proinflammatory or profibrotic effect in NAFLD animal models. In contrast, IFN-γ has been found to antagonize the onset of hepatic fibrosis induced by dimethyl nitrosamine and carbon tetrachloride (CCl4) by inhibiting the activation of hepatic stellate cells (HSCs) and exhibiting an antifibrotic action[160,161]. Many of these studies have suggested that IFN-γ associated with Th1 cells is associated with liver inflammation and fibrosis in NAFLD and with insulin resistance and adipose tissue inflammation in obesity and diabetes that underlie NAFLD.
Peripheral Th1 cells appear to increase in NAFLD
The frequency of peripheral Th1 cells in adult NAFLD patients either remains the same[28,112] or significantly increases[74,110,122,123,126] compared to that in healthy controls. In NASH patients, there is a significantly positive correlation between the frequency of IFN-γ-positive cells in CD4+ T-cells and serum endotoxin levels[74]. The expression of IFN-γ in peripheral blood mononuclear cells (PBMCs) of NASH patients increases significantly compared to that in PBMCs of the NAFL patients; this was significantly positively correlated with histopathological features such as ballooning and fibrosis in liver tissues[162]. Peripheral CD4+ T-cells in NAFLD patients were more likely to produce IFN-γ upon stimulation with leptin, which is a pro-inflammatory adipokine, compared to these cells in the healthy subjects[116]. A study comparing obese and healthy children has shown that Th1 cells are associated with insulin resistance and the development of NASH[163]. In the animal model, there was a significant increase in the frequency of peripheral Th1 cells in C57BL/6 mice fed with an HFD for 12 weeks compared to the control[41,158].
Intrahepatic Th1 cells may increase in NAFLD
A few reports have examined intrahepatic Th1 cells in patients with NAFLD. Intrahepatic Th1 cells in patients with NAFLD are increased compared to those in healthy controls[116]. The gene encoding Th1 cytokine was significantly upregulated in NASH patients compared to that in obese patients with normal liver tissue[115]. Another study found that there were no differences between NAFL and NASH patients[123].
In C57BL/6J mice fed with an HFD for 16 weeks, there was a significant increase in the frequency of Th1 cells in intrahepatic CD4+ T-cells. This was a result of the activation of CD4+ T-cells by intrahepatic B-cells and the promotion of differentiation of CD4+ T-cells into Th1 cells[117]. In C57BL/6 mice fed with an HFD for 12[40,41] or 16 weeks[118], there was a significant increase in the frequency of intrahepatic Th1 cells compared to that in the controls, although this decreased with an improvement in the NAFLD pathology via the administration of antibiotics or Lactobacillus[40]. Moreover, during NASH development, OX40 plays an important role in the activation and proliferation of intrahepatic T-cells and the promotion of differentiation of CD4+ T-cells into Th1 cells[118]. In mice that were fed an MCD diet, the Th1 subset of CD4+ T-cells was activated by oxidative stress-derived antigens, contributing to the progression of NASH[22]. These findings highlight that Th1 cells increased in the liver tissue and were involved in NAFLD pathology. There is also a report mentioning that the frequency of Th1 cells in the liver tissue was maintained in MYC-ON mice that were fed an MCD diet[121].
Although NASH is not traditionally considered a Th1-polarized disease[164], the frequency of peripheral or intrahepatic Th1 cells in NAFLD differs from that in healthy controls, and it is suggested to be involved in the modification of NAFLD and NAFLD-related pathology [Figure 2]. However, there is uncertainty regarding the specific mechanism by which Th1 cells are involved in the formation and progression of the pathological conditions of NAFLD.
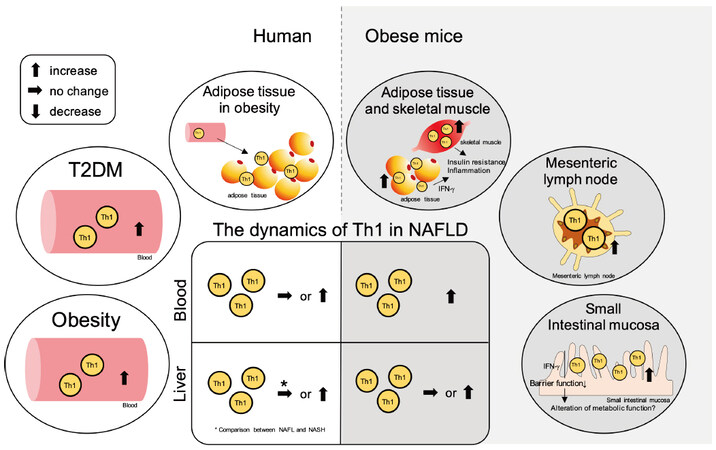
Figure 2. Summary of the contribution of Th1 cells in NAFLD and the related pathologies. The ellipse on the left shows the dynamics of Th1 cells in peripheral blood and adipose tissue in obesity and type 2 diabetes (T2DM) in humans, while the ellipse on the right shows the dynamics of Th1 cells in adipose tissue, skeletal muscle, mesenteric lymph nodes, and small intestinal mucosa in obese mice. The central square summarizes the Th1 cell dynamics in the peripheral blood and liver tissue in NAFLD. The left side shows the dynamics in humans, and the right side shows the dynamics in mice. NAFLD: Non-alcoholic fatty liver disease; NAFL: non-alcoholic fatty liver; NASH: non-alcoholic steatohepatitis; Th: T helper; IFN-γ: interferon-gamma.
TH2 CELLS
Basic characteristics of Th2 cells
Th2 cells are characterized by the production of IL-4, IL-5, IL-9, and IL-13[165] and by the expression of CCR3, CCR4[166], and CCR8[167]. They develop from naïve T-cells under stimulation mainly by IL-2 and
Th2 cells are altered in peripheral blood and adipose tissue in obese patients
In humans, peripheral Th2 cell numbers in obese patients are significantly elevated compared to those in the lean control subjects[102]. Under obese conditions, GATA3/CD3E gene expression significantly increases in the VAT compared to that in the control condition[176]. In contrast, in healthy overweight and obese human patients, the frequency of Th2 cells in the VAT is inversely correlated with plasma hs-CRP levels[144]. In addition, there is a significant negative correlation between the frequency of Th2 cells in adipose tissue and peripheral blood with steady-state plasma glucose concentrations that reflect insulin resistance[144]. These results are representative of the decrease in Th2 cells in peripheral blood and adipose tissue for obese patients, as hs-CRP increase and insulin resistance are associated with obesity[177].
Th2 cells may decrease in VAT and mesenteric lymph node in mice
In C57BL/6J mice that were fed an HFD for 12-16 weeks, there was a significant reduction in the frequency of Th2 cells in VAT compared to that in the control mice[145]. In C57BL/6J mice fed with an HFD for 12 weeks, the frequency of Th2 cells in the mesenteric lymph node was lower than that in the controls, while the frequency recovered with improvement in the pathology of NAFLD upon treatment with antibiotics and probiotics[40]. In C57BL/6J mice that were fed an HFD for 10 weeks, the frequency of Th2 cells in the small intestinal mucosa was the same as that in the controls[146].
Decreased T-cell responsiveness to insulin may be one of the triggers for Th2 changes in obesity
The signaling of the insulin receptor, which is upregulated during T-cell activation, regulates T-cell proliferation and cytokine production[178]. Insulin may promote Th2 differentiation in CD4+ T-cells through extracellular signal-regulated kinase phosphorylation[179]. This indicates that intensive insulin therapy for patients in intensive care and other hyperglycemia scenarios may be dependent on modifying immune cell function. Studies have also reported that, in vitro, supra-physiological insulin does not promote a shift to the Th2 phenotype in insulin-resistant obesity[180]. In addition, a decrease in the Th1:Th2 ratio associated with weight loss and a slimmer waistline at 12 weeks during the 24-week dietary energy restriction with gastric banding surgery were also observed[181]. Therefore, T-cells in obese patients may be resistant to insulin-mediated Th2 differentiation. However, this resistance to Th2 differentiation may be restored by improving insulin sensitivity through weight loss[182].
Th2 cells may have a protective role against obesity and insulin resistance in mice
The transfer of CD4+ T-cells into lymphocyte-free recombination-activating gene (RAG)null mice fed an HFD resulted in suppressed weight gain and improved insulin resistance primarily via Th2 cells. The transfer of STAT6−/−CD4+ T-cells to RAGnull mice fed an HFD did not demonstrate insulin-sensitizing effects[145]. Studies have also found that IL-33 as a treatment for ob/ob mice induces the production of Th2 cytokines[183]. These Th2 cytokines are largely produced in the adipose tissue, resulting in improved insulin sensitivity[184,185]. These results suggest that Th2 may have a protective role against insulin resistance.
The alteration in Th2 in mesenteric lymph nodes may be protective against obesity in mice
Heligomosomoides polygyrus (H. polygyrus) infection is known to cause a Th2-dominated immune response[186]. H. polygyrus infection has been shown to protect C57BL/6 mice from HFD-induced obesity[187] and to increase Th2 cells in mesenteric lymph nodes[188]. In addition, H. polygyrus infection in STAT6−/−mice does not protect against HFD-induced obesity[187]. This phenomenon is considered to be associated with Th2-dependent M2 macrophage-dependent changes in the gut microbiota due to the H. polygyrus infection[187]. These results suggest that changes in the Th2 cells in mesenteric lymph nodes may protect against the pathology of obesity.
IL-13 may have a protective effect on insulin resistance
IL-13 is involved in the inhibition of insulin resistance and low-grade systemic inflammation in C57BL/6J mice fed an HFD. It does this by suppressing adipose inflammation, reducing hepatic gluconeogenesis, and enhancing adaptive thermogenesis[189,190]. In IL-13−/− mice, the dysregulation of glucose metabolism has been observed in the liver, resulting in hepatic insulin resistance and systemic metabolic dysfunction[191].
IL-13 can induce liver fibrosis
IL-13 can induce tissue fibrosis via the stimulation and activation of transforming growth factor-beta1 (TGF-β1)[192]. IL-13 binds to high-affinity IL-13R (IL-13Rα2), which is expressed in activated HSCs, promotes the production of TGF-β1 via TGFB1 promoter activity, and is involved in liver fibrosis[193]. IFN-γ deficient mice fed an HFD rapidly progressed to NASH, resulting in fibrosis dependent on TGF-β and IL-13 signaling[194].
No consensus has been established on the dynamics of peripheral or hepatic Th2 cells in NAFLD
In patients with NAFL/NASH[123] and NAFLD[28,112], there was a significant increase in the frequency of peripheral Th2 cells in CD4+ T-cells compared to that in healthy patients. However, several reports have also indicated that the frequency of peripheral Th2 cells in adult NASH patients[110] and pediatric NASH patients[122] is the same as that in the healthy subjects. In animal models, the percentage of Th2 cells in the peripheral blood of C57BL/6 mice fed an HFD for 12 weeks was the same as control[41].
There are limited studies on Th2 cells in the liver tissue; there was no difference between the frequency of Th2 cells in CD4+ T-cells in the liver tissue of NASH patients and NAFL patients[123]. In C57BL/6 mice fed an HFD for 12 weeks, there was no significant difference in the frequency of Th2 cells in liver tissue compared to that in the controls[41]. In contrast, in C57BL/6 mice fed an HFD for 16 weeks, there was a significant increase in the frequency of Th2 cells in liver tissue compared to that in the controls[118].
The roles of Th2 cells in NAFLD and the related pathologies are summarized in Figure 3. Although there are reports indicating the association of Th2 cytokines with insulin resistance and liver fibrosis as well as changes in the frequency of Th2 cells in obesity, the role of Th2 cells in NAFLD remains unclear.
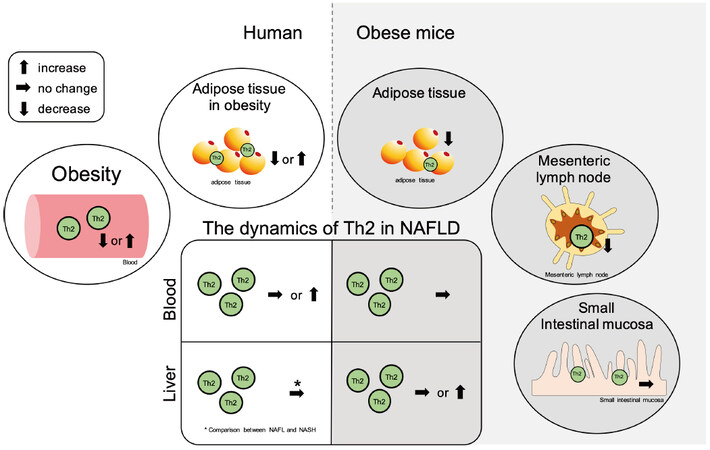
Figure 3. Summary of the contribution of Th2 cells in NAFLD and the related pathologies. The ellipse on the left shows the dynamics of Th2 cells in peripheral blood and adipose tissue in obesity in humans, while the ellipse on the right shows the dynamics of Th2 cells in adipose tissue, mesenteric lymph nodes, and small intestinal mucosa in obese mice. The central square summarizes the Th2 cell dynamics in the peripheral blood and liver tissue in NAFLD. The left side shows the dynamics in humans, and the right side shows the dynamics in mice. NAFLD: Non-alcoholic fatty liver disease; NAFL: non-alcoholic fatty liver; Th: T helper; NASH: non-alcoholic steatohepatitis.
TH9 CELLS
Basic characteristics of Th9 cells
Th9 cells are characterized by the production of IL-9 and IL-10 and by the expression of CCR3, CCR6, and CXCR3[195]. They develop from naïve T-cells and Th2 under stimulation by TGF-β and IL-4[196,197]. In the differentiation of Th9, the important transcription factors are PU.1[198], GATA3[199], and IFN regulatory factor 4 (IRF4)[200], while an important STAT molecule is STAT6[199]. Of note, T-bet and forkhead box P3 (FoxP3) decrease the expression of Il9 in Th9 cells[199] [Figure 1]. As most transcription factors expressed in Th9 cells are also expressed in other Th subsets, the transcription factors in Th9 cells have a highly complex regulatory mechanism[195]. Th9 cells are mainly involved in the pathogenesis of allergic inflammation[198], autoimmune disease[201], and tumor immunity[202]. In terms of the involvement of Th9 cells in liver disease, there have been reports on their association with chronic hepatitis B (CHB)[203], liver fibrosis[204], and HCC[205].
IL-9 may be associated with liver fibrosis
The expression of IL-9 is significantly higher in human liver cirrhosis tissue than that in the normal liver tissue[206]. CCl4 treatment of IL-9-overexpressing mice with recombinant adenovirus vector leads to severe liver fibrosis with increased collagen accumulation and α-smooth-muscle actin (α-SMA) expression levels[206]. Anti-IL-9 antibody treatment in mice treated with CCl4 (hepatic fibrosis mouse model) attenuates the development of hepatic inflammation, necrosis, and fibrosis, accompanied by a marked decrease in the number of activated HSCs[204]. In addition, the in vitro co-culture of HSCs and IL-9 has been shown to upregulate the expression of α-SMA, collagen-I, and collagen-III[207].
The role of Th9 in the pathology of NAFLD remains poorly understood
There are no studies on the involvement of Th9 in obesity, T2DM, dyslipidemia, and inflammation of adipose tissue, and its role is unknown. In contrast, there are several studies on the relationship between
Although there is increasing clarity regarding the relationship between liver fibrosis and IL-9, there are no human or animal model studies on the role of Th9 in NAFLD. As such, further analysis of the relationship between Th9 and NAFLD is warranted.
TH17 CELLS
Basic characteristics of Th17 cells
Th17 cells are characterized by the production of IL-17 (IL-17A and IL-17F), IL-21, and IL-22[213] and by the expression of CCR4 and CCR6[214,215]. They develop from naïve T-cells under stimulation by TGF-β, IL-6,
Obesity may be conducive to Th17 cell differentiation
IL-6 plays an important role in the differentiation of CD4+ T-cells into Th17 cells and is found to increase in blood with the degree of obesity[232]. In diet-induced obese mice, T-cells expand the Th17 cell pool and produce more IL-17 than lean littermates, in an IL-6-dependent process[233]. The expression of miR-326, which promotes Th17 cell differentiation[234], increases in adipose tissue mononuclear cells[235]. A decrease in the adiponectin level, which was found to be low in obese patients[236], may promote the differentiation of naïve T cells into Th17 cells[237]. Under obese conditions, acetyl-CoA carboxylase 1 (ACC1) regulates the function of ROR-γt through fatty acid synthesis in T-cells and is associated with Th17 cell differentiation[238]. ACC1 is typically involved in the fatty acid metabolism of cells by catalyzing the adenosine triphosphate (ATP)-dependent carboxylation of acetyl-CoA to malonyl-CoA[239]. These results suggest that obesity provides a favorable environment for Th17 cell differentiation.
The dynamics of peripheral Th17 cells and serum IL-17 in obesity are poorly understood
Despite this environment, there are few reports on the dynamics of Th17 cells in the peripheral blood of obese individuals. Under obese conditions, there is no difference in the frequency of Th17 cells in peripheral CD4+ T-cells in adults compared to that in the lean control subjects[102]. However, the frequency of peripheral Th17 cells has been reported to increase significantly in children compared to that in the lean control subjects[240]. The dynamics of serum IL-17 in obesity are also unknown[241]; serum IL-17 level has been reported to be significantly higher in obese women than in lean women[242]. Among adolescents, serum IL-17 levels in obese individuals are lower than those in lean control subjects, and a significantly negative correlation between serum IL-17 and BMI has been observed[243]. Although the relationship between peripheral blood Th17 cells and obesity is still unknown, it is becoming apparent that Th17 cells are associated with T2DM and adipose tissue inflammation, as discussed below.
Peripheral Th17 cells tend to increase in T2DM
In T2DM patients who are overweight and obese, the frequency of peripheral Th17 cells is lower than that in non-obese patients[244]. In contrast, there is a significant increase in the frequency of peripheral Th17 cells in T2DM patients compared to that in the lean control subjects[245,246]. There is a significant increase in the frequency of peripheral Th17 cells in patients with T2DM compared to that in the normoglycemic control subjects[143]. There is a significantly negative correlation between the frequency of peripheral Th17 cells and serum high-density lipoprotein (HDL)[143]. Compared with non-diabetic patients, there is a significant increase in the frequency of peripheral Th17 cells in patients with T2DM, and this frequency has a significantly positive correlation with BMI[247].
Furthermore, the productivity of Th17 cytokines in lymphocytes has been observed to be promoted in the context of T2DM. PBMCs in T2DM patients have lower carnitine-acylcarnitine translocase (CACT):carnitine palmitoyltransferase 1A (CPT1A) ratios compared to those in the healthy individuals, indicating the destruction of the mitochondrial long-chain fatty acid import mechanism. This alteration is associated with increased Th17 cytokine production in T2DM[248].
IL-17 may influence glucose metabolism in T2DM
In T2DM patients, serum IL-17 levels have been observed to increase compared to those in healthy subjects[249], decrease compared to normal glucose tolerance[211], and remain the same compared to healthy subjects[250]. No consensus has been established on the dynamics of serum IL-17 in T2DM. However, studies have reported on the molecular mechanisms by which IL-17 causes diabetes. It has been suggested that Th17 cells inhibit the insulin receptor signal via the secretion of IL-17 and IL-22 and cause metabolic disorders[251,252]. In addition, IL-17 activates the nuclear factor-kappa B (NF-κB) pathway[253], regulates the expression of inflammatory cytokine genes, and stimulates the production of IL-1β, IL-6, and TNF-α, resulting in insulin resistance[254]. The treatment of KK-Ay mice with an anti-IL-17 neutralizing antibody significantly increases glucose uptake in the skeletal muscle and decreases concentrations of serum adiponectin and TNF-α[255]. These results indicate that Th17 cells influence glucose metabolism by mediating certain cytokines.
Th17 cells increase in adipose tissue in obese subjects
There is a significant increase in the expression of the IL-17 gene in T-cells[256] and the frequency of Th17 cells[257] in subcutaneous adipose tissue (SAT) for obese patients compared to these parameters in healthy subjects. The frequency of Th17 cells in the VAT increases in obese patients with diabetes compared to that in healthy subjects[258]. In C57BL/6J mice that were fed an HFD for 8-12 weeks, the number of Th17 cells in VAT was the same as that in the controls[145,146], while it significantly increased in SAT[145]. In contrast, in C57BL/6J mice that were fed an HFD for 36 weeks, there was no significant difference in the frequency of Th17 cells in SAT, while there was a significant increase of the frequency of Th17 cells in VAT and a significantly positive correlation of the frequency of Th17 cells with steatosis, ballooning, and lobular inflammation in the liver[259].
The mechanisms of Th17 cell differentiation in adipose tissue are becoming clear
ATP release in stressful environments was increased further in VAT from metabolically unhealthy obese individuals than that in the lean control subjects[260]. Additionally, signal transduction via the P2X7 receptor, which is an extracellular ATP-gated channel, may promote a Th17 cell response in VAT in obese patients[261]. The expression of Rab4b is reduced in adipose tissue T-cells in obesity; Rab4b is a small GTPase that governs endocytic trafficking. Rab4b deficiency in T-cells promotes Th17 cell differentiation in adipose tissue, adipose tissue dysfunction, and insulin resistance[262]. The inflammation of adipose tissue in HFD-fed mice may result from the enhancement of the Th17 cell response by the immature phenotype CD11c+ dendritic cells that are present in adipose tissue[263]. Th17 cells in adipose tissue may be directly involved in adipose tissue inflammation and insulin resistance[264], although this is a substantially complex mechanism, as discussed below.
IL-17 is involved in adipocyte differentiation
Studies have reported that IL-17A contributes to the transmission of inflammation in adipose tissue in human obese patients, although it does not impair adipogenesis and insulin resistance mediated by the inflammatory environment[265]. In contrast, some reports show that Th17 cells may function as a negative regulator of adipogenesis, glucose homeostasis, and obesity via IL-17 secretion[266-268]. Adipose tissue mass is increased compared to that in the control, even in IL-17 knockout mice that were fed an LFD for 14-18 weeks[267]. In vitro experiments have shown that IL-17A inhibits adipocyte differentiation of human bone marrow mesenchymal stem cells (hBM-MSCs) and increases lipolysis of differentiated adipocytes[266]. The inhibitory effect on differentiation of adipocyte was mediated by IL-17A-stimulated upregulation of cyclooxygenase (COX)-2 and elevated levels of prostaglandin (PG) E2[266]. In addition, IL-17 inhibited differentiation into adipocyte from mouse-derived 3T3-L1 preadipocytes and suppressed the expression of the genes encoding pro-adipogenic transcription factors (i.e., peroxisome proliferator-activated receptor γ and CCAAT/enhancer binding protein α), adipokines, and molecules involved in lipid (i.e., fatty acid binding protein 4, perilipin, and adipose triglyceride lipase) and glucose (i.e., glucose transporter-4) metabolism[267]. IL-17 also suppressed the expression of pro-adipogenic Krüppel-like family (KLF) 15, while it enhanced the expression of anti-adipogenic KLF2 and KLF3 in 3T3-L1-cells[268].
IL-17 may be involved in adipose tissue inflammation
IL-17 treatment induced IL-6 mRNA expression and its production in adipocytes differentiated from obese adipose-derived stem cells[265], hBM-MSCs[266], and 3T3-L1 preadipocytes[267]. A recent study observed that adipocyte-derived IL-6 increased macrophage infiltration into adipose tissue, while this was suppressed by myeloid cells and muscle-derived IL-6[269]. This suggests that IL-17 may be involved in the inflammation of adipose tissue via IL-6.
Th17 cells are altered in the mesenteric lymph nodes and small intestine mucosa in obesity
In C57BL/6J mice fed an HFD for 12 weeks, the frequency of Th17 cells in mesenteric lymph nodes was found to increase compared to that in the controls[40,41]. In addition, sleeve gastrectomy and gastric bypass surgery in rats reduced the expression of IL-17 in the jejunum, and changes in IL-17 were strongly correlated with changes in rat body weight and glucose-induced insulin response[270].
In contrast, in a mouse model fed an HFD, changes in the ileum microbiota were found to affect the function of APCs involved in Th17 cell differentiation and reduce the number of Th17 cells in the ileum. This study showed that ROR-γt-deficient mice that were fed a normal diet for 20 weeks experienced impaired glucose tolerance, hyperinsulinemia, and slight insulin resistance. The study suggested that intestinal immune abnormalities, including decreased Th17 cell numbers, were associated with the onset of diabetes[154]. Similarly, in C57BL/6J mice that were fed an HFD for 10 weeks, the frequency of Th17 cells in the mesenteric lymph nodes and small intestinal mucosa was lower than that in the control, and Th17 cells present in the small intestinal mucosa contributed to the development of microbial flora that maintained metabolic homeostasis via IL-17[146]. In IL-17RA−/−mice fed an HFD for 9 weeks, researchers observed metabolic changes such as impaired glucose tolerance and insulin resistance, accompanied by impaired neutrophil migration to intestinal mucosa, increased translocation of commensal bacteria into the bloodstream, and elevated lipopolysaccharide levels in VAT[271]. IL-23-deficient mice that were fed an HFD experienced metabolic alterations such as glucose intolerance and insulin resistance, accompanied by decreased Th17 cell expansion in mesenteric lymph nodes, increased intestinal permeability, the translocation of blood bacteria, and decreased expression of CCL20 in the ileum[272].
IL-17 axis may contribute to the progression of NAFLD
IL-17 causes steatosis in HepG2 cells in vitro in the presence of oleic and palmitic acids by interfering with the insulin signaling pathway[125]. In addition, IL-17 exacerbates palmitic acid-induced hepatocyte lipotoxicity in c-Jun N-terminal kinase (JNK)-dependent mice[119]. Thus, although IL-17 may suppress adipogenesis in adipocytes, it appears to promote fat accumulation in hepatocytes.
The IL-17 axis may contribute to the progression of NAFLD by causing inflammation[273]. In animal models, a defect in the IL-17 axis exerts a protective effect against steatohepatitis. In addition, IL-17RA−/− mice fed an HFD experienced a reduction in immune cell infiltration that was correlated with decreased mRNA expression in the liver for neutrophil chemokines (i.e., CXCL1, CXCL2, CXCL12, and granulocyte-colony-stimulating factor)[274]. In IL-17RA−/−, IL-17A−/−, and IL-17F−/− mice that were fed an MCD, there was a suppression of the infiltration of T-cells and macrophages and the expression of TNF-α in the liver[275]. Treatment with an anti-IL-17mAb in HFD-fed C57BL/6 mice improved liver damage, suppressed Kupffer cell activation, and reduced inflammatory cytokine levels[276].
It has been reported that IL-17 increases the expression of mRNA corresponding to IL-6, α-SMA, collagen, and TGF-β in a concentration-dependent manner in HSCs isolated from naïve C57BL/6 mice; IL-17 may also be involved in liver fibrosis[277]. Therefore, IL-17 may contribute to the progression of NAFLD through its contribution to fat accumulation in hepatocytes and the induction of inflammation and fibrosis in the liver.
Peripheral Th17 cells may be altered in NAFLD
Although many studies have examined the presence of Th17 cells in the disease that underlie NAFLD, only a few studies have examined Th17 cells in the peripheral blood of NAFLD patients. In NAFLD patients, there was either no difference[123] or a significantly higher[28,74] frequency of peripheral Th17 cells in CD4+ T-cells compared to that in healthy individuals. The frequency of Th17 cells in the PBMCs of NAFLD patients did not differ from that in the healthy individuals[112].
In patients with NASH who were experiencing clinical improvements for more than 12 months following bariatric surgery, there was a significant decrease in the frequency of peripheral Th17 cells[123]. There was a significantly positive correlation between the frequency of peripheral Th17 cells in CD4+ T-cells and serum endotoxin[74]. The stimulation of naïve T-cells with endotoxin in vitro led to an increase in the differentiation of naïve T-cells into Th17 cells in NASH patients compared to that in healthy subjects and NAFL patients[74].
In animal models, the frequency of peripheral Th17 cells was the same in C57BL/6J mice fed with an HFD for 36 weeks compared to that in the controls[259]. In contrast, this frequency was higher in C57BL/6J mice fed an HFD for 4 weeks[278] or 12 weeks[41] than that in the controls.
Th17 cells increase in liver tissues in various NASH mouse models and may be associated with inflammation and fibrosis
Although Th17 cells infiltrate the liver and are involved in the pathology of various liver diseases[228-231], only a few studies have examined Th17 cells in the liver tissue of NAFLD patients [Figure 4]. Compared to that in patients with NAFL, the frequency of Th17 cells in CD4+ T-cells in liver tissue is increased in NASH patients[123], and the number of intrahepatic Th17 cells is also increased compared to these parameters in healthy individuals[125].
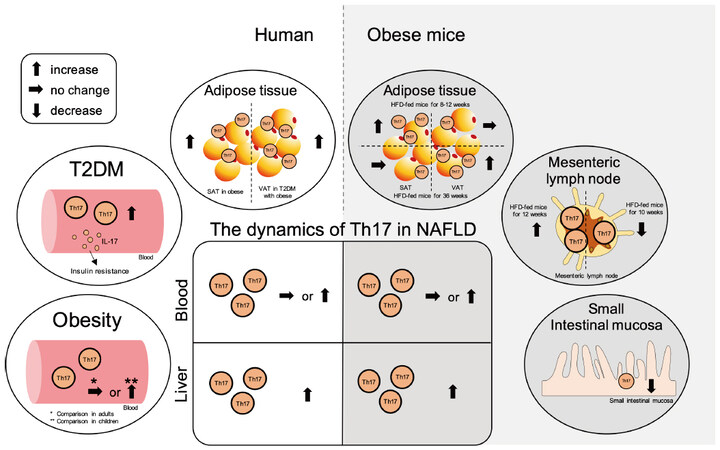
Figure 4. Summary of the contribution of Th17 cells in NAFLD and the related pathologies. The ellipse on the left shows the dynamics of Th17 cells in peripheral blood and adipose tissue in obesity and type 2 diabetes (T2DM) in humans, while the ellipse on the right shows the dynamics of Th17 cells in adipose tissue, mesenteric lymph nodes, and small intestinal mucosa in obese mice. The central square summarizes the Th17 cell dynamics in the peripheral blood and liver tissue in NAFLD. The left side shows the dynamics in humans, and the right side shows the dynamics in mice. NAFLD: Non-alcoholic fatty liver disease; NAFL: non-alcoholic fatty liver; NASH: non-alcoholic steatohepatitis; SAT: subcutaneous adipose tissue; VAT: visceral adipose tissue; Th: T helper; HFD: high-fat diet.
Some studies have shown that the numbers of Th17 cells increase in the liver tissues of various NASH mouse models, and this may be associated with inflammation and fibrosis. In C57BL/6 mice fed an HFD, there was an increased frequency of Th17 cells in CD4+ T-cells in the liver tissue compared to that in the controls[125,259]. The frequency of Th17 cells was positively correlated with histological inflammation[259] as well as with hepatic steatosis and proinflammatory response[125]. In C57BL/6 mice fed an HFD for 12[40,41] and 16 weeks[118], there was a significant increase in the frequency of Th17 cells in liver tissue compared to that in the control. This decreased significantly with an improvement in the pathology of NAFLD upon treatment with antibiotics and Lactobacillus[40]. In mice fed an MCD, there was an increased frequency of Th17 cells in CD4+ T-cells in the liver tissue compared to that in the controls[279]. In this mouse model, the infiltration of Th17 cells in the liver triggered NASH pathogenesis and was important for the progression of liver fibrosis[119]. In MYC-ON mice fed with an MCD, the frequency of Th17 in liver tissue had increased compared to that in the controls[121]. CXCR3 deficiency ameliorates steatohepatitis by attenuating Th1 and Th17 immune responses[280].
TH22
Basic characteristics of Th22 cells
Th22 cells are characterized by the production of IL-22 and by the expression of CCR4, CCR6, and CCR10[281,282]. They develop from naïve T-cells under stimulation by IL-6 and TNF-α[281]. In the differentiation of Th22, an important transcription factor is the aryl hydrocarbon receptor (AHR)[283], while Th22 differentiation is inhibited by high doses of TGF-β[281] [Figure 1]. Th22 cells are associated with various diseases such as skin inflammation[284] and autoimmune disease[285]. In terms of the involvement of Th22 in liver disease, these cells are associated with CHB[286], CHC[287], drug-induced liver injury[288], AIH[289], and HCC[290].
Th22 cells may increase in blood and adipose tissue in obesity
The frequency of peripheral Th22 (IFN-γ−IL-17−IL-22+ CD4+ T-cells) is higher in obese subjects than in healthy subjects[142]. In addition, there is a higher frequency of Th22 (IL-22+ producing CD4+ T-cells) in the SAT in metabolically abnormal obese patients compared to that in healthy or lean individuals[257].
Th22 cells may be associated with the pathology of T2DM
There is an increased frequency of peripheral Th22 cells (IFN-γ−IL-17−IL-22+ CD4+ T-cells) in T2DM patients compared to that in healthy individuals[142,246], and this has been found to be positively correlated with the homeostasis model assessment of insulin resistance (HOMA-IR)[142,246] and BMI[246] and negatively correlated with HOMA-β[142]. The relative mRNA expression of AHR, an important transcriptional factor of Th22 cells[283], in PBMCs in T2DM patients is increased compared to that in lean individuals and is correlated with the frequency of peripheral Th22 cells (IFN-γ−IL-17−IL-22+ CD4+ T-cells)[291]. In addition, Th22 cells (IFN-γ−IL-17−IL-22+ CD4+ T-cells) may be an independent risk factor for cardiovascular complications in diabetes[292]. Th22 cells (IL-17−IL-22+ CD4+ T-cells) are increased in the VAT of T2DM patients, and this is positively correlated with hemoglobin A1c[258]. Based on these findings in obese and diabetic patients, it can be concluded that Th22 cells are increased in the peripheral blood and adipose tissue and are considered to be involved in these diseases. However, based on the effects of IL-22, there is a need for further research regarding the role of Th22 cells in the pathogenesis of obesity.
IL-22 may play a pivotal role in metabolic alterations in obesity
IL-22 is secreted by many lymphoid cells, including Th22 cells, and has a paradoxical dual function of either inhibiting or promoting inflammation in a variety of disease models[293]. A study reported that insulin-mediated glucose uptake in rat muscle with the expression of the IL-22 receptor and insulin sensitivity in the primary hepatocytes with the expression of the IL-22 receptor were reduced by IL-22[257]. Contrastingly, IL-22R1-deficient mice fed an HFD experienced weight gain, impaired glucose tolerance, and insulin resistance. The administration of exogenous IL-22 to genetically obese leptin receptor-deficient (db/db) mice and HFD-fed mice improved hyperglycemia and insulin resistance[294]. Therefore, IL-22 may play a pivotal role in metabolic alterations in obese mice[295].
IL-22 may have a protective role in NAFLD
In liver disease, with activation of the STAT3 pathway, IL-22 is able to protect against various liver disorders, hepatitis, and liver fibrosis[293]. It is also considered to play a crucial role in liver protection and regeneration[296]. IL-22 plays a protective role in hepatic steatosis in HFD-fed mice through the regulation of lipid metabolism in the liver[297]. The IL-22Fc fusion protein suppressed hepatic ROS production, stress kinase activation, and the inflammatory function of hepatocyte-derived extracellular vesicles by inducing hepatic metallothionein; this improved CXCL-1-driven NASH[298]. Blueberry combined with probiotics is a potential therapeutic target for NAFLD, which involves IL-22-mediated activation of Janus kinase 1/STAT3 signaling and the inhibition of the apoptotic factor B-cell lymphoma-2 (Bcl-2)-associated X protein (BAX)[299]. In vitro experiments have demonstrated that IL-22, in the absence of IL-17, prevents palmitate lipotoxicity via the phosphoinositide 3-kinase-mediated inhibition of JNK[119].
The dynamics of Th22 in NAFLD are not known
There are no studies on Th22 cells in the peripheral blood or liver tissue of patients with NAFLD. Although reports on the involvement of IL-22 in NAFLD have increased in recent years, there are limited studies on the animal models of Th22 cells in NAFLD. In C57BL/6 mice fed an MCD, Th22 cells (IL-22+ CD4+ T-cell) increased in number in the liver after 2-4 weeks of being fed an MCD diet[119]. In addition, in IL-17−/− mice fed an MCD, the liver was protected from NASH development and an extensive infiltration of Th22 (IL-22+ CD4+ T-cell) cells occurred[119].
TREG
Basic characteristics of Treg
Treg is characterized by the production of TGF-β, IL-10[300], and IL-35[301] and by the expression of CD25[302] and cytotoxic lymphocyte antigen 4 (CTLA-4)[303]. They develop from naïve T-cells under stimulation by TGF-β[304] and IL-2[305]. One of the important transcription factors involved in Treg differentiation is FoxP3[306], and STAT5 is the important STAT molecule[307]. Treg plays an important role in immune tolerance and the maintenance of immune homeostasis by negatively regulating the immune response through the production of anti-inflammatory cytokines IL-10 and TGF-β[308,309] [Figure 1]. Treg is mainly involved in the pathology of type 1 diabetes[310], allergic disease[311], inflammatory bowel disease[312], and cancer immunity[313]. Furthermore, studies have reported its involvement in viral hepatitis[314], alcoholic liver disease[315], PBC[316], AIH[317], and HCC[318].
Treg may be reduced in obese patients, although its relationship with obesity remains contentions
The relationship between obesity/insulin resistance and Treg, particularly the role of adipose tissue-resident Treg, is becoming clear in animal models; however, the role of peripheral Treg in obese individuals is still being debated. There is an increase in the frequency of peripheral CD25+FoxP3+ Treg in CD4+ T-cells[102] and the frequency of peripheral CD4+CD39+FoxP3+ Treg in obese patients compared to these frequencies in the lean control subjects[244]. In contrast, the frequency of peripheral CD127lowFoxP3+ Treg in CD4+ T-cells remains unaltered in obese patients, while the frequency of CD4+CD45RA−FoxP3high Treg decreases and shows a negative correlation with BMI[319]. In addition, the frequency of peripheral CD25+CD127−FoxP3+ Treg in CD4+ T-cells is lower in obese patients than that in the lean control subjects and is negatively correlated with BMI as well as leptin and hs-CRP levels in plasma[320]. The frequencies of peripheral CD4+CD25+FoxP3+ Treg[321] and CD4+CD25+CD127− Treg[322] were lower in obese patients than that in the lean control subjects.
Peripheral Treg levels decrease in T2DM patients
In T2DM, there is a reduced frequency of peripheral CD4+CD25+FoxP3+ Treg than that in the lean control subjects, and it is significantly negatively correlated with BMI[247]. The frequency of peripheral CD4+CD39+FoxP3+ Treg was decreased in obese patients with T2DM compared to that in obese individuals without T2DM; its frequency was significantly negatively correlated with BMI[244]. The frequency of peripheral CD25highCD127− Tregs in CD4+ T-cells of T2DM patients was lower than that in normoglycemic age-matched controls. This may be attributed to the reduced viability of Tregs in peripheral blood accompanied by a decreased Bcl-2/Bax ratio[143]. A meta-analysis of Treg and proinflammatory immunosuppressive cytokines in T2DM patients showed a reduced frequency of peripheral CD4+CD25+FoxP3+ Treg in T2DM patients compared to that in the healthy controls, and this frequency was further reduced in patients with T2DM-related complications[323].
Tregs in adipose tissue play an important role in maintaining metabolic homeostasis
Functionally specialized Tregs are present in various tissues[324]; one such example is adipose tissue-resident Treg[325]. Its development and maintenance are dependent on the peroxisome proliferator-activated receptor gamma (PPAR-γ)[326], IRF4, basic leucine zipper transcription factor activating transcription factor-like (BATF), and IL-33[327]. In Treg depletion experiments, the loss of Treg exacerbates adipose tissue inflammation, leading to exacerbated metabolic parameters such as increased fasting blood glucose and decreased insulin sensitivity[328,329]. Adoptive transfer of Treg to adipose tissue improves inflammation and insulin resistance[329,330]. In addition, cold exposure and beta-adrenergic stimulation promotes the accumulation of Treg in SAT and brown adipose tissue; this suggests an important role of Treg cells in cold-induced thermogenesis[331]. Thus, Treg in the adipose tissue plays an important role in maintaining metabolic homeostasis through the regulation of adipose inflammation, insulin sensitivity, and thermogenesis[332].
Treg in the adipose tissue may decrease in obesity
Although researchers are continuously attempting to understand the role of Treg in adipose tissue, its role in the context of obesity varies among studies. However, many of the findings are representative of the disruption of the immune homeostasis mechanism due to a decrease in Treg.
In the adipose tissue, there is an increased frequency of CD127lowFoxP3+ Treg in CD4+ T-cells in the VAT in obese patients compared to that in the lean control subjects, and this is significantly positively correlated with BMI. The expression of OX40, which plays an important role in Treg proliferation and survival[333], is significantly enhanced in Tregs in VAT[319]. In addition, FoxP3/CD3E expression is increased in the VAT of obese patients compared to that in the lean control subjects and is significantly positively correlated with CRP and IL-6 levels in plasma[176]. In SAT, the expression of the Foxp3 gene significantly increases in obese patients[334]. In contrast, many studies have reported a decrease in Treg in the adipose tissue of obese individuals. Compared to the parameters in the lean controls, there is a reduced frequency of CD4+CD25+ Treg in the CD3+ T-cells of epididymal adipose in C57BL/6 mice fed an HFD for 12 weeks. The frequency of CD4+CD25+ Treg was negatively correlated with the frequency of CD11b+CD11c+ macrophages in the adipose tissue. Treg differentiation is inhibited by the inflammatory macrophages, as demonstrated by a differentiation assay in vitro[335]. In the VAT of obese patients, there is a lower frequency of FoxP3-positive cells in CD4+ T-cells[145] and CD4+CD25+CD127low Treg[336] than that in the lean controls. In addition, the frequency of CD4+CD45+CD25+FoxP3+ Treg in the omental adipose tissue of obese patients is lower than that in their SAT, and this is negatively correlated with fasting glucose and MCP-1 levels and positively correlated with HOMA-β[337].
Several phenomena have been reported to explain the decrease in Treg in obese adipose tissue. Leptin produced in adipose tissue, the preferred accumulation site for Treg cells, may provide negative control over Treg proliferation[338]. Therefore, elevated leptin levels in obesity[339] may be attributable to the decrease in Tregs in adipose tissue. The expression of IL-21 mRNA was increased in the adipose tissue of C57BL/6J mice fed with an HFD for 16 weeks. In addition, IL-21 knockout mice fed an HFD for 18 weeks experienced greater Treg infiltration in adipose tissue compared to C57BL/6J mice that were fed the same diet. This suggests that IL-21 may be involved in the negative regulation of Tregs in adipose tissue in obesity[340].
Treg is altered in the mesenteric lymph nodes and small intestine mucosa in obesity
In C57BL/6J mice fed an HFD for 12 weeks, the frequency of CD4+CD25+FoxP3+ Treg in the mesenteric lymph nodes was decreased compared to that in the controls[40]. In C57BL/6J mice fed an HFD for 10 weeks, there was little difference in the frequency of FoxP3+CD4+ Treg cells in small intestinal mucosa compared to that in the controls[146]. In contrast, in C57BL/6J mice fed an HFD for 30 days[154] and 12-16 weeks[155], FoxP3+CD4+ Treg was found to be decreased in the small intestinal mucosa.
IL-10 is associated with metabolic syndrome
IL-10 potentially inhibits the production of pro-inflammatory cytokines, such as IL-6[341] and TNF-α[58], that are associated with metabolic syndrome, T2DM, and dyslipidemia[342]. In Sw/Uni mice fed an HFD for 8 weeks, the selective inhibition of IL-10 was associated with increased lipogenesis, TNF-α overexpression, and impaired hepatic insulin sensitivity[343]. Exogenous IL-10 improved insulin action in the skeletal muscle and liver by altering intracellular fat content[344]. In addition, low IL-10 production (i.e., a pro-inflammatory cytokine response) was associated with metabolic syndrome and T2DM[345]. Treg isolated from the VAT of obese hyperinsulinemic mice expressed insulin receptors and experienced specifically impaired IL-10 production; this was attributed to the activation of Treg AKT signaling by insulin. This impaired IL-10 production by Treg promoted macrophage TNF-α production, which may be associated with the chronic inflammation experienced in obesity[346].
IL-10 is associated with liver fibrosis
The importance of Treg in tissue repair has recently been recognized[347]; in the model of CCl4-induced liver injury, IL-10 KO mice developed more extensive fibrosis than C57BL/6 mice[348,349]. In a bile duct ligation mouse model, Treg was decreased, exacerbating liver fibrosis and cholestasis by reducing IL-10 production[350].
Serum IL-10 levels significantly decrease with the progression of NAFLD from simple steatosis to fibrosis[351]. Compared to morbidly obese patients who were not experiencing steatosis, morbidly obese patients with NAFLD showed lower serum IL-10 levels, and serum IL-10 levels tended to decrease depending on the severity of NAFLD[352].
Peripheral Treg may be altered in NAFLD patients
It is suggested that Treg is altered and involved in pathologies associated with NAFLD, such as obesity, T2DM, and the inflammation of adipose tissue. In patients with NAFL/NASH, the frequency of peripheral CD45RA+CD25++ Treg in CD4+ T-cells was significantly reduced compared to that in healthy individuals[123]. In clinical studies, decreased peripheral CD45RA+CD25++ Treg in NASH patients was found to be associated with increased serum cytokeratin-18 fragment M30, and the ratio of Th17 cells to CD45RA+CD25++ Treg may be a risk factor for NASH development[123]. The oral administration of anti-CD3 antibodies may significantly increase the frequency of peripheral CD4+CD25+latency-associated peptide+ Treg and improve the pathology of NASH[353].
In contrast, there was no difference in the frequency of peripheral FoxP3+ Treg[28] and CD25+CD127− Treg[112] in CD4+ T-cells in NAFLD patients compared to that in healthy individuals. In the animal model, there was no change in the frequency of peripheral CD4+CD25+FoxP3+ Treg in C57BL/6 mice that were fed an HFD for 36 weeks compared to that in the controls[259].
Treg in the liver may decrease in NASH mice models
A few reports have examined the role of Treg in the liver tissue of patients with NAFLD. The frequency of FoxP3+ cells in the liver was higher in patients that had an NAFLD activity score of 3-6 points compared to the frequency in those with 0-2 points[124]. The frequency of hepatic CD45RA+CD25++ Treg in CD4+ T-cells tended to decrease, and the frequency of hepatic CD45RA−CD25+++ Treg tended to increase in NASH patients compared to these frequencies in NAFL patients[123].
In C57BL/6 mice fed an HFD for 36 weeks, the frequency of CD25+FoxP3+ Treg in CD4+ T-cells did not change in the liver tissue[259]. In C57BL/6 mice that were fed an HFD for 16 weeks, the frequency of hepatic FoxP3+ Treg cells in CD4+ T-cells tended to increase compared to that in the controls, although there was no significant difference[118]. In contrast, in C57BL/6J mice that were fed an HFD for 12 weeks, the frequency of hepatic CD4+CD25+FoxP3+ Treg cells was significantly decreased compared to that in the controls. The administration of antibiotics or lactic acid bacteria restored this frequency, accompanied by improvements in the condition of NAFLD[40]. In C57BL/6 mice fed an HFD for 8 weeks, the depletion of liver CD4+CD25+FoxP3+ Treg was associated with the low expression of Bcl-2, which protects cells from ROS-induced apoptosis[354]. The depletion of liver Treg results in increased inflammatory signaling and susceptibility to lipopolysaccharide-induced injury[355]. TLR7 signaling in Kupffer and dendritic cells activates TNF-α and type 1 IFN signaling, causing the suppression of intrahepatic Treg and hepatocyte death and ultimately leading to the exacerbation of NASH pathology[356]. In addition, in CD62L−/−mice that were fed an HFD for 24 weeks, liver fibrosis was inhibited due to the increased infiltration of CD45+CD25+FoxP3+ Treg in the liver and the potent activation of the antioxidant stress response[357]. In the context of NAFLD, hepatic Treg tends to decrease; its depletion in the liver may be responsible for disrupting the immune homeostatic mechanism in the liver and causing or maintaining inflammation [Figure 5].
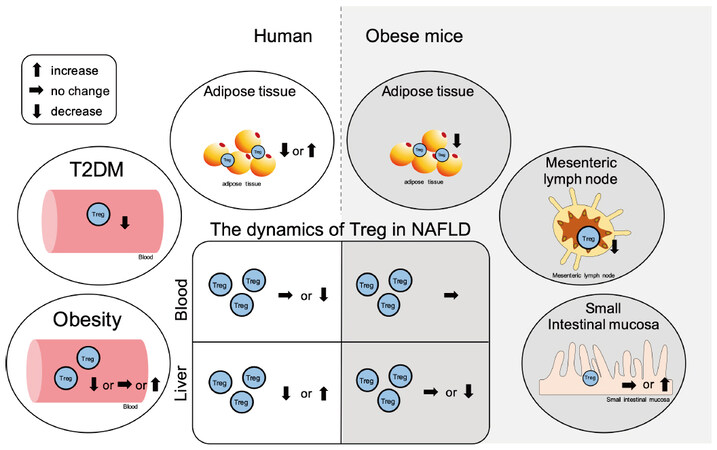
Figure 5. Summary of the contribution of Tregs in NAFLD and the related pathologies. The ellipse on the left shows the dynamics of Tregs in peripheral blood and adipose tissue in obesity and type 2 diabetes (T2DM) in humans, while the ellipse on the right shows the dynamics of Tregs in adipose tissue, mesenteric lymph nodes, and small intestinal mucosa in obese mice. The central square summarizes the Tregs dynamics in the peripheral blood and liver tissue and cytokine dynamics in NAFLD. The left side shows the dynamics in humans, and the right side shows the dynamics in mice. NAFLD: Non-alcoholic fatty liver disease; NAFL: non-alcoholic fatty liver; NASH: non-alcoholic steatohepatitis; Treg: regulatory T-cell.
TFH
Basic characteristics of Tfh
Tfh cells are characterized by the production of IL-4[358], IL-17[359], IL-21[360], and IFN-γ[361] and the expression of CXCR5, programmed cell death 1 (PD1), and inducible costimulatory (ICOS)[362]. They develop from naïve T-cells under stimulation by IL-6, IL-12, IL-21, IL-23, and TGF-β[363,364]. However, the cytokines that regulate murine and human Tfh cell differentiation may be different[365]. Bcl6 was first reported as an important lineage-defining transcription factor of Tfh[366,367], followed by IRF4[368], achaete-scute homolog 2 (Ascl2)[369], T-cell factor 1 (TCF1), and the lymphoid enhancer-binding factor-1 (LEF-1)[370], which positively regulates Tfh cell development. In contrast, Tfh differentiation is inhibited by B lymphocyte-induced maturation protein-1 (Blimp-1)[367]. The key STAT molecules include STAT3 and STAT4[364], which promote Tfh cell differentiation, and STAT5, which inhibits Tfh cell differentiation[371] [Figure 1]. Mature Tfh cells help germinal center (GC) B-cells to promote immunoglobulin affinity maturation, class switch recombination, and development in long-lived plasma cells and memory B-cells[372]. A recent study has reported that the phenotype of Tfh is very diverse and dynamic and that a subset of CD4+ T-cells with “Tfh-like” properties [often termed circulating Tfh (cTfh)] is also present in peripheral blood[372].
The function of Tfh cells may change in T2DM
The frequency of cTfh cells (CXCR5+CD4+ T-cells) was found to be significantly increased in T2DM patients compared to that in controls. In addition, the frequency of cTfh cells was found to be significantly increased in patients with BMI exceeding 24 kg/m2 and in patients with abdominal obesity than in those lacking abdominal obesity[373]. In the small intestinal mucosa of non-obese T2DM patients, IFN-γ production in Tfh cells (CXCR5+CD4+ T-cells) was increased compared to that in healthy subjects. The presence of mucosal Tfh cells may contribute to low-grade inflammation in the intestinal tract in T2DM patients[374]. In T2DM patients with obesity, there was no significant change in the number of cTfh cells (CXCR5+CD4+ T-cells) prior to and after Roux-en-Y gastric bypass (RYGB). However, the expression of ICOS and PD1 in cTfh cells after RYGB was observed to decrease significantly. Furthermore, there was a significantly positive correlation between changes in IL-10 expression in cTfh cells prior to and after RYGB with reductions in glycemia, BMI, and fat mass percentage[375].
B-cells are involved in the pathology of NAFLD
There are no studies on the relationship between Tfh cells and NAFLD; however, recent research has found that B-cells, whose function is assisted by Tfh cells, are involved in metabolic syndrome. B-cells are able to promote insulin resistance through the production of pathogenic IgG[376] and promote obesity and T2DM inflammation through the regulation of inflammatory cytokine profiles and T-cell function[377,378]. Furthermore, the involvement of B-cells in the pathogenesis of NAFLD has also been reported. Intrahepatic B-cells may be involved in NAFLD by inducing the secretion of TNF-α, IL-6, and IgG2a and promoting the activation of CD4+ intrahepatic T-cells and their differentiation into Th1 cells[117]. B-cell activation against oxidative stress-derived epitopes occurs early on during the onset of NASH and contributes to the persistence of hepatitis via the interaction of B-cells with T-cells[157]. In C57BL/6 mice that were fed an HFD, the B-cells induced mesenteric adipose tissue inflammation early during the onset of NAFLD by regulating macrophages; then, they migrated to the liver to induce hepatocyte inflammation[42]. These pathological associations between B-cells and NAFLD may be reflective of the potential contribution of Tfh cells (which support B-cell function) in the pathology of NAFLD.
CHANGES IN IMMUNE DYNAMICS OWING TO THERAPEUTIC INTERVENTION IN NAFLD MODEL
Although steady progress has been made in elucidating the pathogenesis of NAFLD, identifying therapeutic targets, and advancing drug development, there are significant unmet challenges and no drug has been approved for this condition[379]. Here, we summarize the reports centered on animal models demonstrating an improvement in the pathogenesis of NAFLD/NASH by the administration of drugs and the reports that discussed the immunodynamics of CD4+ T-cells [Table 3].
The effect of immune cells due to therapeutic intervention in NAFLD model
| Ref. | Drug | General pharmacological activities | Immunological effect for CD4+ T-cell |
| Inzaugarat et al.[116] | Curcumina | Antioxidant, anti-inflammatory and anti-cancer properties | Ameliorate leptin-induced IFN-γ production in CD4+ T-cells |
| Coia et al.[382] | Theaphenon Eb | Antioxidants | Increase survival of CD4+ T-cell |
| Yue et al.[383] | Kouminec | Antitumor, anti-inflammatory, immunomodulatory activities | Effectively modulate different subtypes of T-cells, such as reducing the Th1 and Th17 cells and increasing Th2 and Treg cells |
| Zhou et al.[384] | Closutrodium butyricum B1 | Probiotics which mainly produced butyrate | Sodium butyrate promote CD4+ T-cell differentiation into Th2 or Treg, and inhibits CD4+ T-cell differentiation into Th1 or Th17 under a cytokine milieu |
| He et al.[386] | Polyene phosphatidylcholined | Antioxidation, anti-inflammation, and immune regulation function | Adjust the imbalance of Th17/Treg cells |
| Alchera et al.[389] | Adenosine A2a receptor agonist | Anti-lipotoxicity | Reduce infiltration to the liver and activation of inflammatory Th subsets and potentiate Treg cell activity |
| Ni et al.[392] | Astaxanthine | Anti-lipid peroxidation and antioxidant | Reduce CD4+ and CD8+ T-cell recruitment in the liver |
| Liu et al.[279] | 3,3’-diindolylmethanef | Anti-inflammation, anti-tumor, anti-mutation, and anti-oxidation | Shift imbalance of Treg/Th17 to Treg dominance and modulate cytokine secretion |
| Kobori et al.[394] | β-Cryptoxanthing | Anti-oxidant | Suppress the accumulation of CD4+ and CD8+ T-cells |
Curcumin, which is known as a natural polyphenol with antioxidant and anti-inflammatory properties, has the potential as an adjunct therapy for the prevention and treatment of NAFLD[380]. Curcumin treatment increased serum IL-13 levels in WD-fed female rats[381]. In C57BL/6 mice that were fed an HFD, the frequency of CD4+ T-cells increased in liver tissue, but oral curcumin administration reduced the accumulation of CD4+ T-cells in hepatic non-parenchymal cells, thereby preventing HFD-induced liver injury, metabolic alterations, and the linoleic acid- and leptin-induced pro-inflammatory and pro-oxidant effects on mouse liver macrophage. Furthermore, curcumin could directly suppress leptin-induced IFN-γ production of CD4+ T-cells[116]. In contrast, in C57BL/6 mice that were fed an HFD, the frequency of CD4+ T-cells in liver tissue decreased, but treatment with Theaphenon E, which is a green tea extract, led to an increase in the number of CD4+ T-cells in the liver tissue with the suppression of weight gain and lipid accumulation in hepatocytes and maintenance of low levels of aspartate aminotransferase and alanine aminotransferase[382]. In an HFD-fed rat model, the proportion of CD4+ T-cells in liver tissue decreased
In NAFLD pathology, changes in the immune system owing to therapeutic interventions suggest that the immune system has some influence on the pathology. Although it is necessary to consider whether the change is the cause or the effect, these results prove that the immune system differs from the normal state at least under NAFLD conditions. In addition, it may be also involved in creating the immune environment[395] that contributes to liver carcinogenesis. Thus, the ability to directly balance the immune system may open new avenues for the treatment of NAFLD.
Recently, the rate of HCC patients with non-viral etiologies continues to increase[396], and there is concern about an increase in the development of HCC in NAFLD patients. Obesity, T2DM, and dyslipidemia in NAFLD patients are associated with the development of HCC[397]. In addition, selective loss of CD4+ T-cell is reported to be involved in NAFLD-associated HCC[121]. Thus, the correction of immunological abnormality, which is caused by NAFLD and NAFLD-associated disease, may further increase the efficacy of immunotherapy for HCC, which is advancing in recent years[398].
IMMUNOMETABOLISM AND NAFLD
We consider the effect of NAFLD pathology on immune cells from the perspective of systemic metabolic diseases, as indicated by the new disease name MAFLD[97-100]. Activated T-cell responses take place via several characteristic phases, such as early cell proliferation, followed by massive clonal expansion and differentiation, contraction or death phases, and establishment and maintenance of immune memory[399]. To exert such complex biological effects, lymphocytes selectively program cellular metabolism[400]. Such metabolic pathways are remarkably influenced by the microenvironment of tissues[96].
Fatty acids are one of the many factors involved in the pathogenesis of NAFLD[401,402]. They also influence the survival of immune cells. In vitro experiments showed cytotoxicity of linoleic acid against lymphocytes[78,79], and a significant negative correlation between linoleic acid and CD4+ T-cell frequency was observed in vivo[28]. Furthermore, it was shown that linoleic acid-induced mitochondrial dysfunction was involved in the selective loss of CD4+ T-cells in the liver tissue in the NASH mouse model[121]. Although these results reflect the direct effects of fatty acids on immune cells, it has recently been shown that fatty acid metabolism is one of the central switches controlling the fate of T-cell differentiation[403]. This suggests that intracellular metabolism disorders may occur in the lymphocytes in the same way that NAFLD causes abnormal lipid metabolism in hepatocytes[404]. It is expected that the dysfunction of lymphocytes owing to intracellular metabolism disorders may modify the pathology of NAFLD. Elucidation of these pathologies may not only result in a potential therapeutic target but also provide a non-invasive diagnostic tool as an initial assessment[405] to identify liver-related complications in NAFLD patients.
CONTRIBUTION OF THE IMMUNE SYSTEM IN THE DEVELOPMENT OF NAFLD-ASSOCIATED HCC
The immune system is able to identify and destroy developing tumor cells in a process known as cancer immunosurveillance; this acts as an important defense against cancer[406]. Senescence surveillance of precancerous hepatocytes is regulated by antigen-specific CD4+ T-cells[407]. CD4+ T-cells have been observed to suppress the formation and progression of diethylnitrosamine-induced liver cancer[408]. Lymphocyte infiltration into the tumor and a high CD4+:CD8+ T-cell ratio have been associated with a reduced risk of tumor recurrence after liver transplantation in HCC[409]. These results suggest that CD4+ T-cells contribute to the development and progression of HCC.
The relationship between CD4+ T-cells with effector functions and HCC is gradually becoming clear. The significantly increased frequency of Th17 cells in the tumors of HCC patients is positively correlated with tumor microvascular density, where many Th17 cells express CCR4 and CCR6. In addition, overall survival and disease-free survival are significantly shorter in HCC patients with a high density of IL-17-producing cells in tumors[231]. Tumor-infiltrating lymphocytes in HCC contain Treg[318,410], wherein the increased proportion and functional expansion of Treg are correlated with the stage of cancer[411]. In hepatitis B virus (HBV)-related HCC, the intratumoral increase of CD4+CD25+FoxP3+ Treg impairs the effector function of CD8+ T-cells in the tumor, and this is involved in the promotion of tumor growth[412]. The infiltration of Th9 cells into HBV-related HCC tumors was involved in tumor promotion through IL-9-mediated phosphorylation of CCL20 and STAT3. The disease-free survival following surgical resection is significantly shorter in HCC, where Th9 cells frequently invade tumors[205]. Dysfunction of cTfh cells (CXCR5+CD4+ T-cells) affects the development of HBV-related HCC, and decrease of cTfh cells is associated with decreased patient survival[413].
The process by which the immune system of the host recognizes and eliminates cancer antigens may be understood through a series of seven steps: (1) the release of cancer antigens; (2) the capture and presentation of cancer antigens by APCs; (3) priming and activation; (4) trafficking of T-cells to tumors; (5) the infiltration of T-cells into tumors; (6) the recognition of cancer cells by T-cells; and (7) the lysis of cancer cells[414]. Any disturbance to these steps will render cancer immunity redundant. The immune response to tumor-related antigens is weaker in NASH-related HCC patients than in HBV and hepatitis C virus-related HCC patients[415]. It has been reported that the selective loss of CD4+ T-cells due to linoleic acid in the liver may contribute to the development of NAFLD-related HCC[121]. This is because PPAR-α, activated by linoleic acid, increases the expression of the CPT enzyme on the mitochondrial membrane. The enhanced mitochondrial uptake ability of linoleic acid has been observed in CD4+ T-cells, and the number of ROS was found to be increased, resulting in the selective apoptosis of CD4+ T-cells[416].
A recent study has found that the tumor microenvironment disrupts the metabolic programs that drive T-cell function and affect antitumor activity[417]. However, in patients with NAFLD, the condition of NAFLD further affects immune function, and the seven steps may be impaired along the way. The clarification of these pathological conditions will help to create new therapeutic strategies and has the potential to further improve the existing therapeutic effects; as such, further research is required.
LIMITATION AND FUTURE PROSPECTIVE
Since T-cells can be classified into various fractions and the classification methods are also diverse[88,90-96], the results of each article should be interpreted with caution. Since the T-cell profile is affected by various factors in the pathophysiology of metabolic syndrome with many organ disorders, its profile is considered to be greatly biased by the disease background of the patient group analyzed. In particular, analysis using human samples is often examined in a small number of cases, and the bias can be more pronounced. In addition, age is an important factor that contributes to the progression of NAFLD, including fibrosis[418] and carcinogenesis[419,420]; the modifier of age also should not be forgotten, as T-cells change with age[421].
Until now, immune cell analysis methods have mainly been evaluated using flow cytometry with fluorochromes[422]. However, the markers that can be evaluated at one time in flow cytometry are limited, and the evaluation of the immune system composed of many phenotypes is also limited. In recent years, with the advent of mass cytometry using mass spectrometry with antibodies that are linked to rare earth metals, it has become possible to comprehensively analyze various cell populations at the same time[423]. In addition, single-cell RNA sequencing has made it possible to analyze gene expression and the mechanisms that control it in detail at the single cell level[424]. By making full use of these techniques and analyzing a large number of cases, if the relationship between the T-cell profile and each disease constituting the metabolic syndrome and the interrelationship between a large number of immune cells are clarified, it is hoped that the role of T-cells in NAFLD will be better understood.
CONCLUSION
This review summarizes the dynamics of CD4+ T-cells in the pathology of NAFLD and the related diseases; peripheral CD4+ T-cells increase in patients with obesity and NAFLD. In NAFLD patients, the gene expression of chemokines and chemokine receptors in the liver increases, enabling lymphocyte infiltration into the liver. In addition, T-cell infiltration increases as fibrosis progresses in patients with NASH. In the NAFLD mouse model, many reports have recently shown that hepatic CD4+ T-cells decrease, contrary to previous reports stating that hepatic CD4+ T-cells increase in such a case.
T-cells may also be divided into various subsets, each of which may be involved in the pathology of obesity, diabetes, and NAFLD. Th1 cells increase in adipose tissue, skeletal muscle, mesenteric lymph node, the small intestine, and colon in obese mice and may also affect metabolic function. In the NAFLD context, peripheral and intrahepatic Th1 cells may increase and may be associated with liver inflammation and fibrosis through IFN-γ. Th2 cells are altered in the peripheral blood and adipose tissue of obese patients and are reduced in number in the adipose tissue and mesenteric lymph nodes of obese mice. However, in the pathophysiology of NAFLD, there are few reports on the dynamics of Th2 in peripheral blood and liver tissue; as such, these details are not yet well understood. Some reports suggest that IL-9 is associated with liver fibrosis and may be altered in obesity and T2DM; however, there are no reports on Th9 cell dynamics in NAFLD or the related pathologies. In obesity, Th17 cells increase in number in the peripheral blood in T2DM and in obese adipose tissue and may be involved in IL-17-mediated glucose metabolism, adipocyte differentiation, and inflammation. In the pathology of NAFLD, Th17 cells increase in number in the liver tissue and are associated with inflammation and fibrosis. Although Th22 cells increase in the peripheral blood and adipose tissue of obese patients and in the peripheral blood of T2DM patients, there are limited reports on NAFLD in Th22 cells. Peripheral Treg in T2DM and adipose tissue Treg in obese individuals may decrease. In NAFLD, the Treg level in the peripheral blood and liver tissue appears to vary; however, the details are unclear. Although Tfh cell function may be altered in T2DM, there are no reports on the direct involvement of Tfh in the pathology of NAFLD.
NAFLD and its associated diseases affect immune cells, either independently or interactively. From the systemic metabolic disease perspective, the function of immune cells may be impaired due to intracellular metabolic disorders. As these effects may be different for each effector T-cell, it is speculated that the environment involved in inflammation, fibrosis, and carcinogenesis is formed through the balance of each immune cell that is modified due to these processes [Figure 6]. Immune cells may have the ability to grasp the state of each organ and exchange information on the immune environment between each organ; this is based on the idea of an inter-organ network as opposed to a single organ. In the context of obesity and NAFLD, it is presumed that this information exchange is also disturbed. Identifying and evaluating the cells that contribute to changes in the balance of immune cells in each organ and information exchange in the immune environment in peripheral blood might lead to the development of new diagnostic tools and therapeutic agents.
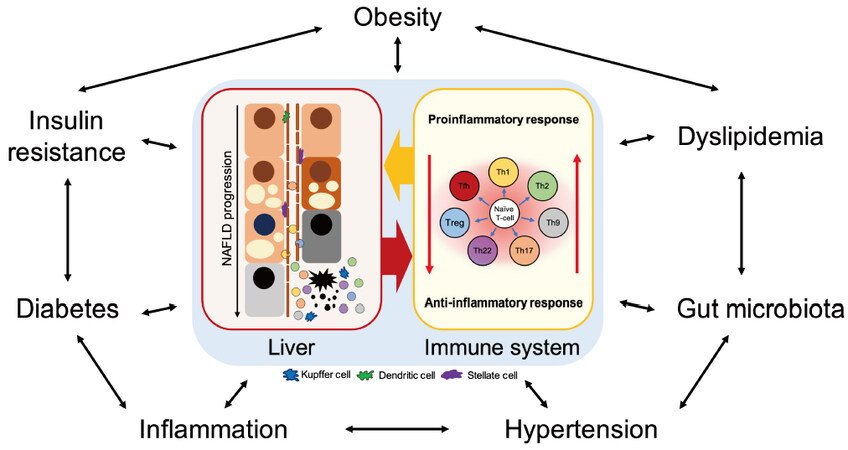
Figure 6. Hypothesis for interpreting the dynamics of CD4+ T-cells in NAFLD. The pathology of NAFLD is affected by various factors, including the immune system. While changes in the liver environment accompanying the development of NAFLD can affect immune cells, changes in the function of immune cells can affect the pathophysiology of NAFLD. From the perspective of systemic metabolic diseases, organ damage associated with NAFLD can affect not only the pathology of NAFLD but also the function of immune cells. As a sum of these effects, we speculate that the balance of the pro- and anti-inflammatory responses of the immune system modifies the pathology of NAFLD. NAFLD: Non-alcoholic fatty liver disease; Th: T helper; Treg: regulatory T-cell; Tfh: T follicular helper.
Finally, the involvement of immune cells in the pathology of NAFLD remains unclear. Hepatitis is inflammation in the liver, and white blood cells play an important role in inflammation. In particular, the marked differentiation of T-cells is important for persistent inflammation[425]. Therefore, understanding the involvement of T-cells in NAFLD is important to better comprehend the pathophysiology and treatment of steatohepatitis.
DECLARATIONS
Authors’ contributionsPerformed the literature review and generated the manuscript: Seike T
Assisted in developing the outline for the manuscript: Mizukoshi E, Kaneko S
Responsible for review of the manuscript: Mizukoshi E
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016;64:73-84.
2. Hamaguchi M, Takeda N, Kojima T, et al. Identification of individuals with non-alcoholic fatty liver disease by the diagnostic criteria for the metabolic syndrome. World J Gastroenterol 2012;18:1508-16.
3. Marchesini G, Brizi M, Bianchi G, et al. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes 2001;50:1844-50.
4. Kim D, Kim WR, Kim HJ, Therneau TM. Association between noninvasive fibrosis markers and mortality among adults with nonalcoholic fatty liver disease in the United States. Hepatology 2013;57:1357-65.
5. Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther 2011;34:274-85.
6. Katsiki N, Mikhailidis DP, Mantzoros CS. Non-alcoholic fatty liver disease and dyslipidemia: An update. Metabolism 2016;65:1109-23.
7. Farrell A, Ryan M, Howell J. Epidemiology of non-alcoholic fatty liver disease-related hepatocellular carcinoma: a western perspective. Hepatoma Res 2020;6:18.
8. Wong RJ, Aguilar M, Cheung R, et al. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015;148:547-55.
9. Boudreau DM, Malone DC, Raebel MA, et al. Health care utilization and costs by metabolic syndrome risk factors. Metab Syndr Relat Disord 2009;7:305-14.
10. Cotter TG, Dong L, Holmen J, Gilroy R, Krong J, Charlton M. Nonalcoholic fatty liver disease: impact on healthcare resource utilization, liver transplantation and mortality in a large, integrated healthcare system. J Gastroenterol 2020;55:722-30.
11. Tiniakos DG, Vos MB, Brunt EM. Nonalcoholic fatty liver disease: pathology and pathogenesis. Annu Rev Pathol 2010;5:145-71.
12. Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology 2010;52:1836-46.
13. Takamura T, Misu H, Ota T, Kaneko S. Fatty liver as a consequence and cause of insulin resistance: lessons from type 2 diabetic liver. Endocr J 2012;59:745-63.
16. Shuai Z, Leung MW, He X, et al. Adaptive immunity in the liver. Cell Mol Immunol 2016;13:354-68.
17. Hu H, Lin A, Kong M, et al. Intestinal microbiome and NAFLD: molecular insights and therapeutic perspectives. J Gastroenterol 2020;55:142-58.
19. Lalor PF, Shields P, Grant A, Adams DH. Recruitment of lymphocytes to the human liver. Immunol Cell Biol 2002;80:52-64.
20. Van Herck MA, Weyler J, Kwanten WJ, et al. The differential roles of T cells in non-alcoholic fatty liver disease and obesity. Front Immunol 2019;10:82.
21. Sutti S, Albano E. Adaptive immunity: an emerging player in the progression of NAFLD. Nat Rev Gastroenterol Hepatol 2020;17:81-92.
22. Sutti S, Jindal A, Locatelli I, et al. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology 2014;59:886-97.
23. Her Z, Tan JHL, Lim YS, et al. CD4+ T cells mediate the development of liver fibrosis in high fat diet-induced NAFLD in humanized mice. Front Immunol 2020;11:580968.
24. Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018;67:328-57.
25. Matteoni C, Younossi Z, Gramlich T, Boparai N, Liu Y, Mccullough A. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology 1999;116:1413-9.
26. Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol 1999;94:2467-74.
27. Kleiner DE, Brunt EM, Van Natta M, et al. Nonalcoholic Steatohepatitis Clinical Research Network. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313-21.
28. Seike T, Mizukoshi E, Yamada K, et al. Fatty acid-driven modifications in T-cell profiles in non-alcoholic fatty liver disease patients. J Gastroenterol 2020;55:701-11.
29. Gadd VL, Skoien R, Powell EE, et al. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014;59:1393-405.
32. Norris S, Collins C, Doherty DG, et al. Resident human hepatitis lymphocytes are phenotypically different from circulating lymphocytes. J Hepatol 1998;28:84-90.
34. Crispe IN, Dao T, Klugewitz K, Mehal WZ, Metz DP. The liver as a site of T-cell apoptosis: graveyard, or killing field? Immunol Rev 2000;174:47-62.
35. Oo YH, Shetty S, Adams DH. The role of chemokines in the recruitment of lymphocytes to the liver. Dig Dis 2010;28:31-44.
36. Pan X, Chiwanda Kaminga A, Liu A, Wen SW, Chen J, Luo J. Chemokines in non-alcoholic fatty liver disease: a systematic review and network meta-analysis. Front Immunol 2020;11:1802.
37. Andrian UH, Mackay CR. T-cell function and migration. Two sides of the same coin. N Engl J Med 2000;343:1020-34.
39. Xu XD, Ueta H, Zhou S, et al. Trafficking of recirculating lymphocytes in the rat liver: rapid transmigration into the portal area and then to the hepatic lymph. Liver Int 2008;28:319-30.
40. Su L, Wu Z, Chi Y, et al. Mesenteric lymph node CD4+ T lymphocytes migrate to liver and contribute to non-alcoholic fatty liver disease. Cell Immunol 2019;337:33-41.
41. Hu Y, Zhang H, Li J, et al. Gut-derived lymphocyte recruitment to liver and induce liver injury in non-alcoholic fatty liver disease mouse model. J Gastroenterol Hepatol 2016;31:676-84.
42. Wu Z, Xu J, Tan J, et al. Mesenteric adipose tissue B lymphocytes promote local and hepatic inflammation in non-alcoholic fatty liver disease mice. J Cell Mol Med 2019;23:3375-85.
43. Tay SS, Wong YC, Roediger B, et al. Intrahepatic activation of naive CD4+ T cells by liver-resident phagocytic cells. J Immunol 2014;193:2087-95.
44. Shetty S, Lalor PF, Adams DH. Lymphocyte recruitment to the liver: molecular insights into the pathogenesis of liver injury and hepatitis. Toxicology 2008;254:136-46.
46. Webb GJ, Hirschfield GM, Krawitt EL, Gershwin ME. Cellular and molecular mechanisms of autoimmune hepatitis. Annu Rev Pathol 2018;13:247-92.
47. Van de Water J, Ansari A, Prindiville T, et al. Heterogeneity of autoreactive T cell clones specific for the E2 component of the pyruvate dehydrogenase complex in primary biliary cirrhosis. J Exp Med 1995;181:723-33.
48. Guidotti LG, Chisari FV. Immunobiology and pathogenesis of viral hepatitis. Annu Rev Pathol 2006;1:23-61.
49. Raza S, Rajak S, Anjum B, Sinha RA. Molecular links between non-alcoholic fatty liver disease and hepatocellular carcinoma. Hepatoma Res 2019;5:42.
50. Komura T, Ohta H, Seike T, et al. The efficacy of corticosteroid therapy in a patient with non-alcoholic steatohepatitis overlapping autoimmune hepatitis. Intern Med 2018;57:807-12.
51. Seike T, Komura T, Shimizu Y, et al. A young man with non-alcoholic steatohepatitis and serum anti-mitochondrial antibody positivity. Intern Med 2018;57:3093-7.
52. Cotler SJ, Kanji K, Keshavarzian A, Jensen DM, Jakate S. Prevalence and significance of autoantibodies in patients with non-alcoholic steatohepatitis. J Clin Gastroenterol 2004;38:801-4.
53. Neuschwander-Tetri BA, Clark JM, Bass NM, et al. NASH Clinical Research Network. Clinical, laboratory and histological associations in adults with nonalcoholic fatty liver disease. Hepatology 2010;52:913-24.
54. Younes R, Govaere O, Petta S, et al. Presence of serum antinuclear antibodies does not impact long-term outcomes in nonalcoholic fatty liver disease. Am J Gastroenterol 2020;115:1289-92.
55. Nishimura S, Manabe I, Nagasaki M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 2009;15:914-20.
56. Cai D, Yuan M, Frantz DF, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med 2005;11:183-90.
57. Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature 2017;542:177-85.
59. Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 2003;112:1821-30.
60. Zhao Y, Lin L, Li J, et al. CD4+ T cells in obesity and obesity-associated diseases. Cell Immunol 2018;332:1-6.
61. Zhou H, Liu F. Regulation, communication, and functional roles of adipose tissue-resident CD4+ T cells in the control of metabolic homeostasis. Front Immunol 2018;9:1961.
62. Tilg H, Zmora N, Adolph TE, Elinav E. The intestinal microbiota fuelling metabolic inflammation. Nat Rev Immunol 2020;20:40-54.
63. Tao L, Liu H, Gong Y. Role and mechanism of the Th17/Treg cell balance in the development and progression of insulin resistance. Mol Cell Biochem 2019;459:183-8.
65. Mailer RKW, Gisterå A, Polyzos KA, Ketelhuth DFJ, Hansson GK. Hypercholesterolemia enhances T cell receptor signaling and increases the regulatory T cell population. Sci Rep 2017;7:15655.
66. Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science 2012;336:1268-73.
67. Aron-Wisnewsky J, Vigliotti C, Witjes J, et al. Gut microbiota and human NAFLD: disentangling microbial signatures from metabolic disorders. Nat Rev Gastroenterol Hepatol 2020;17:279-97.
68. Aron-Wisnewsky J, Warmbrunn MV, Nieuwdorp M, Clément K. Nonalcoholic fatty liver disease: modulating gut microbiota to improve severity? Gastroenterology 2020;158:1881-98.
69. Imajo K, Fujita K, Yoneda M, et al. Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab 2012;16:44-54.
71. González-Navajas JM, Fine S, Law J, et al. TLR4 signaling in effector CD4+ T cells regulates TCR activation and experimental colitis in mice. J Clin Invest 2010;120:570-81.
72. Reynolds JM, Martinez GJ, Chung Y, Dong C. Toll-like receptor 4 signaling in T cells promotes autoimmune inflammation. Proc Natl Acad Sci U S A 2012;109:13064-9.
73. Zanin-Zhorov A, Tal-Lapidot G, Cahalon L, et al. Cutting edge: T cells respond to lipopolysaccharide innately via TLR4 signaling. J Immunol 2007;179:41-4.
74. Wang X, Ji D, Zhu B, et al. Contribution of endotoxin to Th17 bias in patients with non-alcoholic steatohepatitis. Microb Pathog 2020;142:104009.
75. Rolo AP, Teodoro JS, Palmeira CM. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med 2012;52:59-69.
76. Binder CJ, Papac-Milicevic N, Witztum JL. Innate sensing of oxidation-specific epitopes in health and disease. Nat Rev Immunol 2016;16:485-97.
77. Hendrikx T, Binder CJ. Oxidation-specific epitopes in non-alcoholic fatty liver disease. Front Endocrinol (Lausanne) 2020;11:607011.
78. Cury-Boaventura MF, Pompéia C, Curi R. Comparative toxicity of oleic acid and linoleic acid on Jurkat cells. Clin Nutr 2004;23:721-32.
79. Cury-Boaventura MF, Gorjão R, de Lima TM, Newsholme P, Curi R. Comparative toxicity of oleic and linoleic acid on human lymphocytes. Life Sci 2006;78:1448-56.
80. Gorjão R, Cury-Boaventura MF, de Lima TM, Curi R. Regulation of human lymphocyte proliferation by fatty acids. Cell Biochem Funct 2007;25:305-15.
81. Jong AJ, Kloppenburg M, Toes RE, Ioan-Facsinay A. Fatty acids, lipid mediators, and T-cell function. Front Immunol 2014;5:483.
83. Černý J, Stříž I. Adaptive innate immunity or innate adaptive immunity? Clin Sci (Lond) 2019;133:1549-65.
86. Germain RN. MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell 1994;76:287-99.
87. König R, Huang LY, Germain RN. MHC class II interaction with CD4 mediated by a region analogous to the MHC class I binding site for CD8. Nature 1992;356:796-8.
89. Nakayamada S, Takahashi H, Kanno Y, O'Shea JJ. Helper T cell diversity and plasticity. Curr Opin Immunol 2012;24:297-302.
90. Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature 1996;383:787-93.
91. Zhu J, Paul WE. Peripheral CD4+ T-cell differentiation regulated by networks of cytokines and transcription factors. Immunol Rev 2010;238:247-62.
92. Luckheeram RV, Zhou R, Verma AD, Xia B. CD4+ T cells: differentiation and functions. Clin Dev Immunol 2012;2012:925135.
94. Campbell JD, HayGlass KT. T cell chemokine receptor expression in human Th1- and Th2-associated diseases. Arch Immunol Ther Exp (Warsz) 2000;48:451-6.
95. MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol 2013;31:259-83.
96. Joseph AM, Monticelli LA, Sonnenberg GF. Metabolic regulation of innate and adaptive lymphocyte effector responses. Immunol Rev 2018;286:137-47.
97. Younossi ZM, Rinella ME, Sanyal AJ, et al. From NAFLD to MAFLD: implications of a premature change in terminology. Hepatology 2021;73:1194-8.
98. Tilg H, Effenberger M. From NAFLD to MAFLD: when pathophysiology succeeds. Nat Rev Gastroenterol Hepatol 2020;17:387-8.
99. Eslam M, Sanyal AJ, George J. International Consensus Panel. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 2020;158:1999-2014.e1.
100. Eslam M, Newsome PN, Sarin SK, et al. A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J Hepatol 2020;73:202-9.
101. Nieman DC, Henson DA, Nehlsen-cannarella SL, et al. Influence of obesity on immune function. J Am Diet Assoc 1999;99:294-9.
102. van der Weerd K, Dik WA, Schrijver B, et al. Morbidly obese human subjects have increased peripheral blood CD4+ T cells with skewing toward a Treg- and Th2-dominated phenotype. Diabetes 2012;61:401-8.
103. Overwijk WW, Schluns KS. Functions of γC cytokines in immune homeostasis: current and potential clinical applications. Clin Immunol 2009;132:153-65.
104. Wong MM, Fish EN. Chemokines: attractive mediators of the immune response. Semin Immunol 2003;15:5-14.
105. O'Rourke RW, Kay T, Scholz MH, et al. Alterations in T-cell subset frequency in peripheral blood in obesity. Obes Surg 2005;15:1463-8.
106. Fathy SM, Morshed G. Peripheral blood lymphocyte subsets (CD4+, CD8+ T cells), leptin level and weight loss after laparoscopic greater curvature plication in morbidly obese patients. Arch Med Sci 2014;10:886-90.
107. Milner JJ, Beck MA. The impact of obesity on the immune response to infection. Proc Nutr Soc 2012;71:298-306.
108. Si, Isoda F, Yamakawa T, Ishihara M, Sekihara H. T lymphopenia in genetically obese rats. Clin Immunol Immunopathol 1998;86:219-25.
109. Tanaka S, Isoda F, Ishihara Y, Kimura M, Yamakawa T. T lymphopaenia in relation to body mass index and TNF-alpha in human obesity: adequate weight reduction can be corrective. Clin Endocrinol (Oxf) 2001;54:347-54.
110. Inzaugarat ME, Ferreyra Solari NE, Billordo LA, Abecasis R, Gadano AC, Cherñavsky AC. Altered phenotype and functionality of circulating immune cells characterize adult patients with nonalcoholic steatohepatitis. J Clin Immunol 2011;31:1120-30.
111. Maricic I, Marrero I, Eguchi A, et al. Differential activation of hepatic invariant nkt cell subsets plays a key role in progression of nonalcoholic steatohepatitis. J Immunol 2018;201:3017-35.
112. Diedrich T, Kummer S, Galante A, et al. Characterization of the immune cell landscape of patients with NAFLD. PLoS One 2020;15:e0230307.
113. Brunt EM. Nonalcoholic steatohepatitis: definition and pathology. Semin Liver Dis 2001;21:3-16.
115. Bertola A, Bonnafous S, Anty R, et al. Hepatic expression patterns of inflammatory and immune response genes associated with obesity and NASH in morbidly obese patients. PLoS One 2010;5:e13577.
116. Inzaugarat ME, De Matteo E, Baz P, et al. New evidence for the therapeutic potential of curcumin to treat nonalcoholic fatty liver disease in humans. PLoS One 2017;12:e0172900.
117. Zhang F, Jiang WW, Li X, et al. Role of intrahepatic B cells in non-alcoholic fatty liver disease by secreting pro-inflammatory cytokines and regulating intrahepatic T cells. J Dig Dis 2016;17:464-74.
118. Sun G, Jin H, Zhang C, et al. OX40 regulates both innate and adaptive immunity and promotes nonalcoholic steatohepatitis. Cell Rep 2018;25:3786-3799.e4.
119. Rolla S, Alchera E, Imarisio C, et al. The balance between IL-17 and IL-22 produced by liver-infiltrating T-helper cells critically controls NASH development in mice. Clin Sci (Lond) 2016;130:193-203.
120. Hansel C, Erschfeld S, Baues M, et al. The inhibitory T cell receptors PD1 and 2B4 are differentially regulated on CD4 and CD8 T cells in a mouse model of non-alcoholic steatohepatitis. Front Pharmacol 2019;10:244.
121. Ma C, Kesarwala AH, Eggert T, et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016;531:253-7.
122. Ferreyra Solari NE, Inzaugarat ME, Baz P, et al. The role of innate cells is coupled to a Th1-polarized immune response in pediatric nonalcoholic steatohepatitis. J Clin Immunol 2012;32:611-21.
123. Rau M, Schilling AK, Meertens J, et al. Progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis is marked by a higher frequency of Th17 cells in the liver and an increased Th17/resting regulatory T cell ratio in peripheral blood and in the liver. J Immunol 2016;196:97-105.
124. Söderberg C, Marmur J, Eckes K, et al. Microvesicular fat, inter cellular adhesion molecule-1 and regulatory T-lymphocytes are of importance for the inflammatory process in livers with non-alcoholic steatohepatitis. APMIS 2011;119:412-20.
125. Tang Y, Bian Z, Zhao L, et al. Interleukin-17 exacerbates hepatic steatosis and inflammation in non-alcoholic fatty liver disease. Clin Exp Immunol 2011;166:281-90.
126. Alegre NS, Garcia CC, Billordo LA, et al. Limited expression of TLR9 on T cells and its functional consequences in patients with nonalcoholic fatty liver disease. Clin Mol Hepatol 2020;26:216-26.
127. Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015;74:5-17.
128. Loetscher P, Uguccioni M, Bordoli L, et al. CCR5 is characteristic of Th1 lymphocytes. Nature 1998;391:344-5.
129. Bonecchi R, Bianchi G, Bordignon PP, et al. Differential expression of chemokine receptors and chemotactic responsiveness of type 1 T helper cells (Th1s) and Th2s. J Exp Med 1998;187:129-34.
130. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman C, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 2000;100:655-69.
131. Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature 1996;382:174-7.
132. Afkarian M, Sedy JR, Yang J, et al. T-bet is a STAT1-induced regulator of IL-12R expression in naïve CD4+ T cells. Nat Immunol 2002;3:549-57.
133. Ouyang W, Ranganath SH, Weindel K, et al. Inhibition of Th1 development mediated by GATA-3 through an IL-4-independent mechanism. Immunity 1998;9:745-55.
134. Luo XY, Takahara T, Kawai K, et al. IFN-γ deficiency attenuates hepatic inflammation and fibrosis in a steatohepatitis model induced by a methionine- and choline-deficient high-fat diet. Am J Physiol Gastrointest Liver Physiol 2013;305:G891-9.
135. Walker LS, von Herrath M. CD4 T cell differentiation in type 1 diabetes. Clin Exp Immunol 2016;183:16-29.
136. Vyas SP, Hansda AK, Goswami R. Rheumatoid arthritis: 'melting pot' of T helper subsets. Int Rev Immunol 2019;38:212-31.
137. Grigoriadis N, van Pesch V. ParadigMS Group. A basic overview of multiple sclerosis immunopathology. Eur J Neurol 2015;22 Suppl 2:3-13.
138. Kobayashi T, Okamoto S, Hisamatsu T, et al. IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn's disease. Gut 2008;57:1682-9.
139. Bovensiepen CS, Schakat M, Sebode M, et al. TNF-producing Th1 cells are selectively expanded in liver infiltrates of patients with autoimmune hepatitis. J Immunol 2019;203:3148-56.
140. Harada K, Isse K, Kamihira T, Shimoda S, Nakanuma Y. Th1 cytokine-induced downregulation of PPARgamma in human biliary cells relates to cholangitis in primary biliary cirrhosis. Hepatology 2005;41:1329-38.
141. Lin F, Taylor NJ, Su H, et al. Alcohol dehydrogenase-specific T-cell responses are associated with alcohol consumption in patients with alcohol-related cirrhosis. Hepatology 2013;58:314-24.
142. Zhao R, Tang D, Yi S, et al. Elevated peripheral frequencies of Th22 cells: a novel potent participant in obesity and type 2 diabetes. PLoS One 2014;9:e85770.
143. Zeng C, Shi X, Zhang B, et al. The imbalance of Th17/Th1/Tregs in patients with type 2 diabetes: relationship with metabolic factors and complications. J Mol Med (Berl) 2012;90:175-86.
144. McLaughlin T, Liu LF, Lamendola C, et al. T-cell profile in adipose tissue is associated with insulin resistance and systemic inflammation in humans. Arterioscler Thromb Vasc Biol 2014;34:2637-43.
145. Winer S, Chan Y, Paltser G, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med 2009;15:921-9.
146. Hong CP, Park A, Yang BG, et al. Gut-specific delivery of T-helper 17 cells reduces obesity and insulin resistance in mice. Gastroenterology 2017;152:1998-2010.
147. Khan IM, Dai Perrard XY, Perrard JL, et al. Attenuated adipose tissue and skeletal muscle inflammation in obese mice with combined CD4+ and CD8+ T cell deficiency. Atherosclerosis 2014;233:419-28.
148. Rocha VZ, Folco EJ, Sukhova G, et al. Interferon-gamma, a Th1 cytokine, regulates fat inflammation: a role for adaptive immunity in obesity. Circ Res 2008;103:467-76.
149. Stolarczyk E, Vong CT, Perucha E, et al. Improved insulin sensitivity despite increased visceral adiposity in mice deficient for the immune cell transcription factor T-bet. Cell Metab 2013;17:520-33.
150. Kintscher U, Hartge M, Hess K, et al. T-lymphocyte infiltration in visceral adipose tissue: a primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arterioscler Thromb Vasc Biol 2008;28:1304-10.
151. Zhang H, Potter BJ, Cao JM, Zhang C. Interferon-gamma induced adipose tissue inflammation is linked to endothelial dysfunction in type 2 diabetic mice. Basic Res Cardiol 2011;106:1135-45.
152. Wong N, Fam BC, Cempako GR, et al. Deficiency in interferon-gamma results in reduced body weight and better glucose tolerance in mice. Endocrinology 2011;152:3690-9.
153. O'Rourke RW, White AE, Metcalf MD, et al. Systemic inflammation and insulin sensitivity in obese IFN-γ knockout mice. Metabolism 2012;61:1152-61.
154. Garidou L, Pomié C, Klopp P, et al. The gut microbiota regulates intestinal CD4 T cells expressing RORγt and controls metabolic disease. Cell Metab 2015;22:100-12.
155. Luck H, Tsai S, Chung J, et al. Regulation of obesity-related insulin resistance with gut anti-inflammatory agents. Cell Metab 2015;21:527-42.
156. Beaurepaire C, Smyth D, McKay DM. Interferon-gamma regulation of intestinal epithelial permeability. J Interferon Cytokine Res 2009;29:133-44.
157. Bruzzì S, Sutti S, Giudici G, et al. B2-Lymphocyte responses to oxidative stress-derived antigens contribute to the evolution of nonalcoholic fatty liver disease (NAFLD). Free Radic Biol Med 2018;124:249-59.
158. Li Z, Soloski MJ, Diehl AM. Dietary factors alter hepatic innate immune system in mice with nonalcoholic fatty liver disease. Hepatology 2005;42:880-5.
159. Kremer M, Hines IN, Milton RJ, Wheeler MD. Favored T helper 1 response in a mouse model of hepatosteatosis is associated with enhanced T cell-mediated hepatitis. Hepatology 2006;44:216-27.
160. Baroni GS, D'Ambrosio L, Curto P, et al. Interferon gamma decreases hepatic stellate cell activation and extracellular matrix deposition in rat liver fibrosis. Hepatology 1996;23:1189-99.
161. Rockey DC, Chung JJ. Interferon gamma inhibits lipocyte activation and extracellular matrix mRNA expression during experimental liver injury: implications for treatment of hepatic fibrosis. J Investig Med 1994;42:660-70.
162. Kado A, Tsutsumi T, Enooku K, et al. Noninvasive diagnostic criteria for nonalcoholic steatohepatitis based on gene expression levels in peripheral blood mononuclear cells. J Gastroenterol 2019;54:730-41.
163. Pacifico L, Di Renzo L, Anania C, et al. Increased T-helper interferon-gamma-secreting cells in obese children. Eur J Endocrinol 2006;154:691-7.
164. Maher JJ, Leon P, Ryan JC. Beyond insulin resistance: Innate immunity in nonalcoholic steatohepatitis. Hepatology 2008;48:670-8.
165. Gieseck RL 3rd, Wilson MS, Wynn TA. Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol 2018;18:62-76.
166. Sallusto F, Lenig D, Mackay CR, Lanzavecchia A. Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J Exp Med 1998;187:875-83.
167. D'Ambrosio D, Iellem A, Bonecchi R, et al. Selective up-regulation of chemokine receptors CCR4 and CCR8 upon activation of polarized human type 2 Th cells. J Immunol 1998;161:5111-5.
168. Paul WE, Zhu J. How are T(H)2-type immune responses initiated and amplified? Nat Rev Immunol 2010;10:225-35.
169. Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 1997;89:587-96.
170. Zhu J, Cote-sierra J, Guo L, Paul WE. Stat5 activation plays a critical role in Th2 differentiation. Immunity 2003;19:739-48.
171. Hwang ES, Szabo SJ, Schwartzberg PL, Glimcher LH. T helper cell fate specified by kinase-mediated interaction of T-bet with GATA-3. Science 2005;307:430-3.
173. Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol 2014;14:181-94.
174. Liu Y, Munker S, Müllenbach R, Weng HL. IL-13 signaling in liver fibrogenesis. Front Immunol 2012;3:116.
175. Foerster F, Hess M, Gerhold-Ay A, et al. The immune contexture of hepatocellular carcinoma predicts clinical outcome. Sci Rep 2018;8:5351.
176. Zeyda M, Huber J, Prager G, Stulnig TM. Inflammation correlates with markers of T-cell subsets including regulatory T cells in adipose tissue from obese patients. Obesity (Silver Spring) 2011;19:743-8.
177. Festa A, D'Agostino R Jr, Howard G, Mykkänen L, Tracy RP, Haffner SM. Chronic subclinical inflammation as part of the insulin resistance syndrome: the Insulin Resistance Atherosclerosis Study (IRAS). Circulation 2000;102:42-7.
178. Tsai S, Clemente-Casares X, Zhou AC, et al. Insulin receptor-mediated stimulation boosts T cell immunity during inflammation and infection. Cell Metab 2018;28:922-934.e4.
179. Viardot A, Grey ST, Mackay F, Chisholm D. Potential antiinflammatory role of insulin via the preferential polarization of effector T cells toward a T helper 2 phenotype. Endocrinology 2007;148:346-53.
180. Viardot A, Heilbronn LK, Samocha-Bonet D, Mackay F, Campbell LV, Samaras K. Obesity is associated with activated and insulin resistant immune cells. Diabetes Metab Res Rev 2012;28:447-54.
181. Viardot A, Lord RV, Samaras K. The effects of weight loss and gastric banding on the innate and adaptive immune system in type 2 diabetes and prediabetes. J Clin Endocrinol Metab 2010;95:2845-50.
182. Villarreal-Calderón JR, Cuéllar RX, Ramos-González MR, et al. Interplay between the adaptive immune system and insulin resistance in weight loss induced by bariatric surgery. Oxid Med Cell Longev 2019;2019:3940739.
183. Schmitz J, Owyang A, Oldham E, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005;23:479-90.
184. Miller AM, Asquith DL, Hueber AJ, et al. Interleukin-33 induces protective effects in adipose tissue inflammation during obesity in mice. Circ Res 2010;107:650-8.
185. Molofsky AB, Nussbaum JC, Liang HE, et al. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med 2013;210:535-49.
186. Grencis RK. Immunity to helminths: resistance, regulation, and susceptibility to gastrointestinal nematodes. Annu Rev Immunol 2015;33:201-25.
187. Su CW, Chen CY, Jiao L, et al. Helminth-induced and Th2-dependent alterations of the gut microbiota attenuate obesity caused by high-fat diet. Cell Mol Gastroenterol Hepatol 2020;10:763-78.
188. Su CW, Chen CY, Li Y, et al. Helminth infection protects against high fat diet-induced obesity via induction of alternatively activated macrophages. Sci Rep 2018;8:4607.
189. Darkhal P, Gao M, Ma Y, Liu D. Blocking high-fat diet-induced obesity, insulin resistance and fatty liver by overexpression of Il-13 gene in mice. Int J Obes (Lond) 2015;39:1292-9.
190. Kwon H, Laurent S, Tang Y, Zong H, Vemulapalli P, Pessin JE. Adipocyte-specific IKKβ signaling suppresses adipose tissue inflammation through an IL-13-dependent paracrine feedback pathway. Cell Rep 2014;9:1574-83.
191. Stanya KJ, Jacobi D, Liu S, et al. Direct control of hepatic glucose production by interleukin-13 in mice. J Clin Invest 2013;123:261-71.
192. Lee CG, Homer RJ, Zhu Z, et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J Exp Med 2001;194:809-21.
193. Shimamura T, Fujisawa T, Husain SR, Kioi M, Nakajima A, Puri RK. Novel role of IL-13 in fibrosis induced by nonalcoholic steatohepatitis and its amelioration by IL-13R-directed cytotoxin in a rat model. J Immunol 2008;181:4656-65.
194. Hart KM, Fabre T, Sciurba JC, et al. Type 2 immunity is protective in metabolic disease but exacerbates NAFLD collaboratively with TGF-β. Sci Transl Med 2017;9:eaal3694.
195. Chen T, Guo J, Cai Z, et al. Th9 cell differentiation and its dual effects in tumor development. Front Immunol 2020;11:1026.
196. Veldhoen M, Uyttenhove C, van Snick J, et al. Transforming growth factor-beta 'reprograms' the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol 2008;9:1341-6.
197. Noelle RJ, Nowak EC. Cellular sources and immune functions of interleukin-9. Nat Rev Immunol 2010;10:683-7.
198. Chang HC, Sehra S, Goswami R, et al. The transcription factor PU.1 is required for the development of IL-9-producing T cells and allergic inflammation. Nat Immunol 2010;11:527-34.
199. Goswami R, Jabeen R, Yagi R, et al. STAT6-dependent regulation of Th9 development. J Immunol 2012;188:968-75.
200. Staudt V, Bothur E, Klein M, et al. Interferon-regulatory factor 4 is essential for the developmental program of T helper 9 cells. Immunity 2010;33:192-202.
201. Deng Y, Wang Z, Chang C, Lu L, Lau CS, Lu Q. Th9 cells and IL-9 in autoimmune disorders: Pathogenesis and therapeutic potentials. Hum Immunol 2017;78:120-8.
202. Purwar R, Schlapbach C, Xiao S, et al. Robust tumor immunity to melanoma mediated by interleukin-9-producing T cells. Nat Med 2012;18:1248-53.
203. Cui M, Lv Y, Lu J, et al. Decreased frequency of circulating Th9 cells in patients with chronic hepatitis B infection. J Clin Lab Anal 2018;32:e22246.
204. Qin SY, Lu DH, Guo XY, et al. A deleterious role for Th9/IL-9 in hepatic fibrogenesis. Sci Rep 2016;6:18694.
205. Tan H, Wang S, Zhao L. A tumour-promoting role of Th9 cells in hepatocellular carcinoma through CCL20 and STAT3 pathways. Clin Exp Pharmacol Physiol 2017;44:213-21.
206. Guo X, Cen Y, Wang J, Jiang H. CXCL10-induced IL-9 promotes liver fibrosis via Raf/MEK/ERK signaling pathway. Biomed Pharmacother 2018;105:282-9.
207. Zhan T, Ma H, Jiang S, et al. Interleukin-9 blockage reduces early hepatic granuloma formation and fibrosis during Schistosoma japonicum infection in mice. Immunology 2019;158:296-303.
208. Kaplan MH, Hufford MM, Olson MR. The development and in vivo function of T helper 9 cells. Nat Rev Immunol 2015;15:295-307.
209. Dalmas E, Rouault C, Abdennour M, et al. Variations in circulating inflammatory factors are related to changes in calorie and carbohydrate intakes early in the course of surgery-induced weight reduction. Am J Clin Nutr 2011;94:450-8.
210. Hang H, Yuan S, Yang Q, Yuan D, Liu Q. Multiplex bead array assay of plasma cytokines in type 2 diabetes mellitus with diabetic retinopathy. Mol Vis 2014;20:1137-45.
211. Vasanthakumar R, Mohan V, Anand G, Deepa M, Babu S, Aravindhan V. Serum IL-9, IL-17, and TGF-β levels in subjects with diabetic kidney disease (CURES-134). Cytokine 2015;72:109-12.
212. Chen H, Wen F, Zhang X, Su SB. Expression of T-helper-associated cytokines in patients with type 2 diabetes mellitus with retinopathy. Mol Vis 2012;18:219-26.
213. Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity 2008;28:454-67.
214. Hammerich L, Heymann F, Tacke F. Role of IL-17 and Th17 cells in liver diseases. Clin Dev Immunol 2011;2011:345803.
215. Acosta-Rodriguez EV, Rivino L, Geginat J, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol 2007;8:639-46.
216. Mangan PR, Harrington LE, O'Quinn DB, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 2006;441:231-4.
217. Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006;441:235-8.
218. Ivanov II, McKenzie BS, Zhou L, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006;126:1121-33.
219. Durant L, Watford WT, Ramos HL, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity 2010;32:605-15.
220. Ichiyama K, Hashimoto M, Sekiya T, et al. Gfi1 negatively regulates T(h)17 differentiation by inhibiting RORgammat activity. Int Immunol 2009;21:881-9.
221. Cui G, Qin X, Wu L, et al. Liver X receptor (LXR) mediates negative regulation of mouse and human Th17 differentiation. J Clin Invest 2011;121:658-70.
222. Ichiyama K, Sekiya T, Inoue N, et al. Transcription factor Smad-independent T helper 17 cell induction by transforming-growth factor-β is mediated by suppression of eomesodermin. Immunity 2011;34:741-54.
223. Laurence A, Tato CM, Davidson TS, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity 2007;26:371-81.
224. Bystrom J, Clanchy FI, Taher TE, et al. TNFα in the regulation of Treg and Th17 cells in rheumatoid arthritis and other autoimmune inflammatory diseases. Cytokine 2018;101:4-13.
225. Geremia A, Biancheri P, Allan P, Corazza GR, Di Sabatino A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun Rev 2014;13:3-10.
226. Tahmasebinia F, Pourgholaminejad A. The role of Th17 cells in auto-inflammatory neurological disorders. Prog Neuropsychopharmacol Biol Psychiatry 2017;79:408-16.
227. Blauvelt A, Chiricozzi A. The immunologic role of IL-17 in psoriasis and psoriatic arthritis pathogenesis. Clin Rev Allergy Immunol 2018;55:379-90.
228. Zhang JY, Zhang Z, Lin F, et al. Interleukin-17-producing CD4(+) T cells increase with severity of liver damage in patients with chronic hepatitis B. Hepatology 2010;51:81-91.
229. Lemmers A, Moreno C, Gustot T, et al. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology 2009;49:646-57.
230. Lan RY, Salunga TL, Tsuneyama K, et al. Hepatic IL-17 responses in human and murine primary biliary cirrhosis. J Autoimmun 2009;32:43-51.
231. Zhang JP, Yan J, Xu J, et al. Increased intratumoral IL-17-producing cells correlate with poor survival in hepatocellular carcinoma patients. J Hepatol 2009;50:980-9.
232. Weiss R, Dziura J, Burgert TS, et al. Obesity and the metabolic syndrome in children and adolescents. N Engl J Med 2004;350:2362-74.
233. Winer S, Paltser G, Chan Y, et al. Obesity predisposes to Th17 bias. Eur J Immunol 2009;39:2629-35.
234. Du C, Liu C, Kang J, et al. MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol 2009;10:1252-9.
235. Vega-Cárdenas M, Uresti-Rivera EE, Cortés-García JD, et al. Increased levels of adipose tissue-resident Th17 cells in obesity associated with miR-326. Immunol Lett 2019;211:60-7.
236. Arita Y, Kihara S, Ouchi N, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun 1999;257:79-83.
237. Surendar J, Frohberger SJ, Karunakaran I, et al. Adiponectin limits IFN-γ and IL-17 producing CD4 T cells in obesity by restraining cell intrinsic glycolysis. Front Immunol 2019;10:2555.
238. Endo Y, Asou HK, Matsugae N, et al. Obesity drives Th17 cell differentiation by inducing the lipid metabolic kinase, ACC1. Cell Rep 2015;12:1042-55.
239. Wakil SJ, Stoops JK, Joshi VC. Fatty acid synthesis and its regulation. Annu Rev Biochem 1983;52:537-79.
240. Łuczyński W, Grubczak K, Moniuszko M, Głowińska-Olszewska B, Bossowski A. Elevated levels of Th17 cells in children with central obesity. Scand J Clin Lab Invest 2015;75:595-601.
241. Ahmed M, Gaffen SL. IL-17 in obesity and adipogenesis. Cytokine Growth Factor Rev 2010;21:449-53.
242. Sumarac-Dumanovic M, Stevanovic D, Ljubic A, et al. Increased activity of interleukin-23/interleukin-17 proinflammatory axis in obese women. Int J Obes (Lond) 2009;33:151-6.
243. Jung C, Lichtenauer M, Strodthoff D, et al. Alterations in systemic levels of Th1, Th2, and Th17 cytokines in overweight adolescents and obese mice. Pediatr Diabetes 2017;18:714-21.
244. Cortez-Espinosa N, Cortés-Garcia JD, Martínez-Leija E, et al. CD39 expression on Treg and Th17 cells is associated with metabolic factors in patients with type 2 diabetes. Hum Immunol 2015;76:622-30.
245. Chen H, Ren X, Liao N, Wen F. Th17 cell frequency and IL-17A concentrations in peripheral blood mononuclear cells and vitreous fluid from patients with diabetic retinopathy. J Int Med Res 2016;44:1403-13.
246. Guo H, Xu BC, Yang XG, et al. A high frequency of peripheral blood IL-22(+) CD4(+) T cells in patients with new onset type 2 diabetes mellitus. J Clin Lab Anal 2016;30:95-102.
247. Jagannathan-Bogdan M, McDonnell ME, Shin H, et al. Elevated proinflammatory cytokine production by a skewed T cell compartment requires monocytes and promotes inflammation in type 2 diabetes. J Immunol 2011;186:1162-72.
248. Nicholas DA, Proctor EA, Agrawal M, et al. Fatty acid metabolites combine with reduced β oxidation to activate Th17 inflammation in human type 2 diabetes. Cell Metab 2019;30:447-461.e5.
249. Arababadi MK, Nosratabadi R, Hassanshahi G, et al. Nephropathic complication of type-2 diabetes is following pattern of autoimmune diseases? Diabetes Res Clin Pract 2010;87:33-7.
250. Roohi A, Tabrizi M, Abbasi F, et al. Serum IL-17, IL-23, and TGF-β levels in type 1 and type 2 diabetic patients and age-matched healthy controls. Biomed Res Int 2014;2014:718946.
251. O'Rourke RW, Lumeng CN. Obesity heats up adipose tissue lymphocytes. Gastroenterology 2013;145:282-5.
252. Chuang HC, Sheu WH, Lin YT, et al. HGK/MAP4K4 deficiency induces TRAF2 stabilization and Th17 differentiation leading to insulin resistance. Nat Commun 2014;5:4602.
253. Zepp J, Wu L, Li X. IL-17 receptor signaling and T helper 17-mediated autoimmune demyelinating disease. Trends Immunol 2011;32:232-9.
254. Chen L, Chen R, Wang H, Liang F. Mechanisms linking inflammation to insulin resistance. Int J Endocrinol 2015;2015:508409.
255. Ohshima K, Mogi M, Jing F, et al. Roles of interleukin 17 in angiotensin II type 1 receptor-mediated insulin resistance. Hypertension 2012;59:493-9.
256. Bertola A, Ciucci T, Rousseau D, et al. Identification of adipose tissue dendritic cells correlated with obesity-associated insulin-resistance and inducing Th17 responses in mice and patients. Diabetes 2012;61:2238-47.
257. Fabbrini E, Cella M, McCartney SA, et al. Association between specific adipose tissue CD4+ T-cell populations and insulin resistance in obese individuals. Gastroenterology 2013;145:366-74.e1.
258. Dalmas E, Venteclef N, Caer C, et al. T cell-derived IL-22 amplifies IL-1β-driven inflammation in human adipose tissue: relevance to obesity and type 2 diabetes. Diabetes 2014;63:1966-77.
259. Vonghia L, Ruyssers N, Schrijvers D, et al. CD4+ROR γ t++ and Tregs in a mouse model of diet-induced nonalcoholic steatohepatitis. Mediators Inflamm 2015;2015:239623.
260. Pandolfi J, Ferraro A, Lerner M, et al. Purinergic signaling modulates human visceral adipose inflammatory responses: implications in metabolically unhealthy obesity. J Leukoc Biol 2015;97:941-9.
261. Pandolfi JB, Ferraro AA, Sananez I, et al. ATP-induced inflammation drives tissue-resident Th17 cells in metabolically unhealthy obesity. J Immunol 2016;196:3287-96.
262. Gilleron J, Bouget G, Ivanov S, et al. Rab4b deficiency in T cells promotes adipose Treg/Th17 imbalance, adipose tissue dysfunction, and insulin resistance. Cell Rep 2018;25:3329-3341.e5.
263. Chen Y, Tian J, Tian X, et al. Adipose tissue dendritic cells enhances inflammation by prompting the generation of Th17 cells. PLoS One 2014;9:e92450.
264. Wang Q, Wu H. T cells in adipose tissue: critical players in immunometabolism. Front Immunol 2018;9:2509.
265. Pestel J, Chehimi M, Bonhomme M, Robert M, Vidal H, Eljaafari A. IL-17A contributes to propagation of inflammation but does not impair adipogenesis and/or insulin response, in adipose tissue of obese individuals. Cytokine 2020;126:154865.
266. Shin JH, Shin DW, Noh M. Interleukin-17A inhibits adipocyte differentiation in human mesenchymal stem cells and regulates pro-inflammatory responses in adipocytes. Biochem Pharmacol 2009;77:1835-44.
267. Zúñiga LA, Shen WJ, Joyce-Shaikh B, et al. IL-17 regulates adipogenesis, glucose homeostasis, and obesity. J Immunol 2010;185:6947-59.
268. Ahmed M, Gaffen SL. IL-17 inhibits adipogenesis in part via C/EBPα, PPARγ and Krüppel-like factors. Cytokine 2013;61:898-905.
269. Han MS, White A, Perry RJ, et al. Regulation of adipose tissue inflammation by interleukin 6. Proc Natl Acad Sci U S A 2020;117:2751-60.
270. Subramaniam R, Aliakbarian H, Bhutta HY, Harris DA, Tavakkoli A, Sheu EG. Sleeve gastrectomy and Roux-en-Y gastric bypass attenuate pro-inflammatory small intestinal cytokine signatures. Obes Surg 2019;29:3824-32.
271. Pérez MM, Martins LMS, Dias MS, et al. Interleukin-17/interleukin-17 receptor axis elicits intestinal neutrophil migration, restrains gut dysbiosis and lipopolysaccharide translocation in high-fat diet-induced metabolic syndrome model. Immunology 2019;156:339-55.
272. Martins LMS, Perez MM, Pereira CA, et al. Interleukin-23 promotes intestinal T helper type17 immunity and ameliorates obesity-associated metabolic syndrome in a murine high-fat diet model. Immunology ;2018:624-36.
273. Giles DA, Moreno-Fernandez ME, Divanovic S. IL-17 axis driven inflammation in non-alcoholic fatty liver disease progression. Curr Drug Targets 2015;16:1315-23.
274. Harley IT, Stankiewicz TE, Giles DA, et al. IL-17 signaling accelerates the progression of nonalcoholic fatty liver disease in mice. Hepatology 2014;59:1830-9.
275. Giles DA, Moreno-Fernandez ME, Stankiewicz TE, et al. Regulation of inflammation by IL-17A and IL-17F modulates non-alcoholic fatty liver disease pathogenesis. PLoS One 2016;11:e0149783.
276. Xu R, Tao A, Zhang S, Zhang M. Neutralization of interleukin-17 attenuates high fat diet-induced non-alcoholic fatty liver disease in mice. Acta Biochim Biophys Sin (Shanghai) 2013;45:726-33.
277. Tan Z, Qian X, Jiang R, et al. IL-17A plays a critical role in the pathogenesis of liver fibrosis through hepatic stellate cell activation. J Immunol 2013;191:1835-44.
278. Gomes AL, Teijeiro A, Burén S, et al. Metabolic inflammation-associated IL-17A causes non-alcoholic steatohepatitis and hepatocellular carcinoma. Cancer Cell 2016;30:161-75.
279. Liu Y, She W, Wang F, Li J, Wang J, Jiang W. 3, 3'-Diindolylmethane alleviates steatosis and the progression of NASH partly through shifting the imbalance of Treg/Th17 cells to Treg dominance. Int Immunopharmacol 2014;23:489-98.
280. Zhang X, Han J, Man K, et al. CXC chemokine receptor 3 promotes steatohepatitis in mice through mediating inflammatory cytokines, macrophages and autophagy. J Hepatol 2016;64:160-70.
281. Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat Immunol 2009;10:857-63.
282. Trifari S, Kaplan CD, Tran EH, Crellin NK, Spits H. Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from T(H)-17, T(H)1 and T(H)2 cells. Nat Immunol 2009;10:864-71.
283. Ramirez JM, Brembilla NC, Sorg O, et al. Activation of the aryl hydrocarbon receptor reveals distinct requirements for IL-22 and IL-17 production by human T helper cells. Eur J Immunol 2010;40:2450-9.
284. Mirshafiey A, Simhag A, El Rouby NM, Azizi G. T-helper 22 cells as a new player in chronic inflammatory skin disorders. Int J Dermatol 2015;54:880-8.
285. Tian T, Yu S, Ma D. Th22 and related cytokines in inflammatory and autoimmune diseases. Expert Opin Ther Targets 2013;17:113-25.
286. Mo R, Wang P, Lai R, et al. Persistently elevated circulating Th22 reversely correlates with prognosis in HBV-related acute-on-chronic liver failure. J Gastroenterol Hepatol 2017;32:677-86.
287. Jiang BC, Liu X, Liu XH, Li ZS, Zhu GZ. Notch signaling regulates circulating T helper 22 cells in patients with chronic hepatitis C. Viral Immunol 2017;30:522-32.
288. Lai R, Xiang X, Mo R, et al. Protective effect of Th22 cells and intrahepatic IL-22 in drug induced hepatocellular injury. J Hepatol 2015;63:148-55.
289. Liang M, Liwen Z, Yun Z, Yanbo D, Jianping C. The imbalance between Foxp3+Tregs and Th1/Th17/Th22 cells in patients with newly diagnosed autoimmune hepatitis. J Immunol Res 2018;2018:3753081.
290. Qin S, Ma S, Huang X, Lu D, Zhou Y, Jiang H. Th22 cells are associated with hepatocellular carcinoma development and progression. Chin J Cancer Res 2014;26:135-41.
291. Zhao RX, He Q, Sha S, et al. Increased AHR transcripts correlate with pro-inflammatory T-helper lymphocytes polarization in both metabolically healthy obesity and type 2 diabetic patients. Front Immunol 2020;11:1644.
292. Zhao RX, Li WJ, Lu YR, et al. Increased peripheral proinflammatory T helper subsets contribute to cardiovascular complications in diabetic patients. Mediators Inflamm 2014;2014:596967.
293. Wu Y, Min J, Ge C, et al. Interleukin 22 in liver injury, inflammation and cancer. Int J Biol Sci 2020;16:2405-13.
294. Wang X, Ota N, Manzanillo P, et al. Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature 2014;514:237-41.
295. Sabat R, Wolk K. Deciphering the role of interleukin-22 in metabolic alterations. Cell Biosci 2015;5:68.
296. Dudakov JA, Hanash AM, van den Brink MR. Interleukin-22: immunobiology and pathology. Annu Rev Immunol 2015;33:747-85.
297. Yang L, Zhang Y, Wang L, et al. Amelioration of high fat diet induced liver lipogenesis and hepatic steatosis by interleukin-22. J Hepatol 2010;53:339-47.
298. Hwang S, He Y, Xiang X, et al. Interleukin-22 ameliorates neutrophil-driven nonalcoholic steatohepatitis through multiple targets. Hepatology 2020;72:412-29.
299. Zhu J, Zhou M, Zhao X, Mu M, Cheng M. Blueberry, combined with probiotics, alleviates non-alcoholic fatty liver disease via IL-22-mediated JAK1/STAT3/BAX signaling. Food Funct 2018;9:6298-306.
300. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol 2008;8:523-32.
301. Collison LW, Workman CJ, Kuo TT, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 2007;450:566-9.
302. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 1995;155:1151-64.
303. Read S, Malmström V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J Exp Med 2000;192:295-302.
304. Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol 2008;9:632-40.
305. Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol 2005;6:1142-51.
306. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003;299:1057-61.
307. Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol 2007;178:280-90.
308. Shevyrev D, Tereshchenko V. Treg heterogeneity, function, and homeostasis. Front Immunol 2019;10:3100.
309. Ohkura N, Kitagawa Y, Sakaguchi S. Development and maintenance of regulatory T cells. Immunity 2013;38:414-23.
310. Yu H, Paiva R, Flavell RA. Harnessing the power of regulatory T-cells to control autoimmune diabetes: overview and perspective. Immunology 2018;153:161-70.
311. Ling EM, Smith T, Nguyen XD, et al. Relation of CD4+CD25+ regulatory T-cell suppression of allergen-driven T-cell activation to atopic status and expression of allergic disease. Lancet 2004;363:608-15.
312. Barnes MJ, Powrie F. Regulatory T cells reinforce intestinal homeostasis. Immunity 2009;31:401-11.
313. Mizukoshi E, Kaneko S. Immune cell therapy for hepatocellular carcinoma. J Hematol Oncol 2019;12:52.
314. Manigold T, Racanelli V. T-cell regulation by CD4 regulatory T cells during hepatitis B and C virus infections: facts and controversies. Lancet Infect Dis 2007;7:804-13.
315. Almeida J, Polvorosa MA, Gonzalez-Quintela A, et al. Decreased peripheral blood CD4+/CD25+ regulatory T cells in patients with alcoholic hepatitis. Alcohol Clin Exp Res 2013;37:1361-9.
316. Lan RY, Cheng C, Lian ZX, et al. Liver-targeted and peripheral blood alterations of regulatory T cells in primary biliary cirrhosis. Hepatology 2006;43:729-37.
317. Peiseler M, Sebode M, Franke B, et al. FOXP3+ regulatory T cells in autoimmune hepatitis are fully functional and not reduced in frequency. J Hepatol 2012;57:125-32.
318. Zheng C, Zheng L, Yoo JK, et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 2017;169:1342-1356.e16.
319. Donninelli G, Del Cornò M, Pierdominici M, et al. Distinct blood and visceral adipose tissue regulatory T cell and innate lymphocyte profiles characterize obesity and colorectal cancer. Front Immunol 2017;8:643.
320. Wagner NM, Brandhorst G, Czepluch F, et al. Circulating regulatory T cells are reduced in obesity and may identify subjects at increased metabolic and cardiovascular risk. Obesity (Silver Spring) 2013;21:461-8.
321. Yun JM, Jialal I, Devaraj S. Effects of epigallocatechin gallate on regulatory T cell number and function in obese v. lean volunteers. Br J Nutr 2010;103:1771-7.
322. Agabiti-Rosei C, Trapletti V, Piantoni S, et al. Decreased circulating T regulatory lymphocytes in obese patients undergoing bariatric surgery. PLoS One 2018;13:e0197178.
323. Qiao YC, Shen J, He L, et al. Changes of regulatory T cells and of proinflammatory and immunosuppressive cytokines in patients with type 2 diabetes mellitus: a systematic review and meta-analysis. J Diabetes Res 2016;2016:3694957.
324. Burzyn D, Benoist C, Mathis D. Regulatory T cells in nonlymphoid tissues. Nat Immunol 2013;14:1007-13.
325. Cipolletta D. Adipose tissue-resident regulatory T cells: phenotypic specialization, functions and therapeutic potential. Immunology 2014;142:517-25.
326. Cipolletta D, Feuerer M, Li A, et al. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 2012;486:549-53.
327. Vasanthakumar A, Moro K, Xin A, et al. The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat Immunol 2015;16:276-85.
328. Feuerer M, Herrero L, Cipolletta D, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med 2009;15:930-9.
329. Eller K, Kirsch A, Wolf AM, et al. Potential role of regulatory T cells in reversing obesity-linked insulin resistance and diabetic nephropathy. Diabetes 2011;60:2954-62.
330. Ilan Y, Maron R, Tukpah AM, et al. Induction of regulatory T cells decreases adipose inflammation and alleviates insulin resistance in ob/ob mice. Proc Natl Acad Sci U S A 2010;107:9765-70.
331. Kälin S, Becker M, Ott VB, et al. A Stat6/Pten axis links regulatory T cells with adipose tissue function. Cell Metab 2017;26:475-492.e7.
332. Zeng Q, Sun X, Xiao L, Xie Z, Bettini M, Deng T. A unique population: adipose-resident regulatory T cells. Front Immunol 2018;9:2075.
333. Piconese S, Pittoni P, Burocchi A, et al. A non-redundant role for OX40 in the competitive fitness of Treg in response to IL-2. Eur J Immunol 2010;40:2902-13.
334. Travers RL, Motta AC, Betts JA, Bouloumié A, Thompson D. The impact of adiposity on adipose tissue-resident lymphocyte activation in humans. Int J Obes (Lond) 2015;39:762-9.
335. Deiuliis J, Shah Z, Shah N, et al. Visceral adipose inflammation in obesity is associated with critical alterations in tregulatory cell numbers. PLoS One 2011;6:e16376.
336. Esser N, L’homme L, De Roover A, et al. Obesity phenotype is related to NLRP3 inflammasome activity and immunological profile of visceral adipose tissue. Diabetologia 2013;56:2487-97.
337. Gyllenhammer LE, Lam J, Alderete TL, et al. Lower omental t-regulatory cell count is associated with higher fasting glucose and lower β-cell function in adults with obesity. Obesity (Silver Spring) 2016;24:1274-82.
338. De Rosa V, Procaccini C, Calì G, et al. A key role of leptin in the control of regulatory T cell proliferation. Immunity 2007;26:241-55.
339. Frederich RC, Hamann A, Anderson S, Löllmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med 1995;1:1311-4.
340. Fabrizi M, Marchetti V, Mavilio M, et al. IL-21 is a major negative regulator of IRF4-dependent lipolysis affecting Tregs in adipose tissue and systemic insulin sensitivity. Diabetes 2014;63:2086-96.
341. Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 2001;286:327-34.
342. Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol 2001;19:683-765.
343. Cintra DE, Pauli JR, Araújo EP, et al. Interleukin-10 is a protective factor against diet-induced insulin resistance in liver. J Hepatol 2008;48:628-37.
344. Kim HJ, Higashimori T, Park SY, et al. Differential effects of interleukin-6 and -10 on skeletal muscle and liver insulin action in vivo. Diabetes 2004;53:1060-7.
345. Exel E, Gussekloo J, de Craen AJ, Frölich M, Bootsma-Van Der Wiel A, Westendorp RG; Leiden 85 Plus Study. Low production capacity of interleukin-10 associates with the metabolic syndrome and type 2 diabetes: the Leiden 85-Plus Study. Diabetes 2002;51:1088-92.
346. Han JM, Patterson SJ, Speck M, Ehses JA, Levings MK. Insulin inhibits IL-10-mediated regulatory T cell function: implications for obesity. J Immunol 2014;192:623-9.
347. Zhang C, Li L, Feng K, Fan D, Xue W, Lu J. 'Repair' Treg cells in tissue injury. Cell Physiol Biochem 2017;43:2155-69.
348. Thompson K, Maltby J, Fallowfield J, McAulay M, Millward-Sadler H, Sheron N. Interleukin-10 expression and function in experimental murine liver inflammation and fibrosis. Hepatology 1998;28:1597-606.
349. Louis H, Van Laethem JL, Wu W, et al. Interleukin-10 controls neutrophilic infiltration, hepatocyte proliferation, and liver fibrosis induced by carbon tetrachloride in mice. Hepatology 1998;28:1607-15.
350. Katz SC, Ryan K, Ahmed N, et al. Obstructive jaundice expands intrahepatic regulatory T cells, which impair liver T lymphocyte function but modulate liver cholestasis and fibrosis. J Immunol 2011;187:1150-6.
351. Zahran WE, Salah El-Dien KA, Kamel PG, El-Sawaby AS. Efficacy of tumor necrosis factor and interleukin-10 analysis in the follow-up of nonalcoholic fatty liver disease progression. Indian J Clin Biochem 2013;28:141-6.
352. Paredes-Turrubiarte G, González-Chávez A, Pérez-Tamayo R, et al. Severity of non-alcoholic fatty liver disease is associated with high systemic levels of tumor necrosis factor alpha and low serum interleukin 10 in morbidly obese patients. Clin Exp Med 2016;16:193-202.
353. Lalazar G, Mizrahi M, Turgeman I, et al. Oral administration of OKT3 MAb to patients with NASH, promotes regulatory T-cell induction, and alleviates insulin resistance: results of a phase IIa blinded placebo-controlled trial. J Clin Immunol 2015;35:399-407.
354. Hockenbery DM, Oltvai ZN, Yin X, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell 1993;75:241-51.
355. Ma X, Hua J, Mohamood AR, Hamad AR, Ravi R, Li Z. A high-fat diet and regulatory T cells influence susceptibility to endotoxin-induced liver injury. Hepatology 2007;46:1519-29.
356. Roh YS, Kim JW, Park S, et al. Toll-like receptor-7 signaling promotes nonalcoholic steatohepatitis by inhibiting regulatory T cells in mice. Am J Pathol 2018;188:2574-88.
357. Drescher HK, Schippers A, Rosenhain S, et al. L-Selectin/CD62L is a key driver of non-alcoholic steatohepatitis in mice and men. Cells 2020;9:1106.
358. Reinhardt RL, Liang HE, Locksley RM. Cytokine-secreting follicular T cells shape the antibody repertoire. Nat Immunol 2009;10:385-93.
359. Bauquet AT, Jin H, Paterson AM, et al. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nat Immunol 2009;10:167-75.
360. Linterman MA, Beaton L, Yu D, et al. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J Exp Med 2010;207:353-63.
361. Smith KM, Pottage L, Thomas ER, et al. Th1 and Th2 CD4+ T cells provide help for B cell clonal expansion and antibody synthesis in a similar manner in vivo. J Immunol 2000;165:3136-44.
363. Ma CS, Suryani S, Avery DT, et al. Early commitment of naïve human CD4(+) T cells to the T follicular helper (T(FH)) cell lineage is induced by IL-12. Immunol Cell Biol 2009;87:590-600.
364. Schmitt N, Liu Y, Bentebibel SE, et al. The cytokine TGF-β co-opts signaling via STAT3-STAT4 to promote the differentiation of human TFH cells. Nat Immunol 2014;15:856-65.
365. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity 2014;41:529-42.
366. Nurieva RI, Chung Y, Martinez GJ, et al. Bcl6 mediates the development of T follicular helper cells. Science 2009;325:1001-5.
367. Johnston RJ, Poholek AC, DiToro D, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 2009;325:1006-10.
368. Bollig N, Brüstle A, Kellner K, et al. Transcription factor IRF4 determines germinal center formation through follicular T-helper cell differentiation. Proc Natl Acad Sci U S A 2012;109:8664-9.
369. Liu X, Chen X, Zhong B, et al. Transcription factor achaete-scute homologue 2 initiates follicular T-helper-cell development. Nature 2014;507:513-8.
370. Choi YS, Gullicksrud JA, Xing S, et al. LEF-1 and TCF-1 orchestrate T(FH) differentiation by regulating differentiation circuits upstream of the transcriptional repressor Bcl6. Nat Immunol 2015;16:980-90.
371. Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S. STAT5 is a potent negative regulator of TFH cell differentiation. J Exp Med 2012;209:243-50.
372. Song W, Craft J. T follicular helper cell heterogeneity: Time, space, and function. Immunol Rev 2019;288:85-96.
373. Wang Q, Zhai X, Chen X, Lu J, Zhang Y, Huang Q. Dysregulation of circulating CD4+CXCR5+ T cells in type 2 diabetes mellitus. APMIS 2015;123:146-51.
374. Zhou J, Wang Y, He Y, et al. Non-obese type 2 diabetes patients present intestinal B cell dysregulations associated with hyperactive intestinal Tfh cells. Mol Immunol 2018;97:27-32.
375. Zhan J, Huang L, Ma H, et al. Reduced inflammatory responses of follicular helper T cell promote the development of regulatory B cells after Roux-en-Y gastric bypass. Clin Exp Pharmacol Physiol 2017;44:556-65.
376. Winer DA, Winer S, Shen L, et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med 2011;17:610-7.
377. DeFuria J, Belkina AC, Jagannathan-Bogdan M, et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc Natl Acad Sci U S A 2013;110:5133-8.
378. Jagannathan M, Hasturk H, Liang Y, et al. TLR cross-talk specifically regulates cytokine production by B cells from chronic inflammatory disease patients. J Immunol 2009;183:7461-70.
379. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med 2018;24:908-22.
380. Jalali M, Mahmoodi M, Mosallanezhad Z, Jalali R, Imanieh MH, Moosavian SP. The effects of curcumin supplementation on liver function, metabolic profile and body composition in patients with non-alcoholic fatty liver disease: A systematic review and meta-analysis of randomized controlled trials. Complement Ther Med 2020;48:102283.
381. Pickich MB, Hargrove MW, Phillips CN, et al. Effect of curcumin supplementation on serum expression of select cytokines and chemokines in a female rat model of nonalcoholic steatohepatitis. BMC Res Notes 2019;12:496.
382. Coia H, Ma N, Hou Y, et al. Theaphenon E prevents fatty liver disease and increases CD4+ T cell survival in mice fed a high-fat diet. Clin Nutr 2021;40:110-9.
383. Yue R, Jin G, Wei S, et al. Immunoregulatory effect of koumine on nonalcoholic fatty liver disease rats. J Immunol Res 2019;2019:8325102.
384. Zhou D, Pan Q, Liu XL, et al. Clostridium butyricum B1 alleviates high-fat diet-induced steatohepatitis in mice via enterohepatic immunoregulation. J Gastroenterol Hepatol 2017;32:1640-8.
385. Cao M, Li X, Zhang B, et al. The effect of polyene phosphatidyl choline intervention on nonalcoholic steatohepatitis and related mechanism. Am J Transl Res 2016;8:2325-30.
386. He B, Wu L, Xie W, et al. The imbalance of Th17/Treg cells is involved in the progression of nonalcoholic fatty liver disease in mice. BMC Immunol 2017;18:33.
387. Imarisio C, Alchera E, Sutti S, et al. Adenosine A(2a) receptor stimulation prevents hepatocyte lipotoxicity and non-alcoholic steatohepatitis (NASH) in rats. Clin Sci (Lond) 2012;123:323-32.
388. Cai Y, Li H, Liu M, et al. Disruption of adenosine 2A receptor exacerbates NAFLD through increasing inflammatory responses and SREBP1c activity. Hepatology 2018;68:48-61.
389. Alchera E, Rolla S, Imarisio C, et al. Adenosine A2a receptor stimulation blocks development of nonalcoholic steatohepatitis in mice by multilevel inhibition of signals that cause immunolipotoxicity. Transl Res 2017;182:75-87.
390. Wu L, Mo W, Feng J, et al. Astaxanthin attenuates hepatic damage and mitochondrial dysfunction in non-alcoholic fatty liver disease by up-regulating the FGF21/PGC-1α pathway. Br J Pharmacol 2020;177:3760-77.
391. Li S, Takahara T, Fujino M, et al. Astaxanthin prevents ischemia-reperfusion injury of the steatotic liver in mice. PLoS One 2017;12:e0187810.
392. Ni Y, Nagashimada M, Zhuge F, et al. Astaxanthin prevents and reverses diet-induced insulin resistance and steatohepatitis in mice: A comparison with vitamin E. Sci Rep 2015;5:17192.
393. Sugiura M, Nakamura M, Ikoma Y, et al. The homeostasis model assessment-insulin resistance index is inversely associated with serum carotenoids in non-diabetic subjects. J Epidemiol 2006;16:71-8.
394. Kobori M, Ni Y, Takahashi Y, et al. β-Cryptoxanthin alleviates diet-induced nonalcoholic steatohepatitis by suppressing inflammatory gene expression in mice. PLoS One 2014;9:e98294.
395. Song X, Ma C. Mechanisms and immunotherapies of HBV- and NAFLD-related hepatocellular carcinoma. Hepatoma Res 2020;6:27.
396. Tateishi R, Uchino K, Fujiwara N, et al. A nationwide survey on non-B, non-C hepatocellular carcinoma in Japan: 2011-2015 update. J Gastroenterol 2019;54:367-76.
397. Benhammou JN, Lin J, Hussain SK, El-Kabany M. Emerging risk factors for nonalcoholic fatty liver disease associated hepatocellular carcinoma. Hepatoma Res 2020;6:35.
398. Ni L, Feng Y, Dong C. The advancement of immunotherapy in hepatocellular carcinoma. Hepatoma Res 2020;6:25.
400. Geltink RIK, Kyle RL, Pearce EL. Unraveling the complex interplay between T cell metabolism and function. Annu Rev Immunol 2018;36:461-88.
401. Yamada K, Mizukoshi E, Sunagozaka H, et al. Characteristics of hepatic fatty acid compositions in patients with nonalcoholic steatohepatitis. Liver Int 2015;35:582-90.
402. Yamada K, Mizukoshi E, Seike T, et al. Serum C16:1n7/C16:0 ratio as a diagnostic marker for non-alcoholic steatohepatitis. J Gastroenterol Hepatol 2019;34:1829-35.
403. Lochner M, Berod L, Sparwasser T. Fatty acid metabolism in the regulation of T cell function. Trends Immunol 2015;36:81-91.
404. Kohjima M, Enjoji M, Higuchi N, et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int J Mol Med 2007;20:351-8.
405. Wai JW, Fu C, Wong VW. Confounding factors of non-invasive tests for nonalcoholic fatty liver disease. J Gastroenterol 2020;55:731-41.
406. Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol 2011;29:235-71.
407. Kang TW, Yevsa T, Woller N, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011;479:547-51.
408. Schneider C, Teufel A, Yevsa T, et al. Adaptive immunity suppresses formation and progression of diethylnitrosamine-induced liver cancer. Gut 2012;61:1733-43.
409. Unitt E, Marshall A, Gelson W, et al. Tumour lymphocytic infiltrate and recurrence of hepatocellular carcinoma following liver transplantation. J Hepatol 2006;45:246-53.
410. Unitt E, Rushbrook SM, Marshall A, et al. Compromised lymphocytes infiltrate hepatocellular carcinoma: the role of T-regulatory cells. Hepatology 2005;41:722-30.
411. Shen X, Li N, Li H, Zhang T, Wang F, Li Q. Increased prevalence of regulatory T cells in the tumor microenvironment and its correlation with TNM stage of hepatocellular carcinoma. J Cancer Res Clin Oncol 2010;136:1745-54.
412. Fu J, Xu D, Liu Z, et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology 2007;132:2328-39.
413. Jia Y, Zeng Z, Li Y, et al. Impaired function of CD4+ T follicular helper (Tfh) cells associated with hepatocellular carcinoma progression. PLoS One 2015;10:e0117458.
414. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity 2013;39:1-10.
415. Inada Y, Mizukoshi E, Seike T, et al. Characteristics of immune response to tumor-associated antigens and immune cell profile in patients with hepatocellular carcinoma. Hepatology 2019;69:653-65.
416. Brown ZJ, Fu Q, Ma C, et al. Carnitine palmitoyltransferase gene upregulation by linoleic acid induces CD4+ T cell apoptosis promoting HCC development. Cell Death Dis 2018;9:620.
417. Sugiura A, Rathmell JC. Metabolic Barriers to T Cell Function in Tumors. J Immunol 18;200:400-7.
418. Nakahara T, Hyogo H, Yoneda M, et al. Japan Study Group of Nonalcoholic Fatty Liver Disease. Type 2 diabetes mellitus is associated with the fibrosis severity in patients with nonalcoholic fatty liver disease in a large retrospective cohort of Japanese patients. J Gastroenterol 2014;49:1477-84.
419. Kawamura Y, Arase Y, Ikeda K, et al. Large-scale long-term follow-up study of Japanese patients with non-alcoholic Fatty liver disease for the onset of hepatocellular carcinoma. Am J Gastroenterol 2012;107:253-61.
420. Tokushige K, Hashimoto E, Kodama K. Hepatocarcinogenesis in non-alcoholic fatty liver disease in Japan. J Gastroenterol Hepatol 2013;28 Suppl 4:88-92.
422. Chattopadhyay PK, Roederer M. Cytometry: today's technology and tomorrow's horizons. Methods 2012;57:251-8.
423. Atkuri KR, Stevens JC, Neubert H. Mass cytometry: a highly multiplexed single-cell technology for advancing drug development. Drug Metab Dispos 2015;43:227-33.
424. Papalexi E, Satija R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat Rev Immunol 2018;18:35-45.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Seike T, Mizukoshi E, Kaneko S. Role of CD4+ T-cells in the pathology of non-alcoholic fatty liver disease and related diseases. Hepatoma Res 2021;7:46. http://dx.doi.org/10.20517/2394-5079.2021.46
AMA Style
Seike T, Mizukoshi E, Kaneko S. Role of CD4+ T-cells in the pathology of non-alcoholic fatty liver disease and related diseases. Hepatoma Research. 2021; 7: 46. http://dx.doi.org/10.20517/2394-5079.2021.46
Chicago/Turabian Style
Seike, Takuya, Eishiro Mizukoshi, Shuichi Kaneko. 2021. "Role of CD4+ T-cells in the pathology of non-alcoholic fatty liver disease and related diseases" Hepatoma Research. 7: 46. http://dx.doi.org/10.20517/2394-5079.2021.46
ACS Style
Seike, T.; Mizukoshi E.; Kaneko S. Role of CD4+ T-cells in the pathology of non-alcoholic fatty liver disease and related diseases. Hepatoma. Res. 2021, 7, 46. http://dx.doi.org/10.20517/2394-5079.2021.46
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 21 clicks
Cite This Article 21 clicks

 Like This Article 23
likes
Like This Article 23
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.