
p53 functional loss, stemness and hepatocellular carcinoma

 ,
, Abstract
The tumor suppressor p53 is a key player in the control of genomic integrity and homeostasis in connection with p63 and p73, the two other members of the p53 family. Loss of functional p53 leads to the proliferation and survival of mature cells and progenitor or stem cells that accumulate genetic alterations, thus favoring tumorigenesis. p53 loss of function, observed in a wide variety of human tumor types, is frequently caused by missense mutations more frequently found in the DNA binding domain, but can also be due to the expression of a plethora of viral and cellular negative regulators. Human hepatocellular carcinoma (HCC) represents a specific situation, first because the TP53 gene mutations pattern exhibits a “hot spot” rarely found in other tumor types that is linked to Aflatoxin B1 exposure and, second, because many HCCs do not exhibit any TP53 mutation. Here, we provide an overview of the current knowledge about the inhibition of p53 functions by the N-terminal (ΔN) truncated forms of the family, and their role in the emergence and maintenance of pre-malignant cells with stem cell characteristics and in HCC development. We focus in particular on the Nanog-IGF1R-ΔNp73 axis that is associated with stem-like features in HCC cells and that may provide an attractive new therapeutic target and help to develop new biomarkers for HCC risk stratification, as well as preventive strategies.
Keywords
Introduction
Hepatocellular carcinoma (HCC) is a major public health problem, being the fourth most lethal cancer with an increasing incidence around the world[1]. About 90% of HCC cases can be associated with four well-characterized underlying risk factors including chronic infection with hepatitis B and C viruses (HBV, HCV), ethanol consumption, and non-alcoholic fatty liver disease (NAFLD)[2]. Although the risk of developing HCC can be reduced in patients by treatment of the underlying cause - e.g., HCV eradication by direct-acting antivirals (DAAs) and HBV suppression by nucleos(t)ide analogs (NUCs) - strategies to prevent cancer development in patients with advanced fibrosis and established cirrhosis are still lacking[3,4]. Despite the recent improvements, treatment options for HCC remain largely unsatisfactory. Currently, curative treatment options for patients with HCC include surgical resection and loco-regional ablation, frequently associated with tumor recurrence, and orthotopic liver transplantation, a resource-intensive solution[3,4]. However, due to its silent clinical character and the low sensitivity and specificity of currently available diagnostic biomarkers, HCC is commonly diagnosed at an advanced stage, when curative treatments are not feasible, leaving systemic drugs as the only option[3,4]. Patients with untreated advanced HCC carry a very poor prognosis with an expected survival of 4-6 months. Treatment modalities available for patients with advanced HCC not eligible for curative treatment include the multikinase inhibitors (MTKi) sorafenib and lenvatinib as first line treatment, with an increase in survival of approximately 3 months, and MTKi regorafenib or cabozantinib as second or third line treatment that enables extension of survival by an additional 3 months[3,4]. Immune checkpoint inhibitors (ICIs) in monotherapy achieve striking tumor responses in a few patients who have hugely improved outcomes[3,4], but the low rate of responding patients does not allow significant improvements in the median overall survival. More recently, the combination of drugs targeting the liver microenvironment with PD1/PD-L1 inhibitors, such as the association of bevacizumab and atezolizumab, or the combination of ICIs have shown promising results[3-5].
In HCC, as in numerous other tumor types, the chemoresistance is thought to be due to the existence of a sub-population of poorly differentiated cancer cells, widely known as liver cancer stem cells (CSCs) or tumor initiating cells (TICs)[6,7]. The escape mechanisms are diverse, like enzymatic inactivation or increased drug efflux[6,7]. The microenvironment also plays an important role. By secreting growth factors and cytokines, it favors the emergence, maintenance and survival of CSCs and their resistance to chemotherapeutic drugs, resulting in the recurrence of more aggressive tumors[8]. Chronic HBV and HCV infections and alcohol abuse induce liver inflammation, fibrosis, and cirrhosis. This pathological microenvironment exhibits alterations in cytokine secretion, extra-cellular matrix components and stiffness, angiogenesis, and liver resident immune cells. Moreover, the abnormal activation of signaling pathways, several of them strongly involved in stemness, are observed in liver fibrosis and cirrhosis[9,10]. All these alterations could contribute to the emergence of cellular clones with characteristics of liver CSCs, as shown in other tumor types[8] and subsequently to HCC initiation and development.
Understanding the mechanisms responsible for liver CSC maintenance and survival is therefore an important step in the search of efficient therapies for HCC. CSCs exploit signaling pathways essential for self-renewal, proliferation and differentiation that are usually used by stem cells in physiological situations. Wnt/β-catenin, Hedgehog, Notch, and TGFβ are the main pathways found activated in hepatic CSCs[11-14]. For some of them, this activation has been associated with the expression of cell surface markers, such as EpCAM for the Wnt/β-catenin pathway or CK19 for the TGFβ pathway, and with poorly differentiated tumors, drug resistance and worse prognosis[15-17].
In addition to these pathways, another key player of CSCs is the p53 family. In this review, we summarize the main functions of p53 and the p53 family, as well as the mechanisms that lead to their functional inactivation in tumors. We describe the alterations of p53 functions that favor the emergence of CSCs with a particular focus on liver CSCs. Finally, we highlight the close connection between the p53 family members and key players of stemness, which could help developing new approaches to prevent the appearance of liver CSCs in the context of chronic liver diseases, inflammation in the liver, and HCC development.
The p53 family
The p53 family encompasses three members, namely TP53, TP63 and TP73[18,19]. All three genes encode for multiple isoforms generated by both internal promoter usage and alternative splicing [Figure 1]. The full-length isoforms (p53, TAp63 and TAp73) share a N-terminal transactivation domain, followed by a proline-rich sequence, a central DNA-binding domain (DBD) and the C-terminal oligomerization domain involved in the formation of transcriptionally active homo-tetramers [Figure 1]. All three possess the ability to bind sequence specific cognate DNA motifs (p53RE, p53 responsive elements) and thus to transactivate a large number of direct target genes collectively referred to as p53-target genes. Due to the high sequence homology (> 70% sequence identity) in their DBDs[18,19], p73 and p63 regulate many p53-target genes (e.g., WAF1, PUMA, NOXA, BAX and MDM2)[20]. The complete repertoire of common and private target genes regulated by the p53 family members in different physiological and pathological contexts is still unknown[20]. The C-terminus of TAp63 and TAp73 alpha isoforms also contains a sterile alpha motif and a terminal transcription inhibitory domain, not conserved in p53[18,19,21,22]. Additional shorter isoforms (β, γ, δ, and less investigated ε, ζ and η) isoforms, whose specific functions are still poorly characterized, are generated by C-terminus alternative splicing[23,24][Figure 1]. The N-terminal truncated isoforms (ΔTA or ΔN) act as oncogenes, in part through their dominant negative effect (DNE) on TA isoforms[18,25,26][Figure 1].
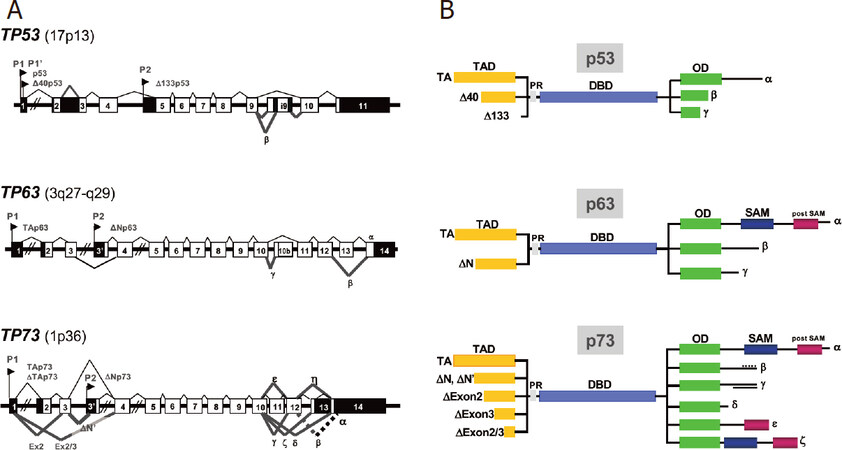
Figure 1. The p53 family - a complex expression strategy. A: schematic representation of the exon structure of TP53, TP63 and TP73 genes. The position of the P1 promoter and the internal P2 promoter relative to the exons in the 3 genes are indicated. p53 gene transcription is initiated from two distinct sites (P1 and P1′); B: p53, p63 and p73 protein domains. TAD: N-terminal transactivation domains (yellow); PR: proline-rich sequence (grey); DBD: DNA-binding domain (purple); OD: C-terminal oligomerization domain (green); SAM: sterile alpha motif (dark blue); post-SAM: post-sterile alpha motif (burgundy). The principal N-terminal truncated isoforms (ΔTA or ΔN), generated by the use of internal promoters (P2), alternative splicing of the first exons or the use of alternative translation start sites, and the COOH-terminal variants α, β, γ, δ, ε, ζ, generated by alternative splicing of p53, p63 and p73 are also indicated
p53 has held its position of master protein in the cancer field for 40 years. A true hub in the cell stress response, p53 is ubiquitously expressed at low levels and in a latent form in physiological conditions. In response to a wide variety of intrinsic and extrinsic stress signals, such as DNA damage, oncogenic activation or hypoxia, p53 is subjected to a number of post-translational modifications, including phosphorylation at multiple N- and C-terminal sites by Chk1/2, CK1/2, DNA-PK and other kinases. Phosphorylation usually blocks the binding of E3-Ubiquitin ligases, resulting in both stabilization and conformational shift of p53 from a latent to an activated state[27]. Stabilization and activation are remarkably fine-tuned processes that depend not only on the type and intensity of the signal, but also on the cell type and its differentiation state[28], leading to the transactivation of canonical p53-target genes. p53 activation translates into various cellular processes, such as cell cycle arrest[29], DNA repair[30], allowing cell survival, or apoptosis[31], autophagy[32] and ferroptosis[33], causing the death of damaged cells. Thereby, p53 response enables the maintenance of genomic stability and prevents the emergence of pre-malignant cells. Accordingly, tp53 knock-out mice or tp53 knock-in mice expressing an inactivated form of p53, are prone to developing spontaneous tumors[26]. Importantly, p53 activities do not depend on transactivation only and the interaction between p53 and other cellular proteins has also been implicated in its tumor suppression function[34]. In addition, besides cell-cycle arrest and cell death the regulation of other cellular processes, including metabolism, cell migration (by blocking epithelia-mesenchymal transition) or induction of differentiation, contributes to the tumor suppressor function of p53[35,36]. The repertoire of functions, pathways and genes regulated by p53, p63 and p73 has progressively widened well beyond cell fate, tumor suppression and development. p53 family members have been so far involved in reproduction, genomic repair, fidelity and recombination, metabolic processes, longevity, stem cell biology, adaptive immunity and T cell functions and changes in epigenetic marks[26,37].
TP53 mutations and their consequences on liver tumorigenesis
Unlike TP63 and TP73 genes, which are rarely lost or mutated in tumors, genetic alterations of the TP53 gene are frequently observed, but in contrast to other tumor suppressor genes, the deletion of the two alleles is a rare event[38]. Strikingly, more than 50% of human tumors present a missense (or less frequently a nonsense or frame shift) mutation in one TP53 allele, frequently accompanied by the deletion of the other one (loss of heterozygosity, LOH)[39]. The location of the mutation depends on numerous parameters, such as cell type and the tumor-inducing agent. Nevertheless, some codons (175, 245, 248, 249, 273 and 282) have been found to be preferentially mutated and defined as “hot spot” codons. Mutations of these hot spots can be classified in two categories, those affecting p53 conformation (175, 245, 249, 282) and those that lie in the DNA contact domain (248, 273)[40]. All block p53 binding to the cognate p53REs. Altogether, these six mutations account for more than 25% of the missense and nonsense mutations listed in the IARC TP53 database R20, (https://p53.iarc.fr/)[41]. Figure 2A illustrates the distribution of the resulting substituted amino acids along the p53 protein (only those for which more than 100 mutations have been reported in the database are shown). Most of the mutations are located in the DBD, mostly resulting in the inability of mutant p53 (mutp53) to bind p53RE and thus to transactivate p53-target genes (Figure 3, left upper panel). The mutation at codon 249 represents a particular case among the hot spots, because it is overrepresented in HCC. Figure 2B clearly shows the high prevalence of this mutation in HCC (around 30%), whereas the five other hot spots account for only 9%. Interestingly, the arginine residue at codon 249 is almost exclusively changed to a serine residue (96%) in HCC, due to a G/C to T/A transversion. Transversions in the TP53 gene are also found in several other types of cancer, such as those affecting the respiratory and urinary tracts, whereas G/C to A/T transitions are mainly present in colorectal carcinomas or brain tumors. Whereas transitions are expected to be due to intrinsic factors, transversions are correlated with exposure to environmental mutagens that form DNA adducts and often affect particular codons, as in the TP53 gene[42]. For example, TP53 codons 157 and 158 are targets of tobacco smoke compounds in lung adenocarcinoma. Similarly, in HCC the G/C to T/A transversion at codon 249 is caused by consumption of AFB1-contamined food[43-46]. AFB1 exposure is mainly observed in sub-Saharan Africa and Southeast Asia, two regions in which HBV chronic infection is frequent. The combination of these two extrinsic factors has been shown to be responsible for the high incidence of HCC in these regions and to be associated with the presence of the p53R249S mutant. More than 50% of HCC from these regions express a p53R249S mutant[47-49]. It is important to underline that TP53 is the most frequently mutated gene and is associated with a shorter survival in HBV-related HCC also independently from AFB1 exposure[50,51].
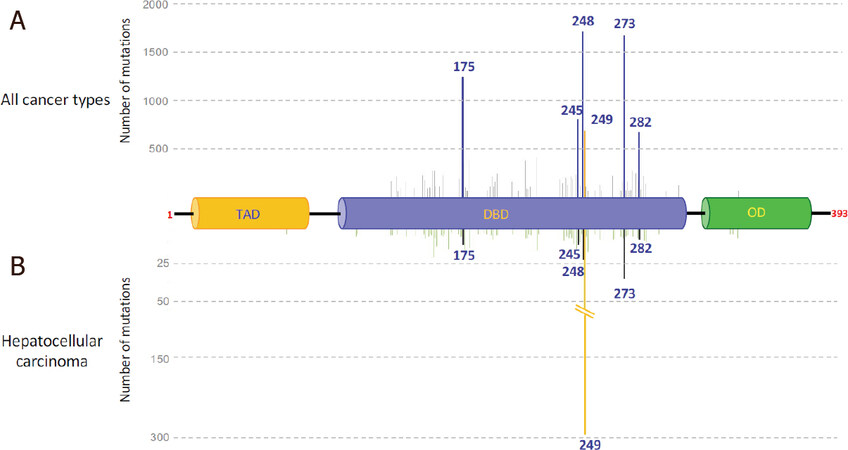
Figure 2. TP53 missense mutations found in human cancers and liver cancer. The main mutated codons were placed on the p53 protein. A: main somatic mutations found in all cancer types (more than 100 mutations listed); B: main mutations found in HCC (more than 4 mutations listed). Source, IARC TP53 database R20. TA: transactivation domain; DBD: DNA binding domain; OD: oligomerization domain. The position of the first and the last p53 aminoacids are noted in red. Deletions, insertions and mutations in introns have not been included in the cartoon
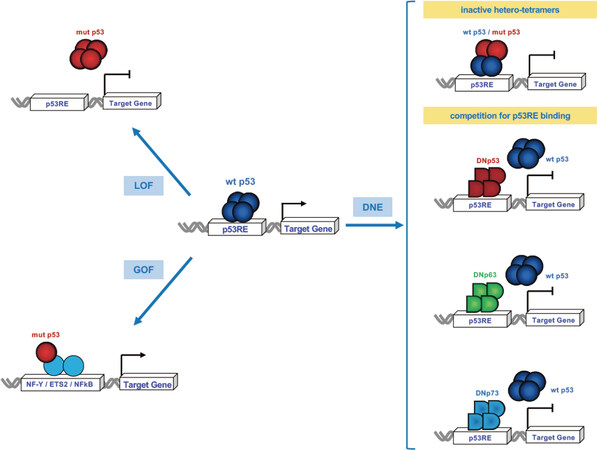
Figure 3. Wtp53 inactivation by genetic mutations and dominant negative p53 family proteins. The wtp53, once activated, works as a tetramer to bind the p53-regulatory elements (p53RE) of its direct target genes and induce transcription. At a late stage of tumor cells evolution mutant p53 forms tetramers and/or aggregates that are unable to bind to canonical target sequences to turn on its targets (LOF) (upper left panel). Some mutant p53 (p53R175H, p53R248Q, p53R273H) acquire novel gain of functions (GOFs) to activate (or repress) transcription independently of their binding to p53RE by cooperating with other transcription factors, such as NF-Y, ETS2 or NFkB, and drive the growth, survival and invasion of tumor cells. At earlier stages, when one allele is mutated there is reduced overall function resulting in haploinsufficieny and also the DNE of the mutant p53 on the wtp53 due to the formation of transcriptionally inactive heterotetramers (upper right panel). The N-terminal truncated isoforms (ΔTA or ΔN) of p53, p63 and p73 also exert a DNE on the wtp53. ΔN-tetramers compete with p53, TAp63 and TAp73 on the same p53RE leading to the abrogation of the wtp53 transcriptional program (middle and lower right panels)
Most p53 mutants have lost wild-type p53 (wtp53) functions (loss of function, LOF). Moreover, when expressed with wtp53 encoded from the non-mutated remaining allele, they form heterotetramers, or aggregates, and thus reduce or eliminate the ability of wtp53 to maintain genomic integrity by a dominant negative effect[52]. In the context of response to stress of premalignant cells (e.g., hepatocytes with p53R249S), the expression of mutant p53 may allow cells to survive despite the accumulation of genetic alterations, thus favoring the emergence of potential tumor cells. Many tumors harboring a missense mutation in one TP53 allele have lost the wild-type one, suggesting that LOH provides a selective advantage in the transformation process for cells expressing at least some of the p53 mutants.
In an attempt to better understand the properties of the different p53 mutants, many functional experiments both in cells, yeast and mouse models have led to the classification of these mutants in two main categories. The first one includes LOF mutants whose expression has biological consequences similar to the homozygous deletion of the TP53 gene. The second one includes mutants that enhance cell transformation, tumor growth and aggressiveness compared to their p53-null counterparts. This last class is expected to acquire new functions (gain of function, GOF). Numerous studies since the 1990s have shown that GOF mutp53 are involved in transcriptional activation or repression, independently of their binding to p53RE, but mediated by cooperation with other transcription factors, such as NF-Y, ETS2 or NF-κB[53-55]. Among the hot spot mutants, p53R175H, p53R248Q and p53R273H have been shown to be GOF mutants, whereas p53G245S is part of the LOF category with a DNE on wtp53[53,56,57]. p53R249S has been considered a LOF mutation, mainly according to the results of in vitro and in vivo experiments performed in non-hepatic cells or tissues other than the liver[56], whereas a GOF phenotype was observed in the liver and in the context of HCC[58-60].
Why p53R249S is selected in HCC compared to other AFB1-induced TP53 mutations remains an open question. In vitro mutagenesis with AFB1 generates a transversion at codon 249, but also at hot spot codons 245 and 248[44] and the preferential formation of AFB1 adducts alone cannot explain the high frequency of p53R249S in HCC[61]. The other extrinsic factor that could participate in the strong selection of this mutation is HBV. Indeed, we showed that p53R249S interacts with the viral HBx protein and may contribute to cell proliferation and survival[58]. Together with its DNE on wtp53, one can hypothesize that this mutant could provide a selective advantage to human hepatocytes chronically exposed to AFB1 and HBV infection. Results from knock-in mice have shown that the expression of the mouse equivalent of the human p53R249S mutant (p53R246S) under the control of the albumin promoter enhances the carcinogenic effect of AFB1 exposure alone and the cooperative carcinogenic effect together with HBsAg expression[59,62]. Altogether, p53R249S represents a very peculiar p53 mutant with a cell- and genotoxic-specific GOF.
Recently, Liao and colleagues reported new insights in the preferential selection of p53R249S[60]. They found that CDK4 is able to phosphorylate p53R249S. Phosphorylated p53R249S interacts with Pin1 and then the complex is targeted to the nucleus, where it interacts with c-Myc. This interaction stabilizes c-Myc and enhances c-Myc-driven ribosomal biogenesis and HCC cell growth. Interestingly, HBx viral protein has been shown to interplay with several of these p53R249S partners. HBx is able to increase CDK4 activity and also to interact with Pin1 and thus enhances HBV-related hepatocarcinogenesis[63,64]. The identification of this CDK4-p53R249S-PIN1-c-Myc pathway could explain the high frequency of p53R249S mutant in HCC associated with HBV chronic infection and its low frequency in other tumor types. Moreover, the ability of p53R249S to increase c-Myc activity allows its classification among the GOF mutants, at least in the particular context of HCC.
Additional mechanisms responsible for p53 functional loss in hepatocellular carcinoma
In North America and Europe, the TP53 mutation rate in HCC is around 25%, meaning that the majority of HCC cells express the wild-type form. This indicates that mechanisms other than genetic inactivation must ensure the abrogation of wtp53 tumor suppressor functions. Several viral (SV40 LargeT Ag, Ad12 E1B, HPV16-18 E6, HBx) and cellular proteins have been reported to be able to inactivate wtp53 by direct interaction, possibly followed by degradation[65-68], the prototype being MDM2, a direct p53-target gene. MDM2 protein is an E3 ubiquitin ligase able to bind p53 and to drive the complex to proteasomal degradation[69]. In physiological conditions, MDM2 is the main regulator of p53 intracellular levels, but it has been found to be overexpressed, mainly in sarcomas, but also in HCC (around 25% positivity), leading to wtp53 loss in the tumor cells[70,71].
In addition to MDM2, 18 other E3 ubiquitin ligases leading to p53 proteasome-dependent degradation have been reported[72]. Some of them are of particular interest, since their expression has been found to be altered in HCCs. PIRH2 overexpression in human HCCs is associated with a worse prognosis[73], while COP1 silencing results in the inhibition of proliferation and the induction of apoptosis in human HCC cells, as well as in the suppression of tumor growth in mouse liver[74]. Notably, PIRH2 and COP1, like MDM2, are encoded by genes inducible by p53 and, although they act independently, all participate in the autoregulatory feedback loop that controls p53 function[75,76]. RING1 and CUL4A E3 ubiquitin ligases are also overexpressed in HCCs[77,78] and play a role in stem cell maintenance. RING1, by activating the Wnt/β-catenin pathway, promotes the transformation of hepatic progenitor cells into CSCs[79]. The CUL4A/DDB1 complex that plays a key role in HBV replication controls embryonic and hematopoietic stem cell differentiation and homeostasis[80]. Although each of these ubiquitin ligases can, in principle, favor hepatocarcinogenesis by inducing wtp53 degradation, their contribution to HCC development is not known and the prevalent mechanism of hepatocarcinogenesis in the absence of TP53 mutations in human HCCs may be, according to the data available so far, the overexpression of ΔNp73 (see below).
Truncated members of the p53 family act as functional inhibitors of wtp53
The N-terminal truncated isoforms (ΔTA or ΔN) of p53, p63 and p73, generated by the use of internal promoters (P2), alternative splicing of the first exons or the use of alternative translation start sites, exert a DNE on TA isoforms[18,19,81]. Two major mechanisms underlie the DNE of ΔTA/ΔN p53, p63 and p73 [Figure 3]. The first is a direct competition of ΔN-tetramers with p53, TAp63 and TAp73 on the same p53REs and the consequent inhibition of p53/TAp73-mediated activation (right middle and lower panels). The second is the oligomerization with TA proteins to form transcriptionally ineffective heterocomplexes (right upper panel). Notably, since the oligomerization domains are only partially conserved, p73 and p63 form hetero-oligomers with each other but not, or to a very limited extent, with p53[82].
The mechanisms responsible for the differential expression of TA and ΔN isoforms in the different tissues and pathophysiological conditions are far from being fully elucidated. Several signaling and/or oncogenic pathways affect TA- and/or ΔN-p73 levels by affecting transcription or protein stability. TAp73 isoform transcription from the P1 promoter is primarily driven by E2F1[83-88], but its activity can be also modulated by other factors such as C-EBPα[89], ZEB[90] and Ying Yang 1 (YY1)[91]. The regulation of the P2 promoter is much less clear. Notably, both p53 and TAp73 bind the internal P2 promoter of TP73, activate the transcription of P2 ΔNp73 isoforms to create a negative feedback loop between ΔNp73 and p53/TAp73 that may self-restrict their transcriptional activities[92-95].
The ratio between ΔN- and TA isoforms has been shown to determine the net effect and, in the case of p73, to predict the effectiveness of chemotherapy[96-98]. The TA pro-oncogenic role of dominant negative ΔTA/ΔN p53, p63 and p73 is supported by several observations. ΔNp73 overexpression in fibroblasts increases their colony formation capacity[99] and cooperates with RAS, c-Myc and E1A in promoting transformation and tumorigenicity[21,100]. In melanoma xenografts ΔNp73 expression is associated with upregulation of Slug, downregulation of the actin binding protein EPLIN, activation of the IGF1R-AKT/STAT3 pathway, loss of E-cadherin and a higher ability to invade and metastasize[101]. Notably, 83% of 12-20 month-old transgenic mice expressing ΔNp73 under the control of the albumin promoter develop HCC, confirming the oncogenic potential of ΔNp73 and its capability to drive hepatocarcinogenesis in vivo[102]. In the case of ΔNp63, in addition to the mechanisms already described, its oncogenic potential in squamous cell carcinomas is related to its ability to control transcription of genes involved in skin developmental and tumorigenic pathways, such as IRF6, IKKα, and FGFR2[103-105], and to act as a regulator of p53 tumor suppressive functions both in a cell-autonomous way and as a mediator of activation of FGF signaling pathway in a paracrine way[26].
The role of p53 family in the emergence, maintenance and immature phenotype of CSCs
Since their discovery, p63 and p73 have been associated with tissue development, because of the multiple developmental abnormalities observed in tp63 and tp73 knock-out mice[26]. tp63 knock-out mice are devoid of stratified and glandular epithelia and die few hours after birth, whereas tp73 knock-out mice exhibit defects in central nervous system neurogenesis and fertility, but do not develop tumors[18,106-109]. On the other hand, tp53 knock-out mice exhibit only few developmental abnormalities, but are prone to the spontaneous development of a variety of neoplasms, mainly lymphomas and sarcomas by 6 months of age[110,111]. Therefore, due to this prevailing tumor suppressor function, p53 has been extensively studied in the context of cancer. The role of TAp73 and TAp63 in DNA damage response (DDR) and cancer cell chemosensitivity[21,96] was also described, whereas their contribution to tumor suppression was established only later through the analysis of p73 and p63 heterozygous mutation in mice[112] and the generation of mice selectively lacking TAp73 or TA63 isoforms[113,114]. Fifteen years ago, several groups reported p53 ability to induce the differentiation of embryonic stem (ES) cells and to negatively regulate the proliferation and survival of adult neural stem cells[115,116]. Consequently, adult bone marrow of tp53 knock-out mice harbors less quiescent- and more proliferative- hematopoietic stem cells[117]. More recently, p53 has been shown to control genomic integrity during reprogramming and to dramatically reduce reprogramming efficiency in several cell types[118,119]. p53 is also essential for the maintenance of DNA methylation homeostasis in ES cells[120]. Based on these properties, p53 - known as the “guardian of the genome” since 1992[121] - has also been named “guardian of reprogramming”[122]. Notably, some p53 mutants, such as p53R175H and p53R273H, enhance reprogramming to a level that is higher than that observed in the case of p53 loss, thus supporting an additional GOF for these mutants[119,123].
The association of TP53 mutations with poorly differentiated human tumors has been widely reported. In breast and lung cancers, the presence of p53 mutants has been linked to stem cells and iPSCs (induced pluripotent stem cells) transcriptional signatures and downregulation of differentiation genes regulated by the polycomb repressor complex 2[124], a complex required for maintaining ES cell pluripotency and plasticity during embryonic development[124-126]. A direct link between some p53 mutants, like p53R248W in osteosarcoma cells, and the acquisition of CSCs features (high proliferation rate, sphere formation, clonogenic growth, high migration and invasive ability) and increased aggressiveness has been established[127]. Truncated forms of p53 have also been shown to favor cancer stemness. For example, the Δ133p53β isoform enhances the expression of the pluripotency factors SOX2, OCT3/4 and NANOG, promotes mammosphere formation and is associated with metastatic potential and chemoresistance in breast cancer cell lines[128]. The N-terminal truncated isoforms of p63 and p73 are also involved in the emergence and maintenance of CSCs. ΔNp63, which has been described as master regulator of normal epithelial stem cell maintenance in stratified and glandular epithelia[129,130], promotes stem cell properties by activating signaling pathways, like the Wnt/FZD7 pathway in basal-like mammary cell lines[130], or components of the Hedgehog (Hh) pathway (SHH, PTCH1, and GLI2) and the Hh-target gene BMI1 in mammary cell lines with a luminal-like phenotype[131]. Notably, BMI1 plays important roles in the self-renewal of normal and CSCs[132], as well as in the chemoresistance and survival of CSCs from several tumor types[133]. ΔNp73 also favors CSCs properties. Its overexpression in a non-tumorigenic melanoma cell line exerts a DNE on TAp73-mediated miR-885-5p expression, a negative regulator of the IGF1 receptor (IGF1R)[134], leading to an increased expression of IGF1R and, consequently, of CD133, NANOG and OCT4 and an enhanced ability to form tumors in xenografts[134]. ΔNp73 also forms complexes with Smad3/4 on Smad binding elements, leading to the enhanced transactivation of stemness-related TGFβ-target genes[135]. Finally, both ΔNp63 and ΔNp73 are able to enhance reprogramming by favoring the mesenchymal-epithelial transition (MET) required during iPSCs generation[136,137] and in the case of ΔNp73, by inhibiting wtp53[138].
Altogether, there is enough evidence to affirm that the truncated forms of the p53 family members and some p53 mutants not only act as functional inhibitors of wtp53 but also exhibit new additional functions, some of them contributing to stemness. It is more difficult, however, to define the relative contribution of the different isoforms to the stem-related phenotype, due to their variable expression (not having discussed the C-terminal variants), the complexity of their crosstalk and their largely redundant functions.
P53 family members in HCC
The expression and activities of p53 family members have been studied in normal hepatocytes and HCC cells. In a small series of 16 cholangiocarcinomas (CCAs), p63 overexpression correlated with CK19 positivity and low tumor differentiation, whereas no expression was found in 37 HCCs[139]. Similarly, in hepatocytes and HCC cell lines, TAp63 is barely expressed and the detection of ΔNp63, generated from the P2 promoter, is restricted to p53-null cells[140], because its expression is repressed by p53, as previously reported in other cell types[141,142]. Conversely, ΔNp73 expression is activated by p53 and to a lesser extent by TAp63 and TAp73[141,143]. TAp73 and several p73 N-terminal truncated isoforms are highly expressed in HCC samples, independently of the presence of wild type or mutant p53[144,145]. The overexpression of ΔNp73 and/or a high ΔNp73/TAp73 ratio are associated with a reduced survival in patients with HCC[146-149]. Moreover, in patients with HCC undergoing OLT, recurrence and reduced survival were correlated with an increased expression of ΔNp63 and ΔNp73, and a reduction of TAp63 and TAp73 expression[150]. All these observations indicate that ΔNp73/TAp73 ratio could be a potentially relevant prognostic factor in HCC.
As already mentioned, the expression of p53 family members is highly dependent on the differentiation state of cells. Thus, ΔNp73 is detected in proliferative immature HepaRG non-transformed bi-potent liver cells, but not in the differentiated state[151], whereas p53 and TAp73 are expressed in mature hepatocytes. The presence of p53 in mature hepatocytes has been shown to play an important role in the maintenance of their cell identity when exposed to an oncogenic stress. The expression of c-MYC at low levels is able to induce the expression of NANOG, OCT4 and EpCAM and to increase both sphere formation and tumorigenicity of HCC cell lines[152]. c-Myc-induced reprogramming and the acquisition of CSCs features are readily observed in p53-null cells but the functional inactivation of p53 is needed in cells with wtp53 alleles[152]. HCC cells in which p53 functional inactivation is achieved by genetic alteration or expression of proteins that exert a DNE are prone to dedifferentiation if exposed to oncogenic stimuli. Similar results have been obtained in mouse models. Tschaharganeh and colleagues have shown that in the presence of high levels of YAP, an oncogene overexpressed in human HCC and CCA[153], tp53 deletion in mature hepatocytes facilitates their dedifferentiation in vivo, the emergence of progenitor tumor cells with high NESTIN expression and the development of tumors sharing HCC and CCA characteristics[154]. NESTIN was required for the dedifferentiation and malignant expansion of p53-deficient cells and the activation of Wnt or Notch pathways in the p53-null progenitor tumors drive the development of HCC and CCA, respectively. NESTIN was also overexpressed in 17% of human HCCs and 40% of CCAs, associated with a reduced survival and the presence of mutations or LOH in the TP53 gene[154]. Altogether, these results suggest that in mature liver cells the p53-mediated inhibition of NESTIN restricts plasticity and tumorigenesis in response to oncogene activation.
Liver CSCs and the crosstalk between Nanog, IGF1R and the p53 family
Cancer tissues contain subpopulations of cells known as stem-like TICs (sl-TICs or CSCs) that have been identified as key drivers of tumor growth and malignant progression with drug resistance. Liver CSCs are bi-potent and give rise to two different lineage types, HCC and CCA. Indeed, combined hepatocellular-cholangiocarcinomas (cHCC-CCAs) with both hepatocytic and cholangiocytic phenotypes account for 1% to 5% of all primary liver cancers[155], and about one third of HCCs express progenitor cell markers such as CK7 and CK19[156], suggesting that at least a portion of HCC cells have intermediate characteristics between the bi-potent hepatic progenitor cells (HPCs) and differentiated mature hepatocytes[157,158]. The origin of liver CSCs and liver cancer cells is still controversial[159] and there is evidence for both their derivation from HPCs chronically activated by the persistent intrahepatic inflammation sustained by chronic HBV, HDV and HCV infections, alcohol consumption and metabolic disorders[160] and the dedifferentiation of mature hepatocytes that acquire the expression of stem-related genes when exposed to inflammatory cytokines, lipid overload or epigenetic reprogramming[161-163][Figure 4A].
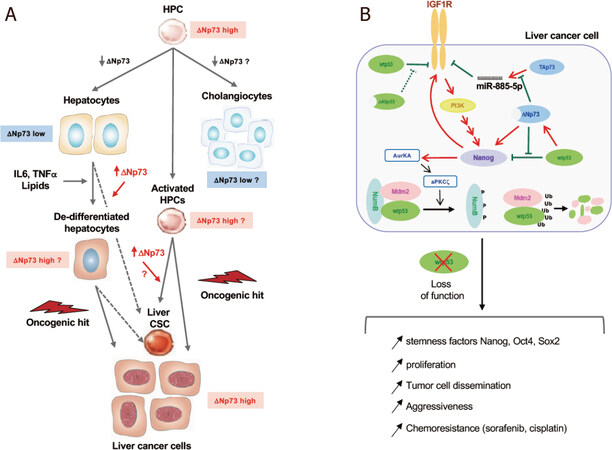
Figure 4. p53 family proteins and liver progenitor/stem cell transformation. A: the origin of cancer stem cells (CSCs) in HCC. CSCs and liver cancer cells can originate from: (1) hepatic progenitor cells (HPCs) chronically activated by the persistent intrahepatic inflammation sustained by viral infections (HBV, HDV, HCV), alcohol consumption and metabolic disorders; (2) hepatocytes undergoing dedifferentiation and expressing stem-related genes (such as Nanog). According to the currently available knowledge, ΔNp73 expression levels and its role in the different steps are highlighted; B: crosstalk between IGF1R, Nanog and p53 family in liver cancer cells. IGF1R repression by wtp53 and TAp73 and NANOG repression by wtp53 inhibit the dedifferentiation of liver cancer cells. In presence of the N-terminal truncated isoforms Δ40p53 or ΔNp73, wtp53 activity is inhibited, IGF1R and NANOG are expressed and activate a positive feedback loop. By activating AURKA-aPKCζ kinase cascade Nanog induces NumB phosphorylation and disrupts the p53-NumB interaction thus allowing Mdm2-mediated polyubiquinilation and proteasome degradation of p53. The loss of p53 function, achieved either by expression of ΔN isoforms or p53 degradation, favors stemness features, increased proliferation, dissemination and chemoresistance of liver cancer cells
p53 has been shown to repress many key transcription regulators, such as Oct4, Nanog, Sox2, Zic3, Jmjd1c, Esrrb, Tcfcp2l1, Utf1, n-Myc, c-Myc, and Prdm14 in mouse ES cells[164,165]. Among them, Nanog is of particular interest with respect to the p53 family in the context of HCC development. Nanog is overexpressed in about 30% of HCCs as shown by immunohistochemistry[166-168], regulates the expression of genes involved in mitochondrial metabolic pathways[169], promotes CSC properties[169] and enhances resistance to drugs, such as sorafenib or cisplatin, as well as tumor invasion and metastasis[169,170].
The wtp53 represses NANOG expression either by direct binding to the Nanog promoter in mouse cells[115,164] or by transactivating miR34a-c, a direct p53-target gene that in turn represses NANOG[171] in human cells[172]. In a recent study, Liu and colleagues have shown that the enhanced mitophagy observed in HCCs increases p53 localization at the mitochondrial membrane[173-176] and leads to its subsequent degradation. In contrast, when mitophagy is impaired, mitochondrial p53 is phosphorylated by Pink1 and translocated into the nucleus, where it represses NANOG expression resulting in a decrease of liver CSCs[176,177].
Two p53 family isoforms are able to exert a DNE on p53 and TAp73 regulation of NANOG. Δ40p53, by titrating full-length p53, regulates the switch from pluripotency to differentiation by increasing the expression of Nanog and Igf1R in mouse ES cells[178] and ΔNp73, as already mentioned, interferes with TAp73 in the regulation of IGF1R expression in human melanoma cells[134]. Furthermore, we and others demonstrated that ΔNp73 transactivates NANOG expression independently of p53[138,179]. The overexpression/accumulation of NANOG leads to p53 proteolysis. By activating the Aurora A kinase and aPKCζ, Nanog allows the phosphorylation of NumB and the dissociation of the p53-NumB complex, thus relieving Numb inhibition and freeing Mdm2 to degrade p53 and to potentiate the proliferation of CSCs[180]. Based on all these results, the fine-tuned crosstalk between p53 and Nanog plays a key role in the emergence, maintenance and chemoresistance of CSCs [Figure 4B].
Conclusion
In HCC, in contrast to other tumor types, the loss of functional p53 seems to be achieved by several other mechanisms in addition to genetic mutation. p53 degradation mediated by MDM2 or other E3 ubiquitin ligases and the overexpression of N-terminal truncated isoforms of the family have a prominent place due to their important role in the emergence and regulation of liver CSCs. ΔNp73 is of particular interest because, by inhibiting p53 and TAp73 activities, it activates the expression of NANOG both in a p53-dependent and -independent manner and enhances IGF1R expression. The ΔNp73-IGF1R-NANOG axis may prove to be an attractive therapeutic target for HCC patients, as already proposed in melanoma[134]. The characterization of how this axis is regulated in the viral and metabolic inflammatory chronic liver diseases that precede HCC development might also help to develop new biomarkers for risk-stratification and new preventive strategies.
The identification of at least three main mechanisms of wtp53 inactivation in HCCs may have therapeutic implications and lead to new strategies for treatment personalization. Besides p53-based gene therapy approaches, there are very few reports of drugs targeting the p53 family in HCC, either in preclinical or clinical settings. PRIMA-1 (p53 Reactivation and Induction of Massive Apoptosis), a small molecule capable of restoring a wild-type activity in some p53 mutants, has been shown to slow down tumor growth in immunocompromised mice xenografted with p53R249S-expressing human cells[181]. Therefore, it might be valuable to test the potential of PRIMA-Met (an improved version of PRIMA-1) and MIRA-1 (mutant p53-dependent induction of rapid apoptosis), a small molecule targeting p53 structurally distinct from PRIMA-1[182], to reactivate wtp53 functions in HCCs with p53 mutants, or of Nutlins to counteract MDM2-mediated degradation of wtp53[183]. IFNα has been shown to inhibit the expression of ΔNp73 by inducing chromatin remodeling at the P2p73 promoter and increasing apoptosis of human HCC cells[184]. These results provide a proof of concept of the possibility to target the third mechanism of wtp53 inactivation frequently engaged in HCCs, i.e., the DNE of N-terminal truncated members of the p53 family. However, IFNα has no indication in current HCC treatment algorithms and the identification of potent and selective drugs capable to target ΔNp73 expression might prove of value to increase HCC chemosensitivity and to enhance tumor and cancer stem cells apoptosis, either alone or in combination with TKIs.
Declarations
AcknowledgmentsWe thank Mirjam Zeisel and Giovanni Blandino for critical reading of the manuscript.
Authors’ contributionsResearched public databases for TP53 mutations: Caron de Fromentel C
Designed and wrote the manuscript: Caron de Fromentel C, Levrero M
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by grants from «Agence Nationale pour la Recherche sur le SIDA et les hepatites virales» (ANRS) (ECTZ117561) to Levrero M, from the Agence Nationale de la Recherche (ANR@TRACTION) to Levrero M; from the EU project (667273 HEP-CAR) to Levrero M; from the Ligue Nationale Contre le Cancer, Coordination Auvergne-Rhône-Alpes & Saône-et-Loire to Caron de Fromentel C and Levrero M.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Ferlay J, Colombet M, Soerjomataram I, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer 2019;144:1941-53.
2. Llovet JM, Zucman-Rossi J, Pikarsky E, et al. Hepatocellular carcinoma. Nat Rev Dis Primers 2016;2:16018.
3. Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol 2018;15:599-616.
4. Nault JC, Cheng AL, Sangro B, Llovet JM. Milestones in the pathogenesis and management of primary liver cancer. J Hepatol 2020;72:209-14.
5. Finn RS, Qin S, Ikeda M, et al; IMbrave150 Investigators. Atezolizumab plus Bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med 2020;382:1894-905.
6. Cojoc M, Mäbert K, Muders MH, Dubrovska A. A role for cancer stem cells in therapy resistance: cellular and molecular mechanisms. Semin Cancer Biol 2015;31:16-27.
7. Fekir K, Dubois-Pot-Schneider H, Désert R, et al. Retrodifferentiation of human tumor hepatocytes to stem cells leads to metabolic reprogramming and chemoresistance. Cancer Res 2019;79:1869-83.
8. Barbato L, Bocchetti M, Di Biase A, Regad T. Cancer stem cells and targeting strategies. Cells 2019;8:926.
9. Philips GM, Chan IS, Swiderska M, et al. Hedgehog signaling antagonist promotes regression of both liver fibrosis and hepatocellular carcinoma in a murine model of primary liver cancer. PLoS One 2011;6:e23943.
10. Khosla R, Rastogi A, Ramakrishna G, et al. EpCAM+ liver cancer stem-like cells exhibiting autocrine wnt signaling potentially originate in cirrhotic patients. Stem Cells Transl Med 2017;6:807-18.
11. Yang W, Yan HX, Chen L, et al. Wnt/beta-catenin signaling contributes to activation of normal and tumorigenic liver progenitor cells. Cancer Res 2008;68:4287-95.
12. Malfettone A, Soukupova J, Bertran E, et al. Transforming growth factor-β-induced plasticity causes a migratory stemness phenotype in hepatocellular carcinoma. Cancer Lett 2017;392:39-50.
13. Luo J, Wang P, Wang R, et al. The Notch pathway promotes the cancer stem cell characteristics of CD90+ cells in hepatocellular carcinoma. Oncotarget 2016;7:9525-37.
14. Della Corte CM, Viscardi G, Papaccio F, et al. Implication of the Hedgehog pathway in hepatocellular carcinoma. World J Gastroenterol 2017;23:4330-40.
15. Kawai T, Yasuchika K, Ishii T, et al. Keratin 19, a cancer stem cell marker in human hepatocellular carcinoma. Clin Cancer Res 2015;21:3081-91.
16. Terris B, Cavard C, Perret C. EpCAM, a new marker for cancer stem cells in hepatocellular carcinoma. J Hepatol 2010;52:280-1.
17. Yamashita T, Ji J, Budhu A, et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology 2009;136:1012-24.
18. Levrero M, De Laurenzi V, Costanzo A, et al. The p53/p63/p73 family of transcription factors: overlapping and distinct functions. J Cell Sci 2000;113:1661-70.
19. Murray-Zmijewski F, Lane DP, Bourdon JC. p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ 2006;13:962-72.
20. Harms K, Nozell S, Chen X. The common and distinct target genes of the p53 family transcription factors. Cell Mol Life Sci 2004;61:822-42.
22. Sauer M, Bretz AC, Beinoraviciute-Kellner R, et al. C-terminal diversity within the p53 family accounts for differences in DNA binding and transcriptional activity. Nucleic Acids Res 2008;36:1900-12.
23. De Laurenzi V, Costanzo A, Barcaroli D, et al. Two new p73 splice variants, gamma and delta, with different transcriptional activity. J Exp Med 1998;188:1763-8.
24. De Laurenzi VD, Catani MV, Terrinoni A, et al. Additional complexity in p73: induction by mitogens in lymphoid cells and identification of two new splicing variants epsilon and zeta. Cell Death Differ 1999;6:389-90.
25. Courtois S, Caron de Fromentel C, Hainaut P. p53 protein variants: structural and functional similarities with p63 and p73 isoforms. Oncogene 2004;23:631-8.
26. Costanzo A, Pediconi N, Narcisi A, et al. TP63 and TP73 in cancer, an unresolved “family” puzzle of complexity, redundancy and hierarchy. FEBS Lett 2014;588:2590-9.
27. Liu Y, Tavana O, Gu W. p53 modifications: exquisite decorations of the powerful guardian. J Mol Cell Biol 2019;11:564-77.
29. El-Deiry WS. p21(WAF1) mediates cell-cycle inhibition, relevant to cancer suppression and therapy. Cancer Res 2016;76:5189-91.
30. Sengupta S, Harris CC. p53: traffic cop at the crossroads of DNA repair and recombination. Nat Rev Mol Cell Biol 2005;6:44-55.
31. Aubrey BJ, Kelly GL, Janic A, Herold MJ, Strasser A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ 2018;25:104-13.
32. Crighton D, Wilkinson S, O’Prey J, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006;126:121-34.
33. Jiang L, Kon N, Li T, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015;520:57-62.
34. Ho T, Tan BX, Lane D. How the other half lives: what p53 does when it is not being a transcription factor. Int J Mol Sci 2019;21:13.
35. Valente LJ, Gray DH, Michalak EM, et al. p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell Rep 2013;3:1339-45.
36. Pitolli C, Wang Y, Candi E, et al. p53-mediated tumor suppression: DNA-damage response and alternative mechanisms. Cancers (Basel) 2019;11:1983.
37. Barili V, Fisicaro P, Montanini B, et al. Targeting p53 and histone methyltransferases restores exhausted CD8+ T cells in HCV infection. Nat Commun 2020;11:604.
39. Parikh N, Hilsenbeck S, Creighton CJ, et al. Effects of TP53 mutational status on gene expression patterns across 10 human cancer types. J Pathol 2014;232:522-33.
40. Seemann S, Maurici D, Olivier M, Caron de Fromentel C, Hainaut P. The tumor suppressor gene TP53: implications for cancer management and therapy. Crit Rev Clin Lab Sci 2004;41:551-83.
41. Bouaoun L, Sonkin D, Ardin M, et al. TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum Mutat 2016;37:865-76.
42. Puisieux A, Lim S, Groopman J, Ozturk M. Selective targeting of p53 gene mutational hotspots in human cancers by etiologically defined carcinogens. Cancer Res 1991;51:6185-9.
43. Pfeifer GP, Denissenko MF, Olivier M, et al. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 2002;21:7435-51.
44. Aguilar F, Hussain SP, Cerutti P. Aflatoxin B1 induces the transversion of G-->T in codon 249 of the p53 tumor suppressor gene in human hepatocytes. Proc Natl Acad Sci U S A 1993;90:8586-90.
45. Olivier M, Hussain SP, Caron de Fromentel C, Hainaut P, Harris CC. TP53 mutation spectra and load: a tool for generating hypotheses on the etiology of cancer. IARC Sci Publ 2004:247-70.
46. Bressac B, Kew M, Wands J, Ozturk M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature 1991;350:429-31.
47. Gouas DA, Villar S, Ortiz-Cuaran S, et al. TP53 R249S mutation, genetic variations in HBX and risk of hepatocellular carcinoma in The Gambia. Carcinogenesis 2012;33:1219-24.
48. Marchio A, Amougou Atsama M, Béré A, et al. Droplet digital PCR detects high rate of TP53 R249S mutants in cell-free DNA of middle African patients with hepatocellular carcinoma. Clin Exp Med 2018;18:421-31.
49. Hussain SP, Schwank J, Staib F, Wang XW, Harris CC. TP53 mutations and hepatocellular carcinoma: insights into the etiology and pathogenesis of liver cancer. Oncogene 2007;26:2166-76.
50. Amaddeo G, Cao Q, Ladeiro Y, et al. Integration of tumour and viral genomic characterizations in HBV-related hepatocellular carcinomas. Gut 2015;64:820-9.
51. Levrero M, Zucman-Rossi J. Mechanisms of HBV-induced hepatocellular carcinoma. J Hepatol 2016;64:S84-101.
52. Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer 2009;9:701-13.
53. Hanel W, Marchenko N, Xu S, et al. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ 2013;20:898-909.
54. Aschauer L, Muller PA. Novel targets and interaction partners of mutant p53 gain-of-function. Biochem Soc Trans 2016;44:460-6.
55. Dell’Orso S, Fontemaggi G, Stambolsky P, et al. ChIP-on-chip analysis of in vivo mutant p53 binding to selected gene promoters. OMICS 2011;15:305-12.
56. Lee MK, Teoh WW, Phang BH, et al. Cell-type, dose, and mutation-type specificity dictate mutant p53 functions in vivo. Cancer Cell 2012;22:751-64.
57. Sabapathy K, Lane DP. Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat Rev Clin Oncol 2018;15:13-30.
58. Gouas DA, Shi H, Hautefeuille AH, et al. Effects of the TP53 p.R249S mutant on proliferation and clonogenic properties in human hepatocellular carcinoma cell lines: interaction with hepatitis B virus X protein. Carcinogenesis 2010;31:1475-82.
59. Ghebranious N, Sell S. The mouse equivalent of the human p53ser249 mutation p53ser246 enhances aflatoxin hepatocarcinogenesis in hepatitis B surface antigen transgenic and p53 heterozygous null mice. Hepatology 1998;27:967-73.
60. Liao P, Zeng SX, Zhou X, et al. Mutant p53 gains its function via c-Myc activation upon CDK4 phosphorylation at serine 249 and consequent PIN1 binding. Mol Cell 2017;68:1134-46.e6.
61. Besaratinia A, Kim SI, Hainaut P, Pfeifer GP. In vitro recapitulating of TP53 mutagenesis in hepatocellular carcinoma associated with dietary aflatoxin B1 exposure. Gastroenterology 2009;137:1127-37. 1137.e1-5
62. Sell S. Mouse models to study the interaction of risk factors for human liver cancer. Cancer Res 2003;63:7553-62.
63. Gearhart TL, Bouchard MJ. The hepatitis B virus X protein modulates hepatocyte proliferation pathways to stimulate viral replication. J Virol 2010;84:2675-86.
64. Pang R, Lee TK, Poon RT, et al. Pin1 interacts with a specific serine-proline motif of hepatitis B virus X-protein to enhance hepatocarcinogenesis. Gastroenterology 2007;132:1088-103.
65. Truant R, Antunovic J, Greenblatt J, Prives C, Cromlish JA. Direct interaction of the hepatitis B virus HBx protein with p53 leads to inhibition by HBx of p53 response element-directed transactivation. J Virol 1995;69:1851-9.
66. Wang XW, Forrester K, Yeh H, et al. Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding, transcriptional activity, and association with transcription factor ERCC3. Proc Natl Acad Sci U S A 1994;91:2230-4.
67. Levine AJ. The common mechanisms of transformation by the small DNA tumor viruses: The inactivation of tumor suppressor gene products: p53. Virology 2009;384:285-93.
68. Sanz G, Singh M, Peuget S, Selivanova G. Inhibition of p53 inhibitors: progress, challenges and perspectives. J Mol Cell Biol 2019;11:586-99.
69. Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature 1997;387:299-303.
70. Cordon-Cardo C, Latres E, Drobnjak M, et al. Molecular abnormalities of mdm2 and p53 genes in adult soft tissue sarcomas. Cancer Res 1994;54:794-9.
71. Endo K, Ueda T, Ohta T, Terada T. Protein expression of MDM2 and its clinicopathological relationships in human hepatocellular carcinoma. Liver 2000;20:209-15.
72. Bang S, Kaur S, Kurokawa M. Regulation of the p53 family proteins by the ubiquitin proteasomal pathway. Int J Mol Sci 2019;21:261.
73. Huang X, Qian X, Cheng C, et al. Expression of Pirh2, a p27(Kip1) ubiquitin ligase, in hepatocellular carcinoma: correlation with p27(Kip1) and cell proliferation. Hum Pathol 2011;42:507-15.
74. Lee YH, Andersen JB, Song HT, et al. Definition of ubiquitination modulator COP1 as a novel therapeutic target in human hepatocellular carcinoma. Cancer Res 2010;70:8264-9.
75. Leng RP, Lin Y, Ma W, et al. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell 2003;112:779-91.
76. Dornan D, Wertz I, Shimizu H, et al. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 2004;429:86-92.
77. Shen J, Li P, Shao X, et al. The E3 ligase RING1 targets p53 for degradation and promotes cancer cell proliferation and survival. Cancer Res 2018;78:359-71.
78. Pan Y, Wang B, Yang X, et al. CUL4A facilitates hepatocarcinogenesis by promoting cell cycle progression and epithelial-mesenchymal transition. Sci Rep 2015;5:17006.
79. Zhu K, Li J, Li J, et al. Ring1 promotes the transformation of hepatic progenitor cells into cancer stem cells through the Wnt/β-catenin signaling pathway. J Cell Biochem 2019. Online ahead of print
80. Gao J, Buckley SM, Cimmino L, et al. The CUL4-DDB1 ubiquitin ligase complex controls adult and embryonic stem cell differentiation and homeostasis. Elife 2015;4:e07539.
81. Surget S, Khoury MP, Bourdon JC. Uncovering the role of p53 splice variants in human malignancy: a clinical perspective. Onco Targets Ther 2013;7:57-68.
82. Davison TS, Vagner C, Kaghad M, et al. p73 and p63 are homotetramers capable of weak heterotypic interactions with each other but not with p53. J Biol Chem 1999;274:18709-14.
83. Irwin M, Marin MC, Phillips AC, et al. Role for the p53 homologue p73 in E2F-1-induced apoptosis. Nature 2000;407:645-8.
84. Seelan RS, Irwin M, van der Stoop P, et al. The human p73 promoter: characterization and identification of functional E2F binding sites. Neoplasia 2002;4:195-203.
85. Stiewe T, Pützer BM. Role of the p53-homologue p73 in E2F1-induced apoptosis. Nat Genet 2000;26:464-9.
86. Costanzo A, Merlo P, Pediconi N, et al. DNA damage-dependent acetylation of p73 dictates the selective activation of apoptotic target genes. Molecular Cell 2002;9:175-86.
87. Pediconi N, Ianari A, Costanzo A, et al. Differential regulation of E2F1 apoptotic target genes in response to DNA damage. Nat Cell Biol 2003;5:552-8.
88. Pediconi N, Guerrieri F, Vossio S, et al. hSirT1-dependent regulation of the PCAF-E2F1-p73 apoptotic pathway in response to DNA damage. Mol Cell Biol 2009;29:1989-98.
89. Marabese M, Vikhanskaya F, Rainelli C, Sakai T, Broggini M. DNA damage induces transcriptional activation of p73 by removing C-EBPalpha repression on E2F1. Nucleic Acids Res 2003;31:6624-32.
90. Fontemaggi G, Gurtner A, Strano S, et al. The transcriptional repressor ZEB regulates p73 expression at the crossroad between proliferation and differentiation. Mol Cell Biol 2001;21:8461-70.
91. Wu S, Murai S, Kataoka K, Miyagishi M. Yin Yang 1 induces transcriptional activity of p73 through cooperation with E2F1. Biochem Biophys Res Commun 2008;365:75-81.
92. Grob TJ, Novak U, Maisse C, et al. Human delta Np73 regulates a dominant negative feedback loop for TAp73 and p53. Cell Death Differ 2001;8:1213-23.
93. Vossio S, Palescandolo E, Pediconi N, et al. DN-p73 is activated after DNA damage in a p53-dependent manner to regulate p53-induced cell cycle arrest. Oncogene 2002;21:3796-803.
94. Kartasheva NN, Contente A, Lenz-Stöppler C, Roth J, Dobbelstein M. p53 induces the expression of its antagonist p73 Delta N, establishing an autoregulatory feedback loop. Oncogene 2002;21:4715-27.
95. Nakagawa T, Takahashi M, Ozaki T, et al. Autoinhibitory regulation of p73 by Delta Np73 to modulate cell survival and death through a p73-specific target element within the Delta Np73 promoter. Mol Cell Biol 2002;22:2575-85.
96. Lunghi P, Costanzo A, Mazzera L, et al. The p53 family protein p73 provides new insights into cancer chemosensitivity and targeting. Clin Cancer Res 2009;15:6495-502.
97. Irwin MS, Kondo K, Marin MC, et al. Chemosensitivity linked to p73 function. Cancer Cell 2003;3:403-10.
98. Leong CO, Vidnovic N, DeYoung MP, Sgroi D, Ellisen LW. The p63/p73 network mediates chemosensitivity to cisplatin in a biologically defined subset of primary breast cancers. J Clin Invest 2007;117:1370-80.
99. Ishimoto O, Kawahara C, Enjo K, et al. Possible oncogenic potential of DeltaNp73: a newly identified isoform of human p73. Cancer Res 2002;62:636-41.
100. Petrenko O, Zaika A, Moll UM. deltaNp73 facilitates cell immortalization and cooperates with oncogenic Ras in cellular transformation in vivo. Mol Cell Biol 2003;23:5540-55.
101. Beitzinger M, Hofmann L, Oswald C, et al. p73 poses a barrier to malignant transformation by limiting anchorage-independent growth. EMBO J 2008;27:792-803.
102. Tannapfel A, John K, Mise N, et al. Autonomous growth and hepatocarcinogenesis in transgenic mice expressing the p53 family inhibitor DNp73. Carcinogenesis 2008;29:211-8.
103. Botti E, Spallone G, Moretti F, et al. Developmental factor IRF6 exhibits tumor suppressor activity in squamous cell carcinomas. Proc Natl Acad Sci U S A 2011;108:13710-5.
104. Marinari B, Moretti F, Botti E, et al. The tumor suppressor activity of IKKalpha in stratified epithelia is exerted in part via the TGF-beta antiproliferative pathway. Proc Natl Acad Sci U S A 2008;105:17091-6.
105. Ramsey MR, Wilson C, Ory B, et al. FGFR2 signaling underlies p63 oncogenic function in squamous cell carcinoma. J Clin Invest 2013;123:3525-38.
106. Koster MI, Kim S, Mills AA, DeMayo FJ, Roop DR. p63 is the molecular switch for initiation of an epithelial stratification program. Genes Dev 2004;18:126-31.
107. De Laurenzi V, Raschellá G, Barcaroli D, et al. Induction of neuronal differentiation by p73 in a neuroblastoma cell line. J Biol Chem 2000;275:15226-31.
108. Yang A, Walker N, Bronson R, et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 2000;404:99-103.
109. Yang A, Schweitzer R, Sun D, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999;398:714-8.
110. Donehower LA, Harvey M, Slagle BL, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992;356:215-21.
111. Jacks T, Remington L, Williams BO, et al. Tumor spectrum analysis in p53-mutant mice. Current Biology 1994;4:1-7.
112. Flores ER, Sengupta S, Miller JB, et al. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell 2005;7:363-73.
113. Guo X, Keyes WM, Papazoglu C, et al. TAp63 induces senescence and suppresses tumorigenesis in vivo. Nat Cell Biol 2009;11:1451-7.
114. Tomasini R, Tsuchihara K, Wilhelm M, et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev 2008;22:2677-91.
115. Lin T, Chao C, Saito S, et al. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol 2005;7:165-71.
116. Meletis K, Wirta V, Hede SM, et al. p53 suppresses the self-renewal of adult neural stem cells. Development 2006;133:363-9.
117. Liu Y, Elf SE, Miyata Y, et al. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell 2009;4:37-48.
118. Kim J, Lengner CJ, Kirak O, et al. Reprogramming of postnatal neurons into induced pluripotent stem cells by defined factors. Stem Cells 2011;29:992-1000.
119. Yi L, Lu C, Hu W, Sun Y, Levine AJ. Multiple roles of p53-related pathways in somatic cell reprogramming and stem cell differentiation. Cancer Res 2012;72:5635-45.
120. Tovy A, Spiro A, McCarthy R, et al. p53 is essential for DNA methylation homeostasis in naïve embryonic stem cells, and its loss promotes clonal heterogeneity. Genes Dev 2017;31:959-72.
122. Menendez S, Camus S, Izpisua Belmonte JC. p53: guardian of reprogramming. Cell Cycle 2010;9:3887-91.
123. Sarig R, Rivlin N, Brosh R, et al. Mutant p53 facilitates somatic cell reprogramming and augments the malignant potential of reprogrammed cells. J Exp Med 2010;207:2127-40.
124. Mizuno H, Spike BT, Wahl GM, Levine AJ. Inactivation of p53 in breast cancers correlates with stem cell transcriptional signatures. Proc Natl Acad Sci U S A 2010;107:22745-50.
125. Lee TI, Jenner RG, Boyer LA, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 2006;125:301-13.
126. Boyer LA, Plath K, Zeitlinger J, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 2006;441:349-53.
127. Di Fiore R, Marcatti M, Drago-Ferrante R, et al. Mutant p53 gain of function can be at the root of dedifferentiation of human osteosarcoma MG63 cells into 3AB-OS cancer stem cells. Bone 2014;60:198-212.
128. Arsic N, Gadea G, Lagerqvist EL, et al. The p53 isoform Δ133p53β promotes cancer stem cell potential. Stem Cell Reports 2015;4:531-40.
129. Senoo M, Pinto F, Crum CP, McKeon F. p63 is essential for the proliferative potential of stem cells in stratified epithelia. Cell 2007;129:523-36.
130. Chakrabarti R, Wei Y, Hwang J, et al. ΔNp63 promotes stem cell activity in mammary gland development and basal-like breast cancer by enhancing Fzd7 expression and Wnt signalling. Nat Cell Biol 2014;16:1004-15. 1-13
131. Memmi EM, Sanarico AG, Giacobbe A, et al. p63 Sustains self-renewal of mammary cancer stem cells through regulation of Sonic Hedgehog signaling. Proc Natl Acad Sci U S A 2015;112:3499-504.
132. Liu S, Dontu G, Mantle ID, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res 2006;66:6063-71.
133. Siddique HR, Saleem M. Role of BMI1, a stem cell factor, in cancer recurrence and chemoresistance: preclinical and clinical evidences. Stem Cells 2012;30:372-8.
134. Meier C, Hardtstock P, Joost S, Alla V, Pützer BM. p73 and IGF1R regulate emergence of aggressive cancer stem-like features via miR-885-5p Control. Cancer Res 2016;76:197-205.
135. Niemantsverdriet M, Nagle P, Chiu RK, et al. ΔNp73 enhances promoter activity of TGF-β induced genes. PLoS One 2012;7:e50815.
136. Alexandrova EM, Petrenko O, Nemajerova A, et al. ΔNp63 regulates select routes of reprogramming via multiple mechanisms. Cell Death Differ 2013;20:1698-708.
137. Martin-Lopez M, Maeso-Alonso L, Fuertes-Alvarez S, et al. p73 is required for appropriate BMP-induced mesenchymal-to-epithelial transition during somatic cell reprogramming. Cell Death Dis 2017;8:e3034.
138. Lin Y, Cheng Z, Yang Z, Zheng J, Lin T. DNp73 improves generation efficiency of human induced pluripotent stem cells. BMC Cell Biol 2012;13:9.
139. Ramalho FS, Ramalho LN, Della Porta L, Zucoloto S. Comparative immunohistochemical expression of p63 in human cholangiocarcinoma and hepatocellular carcinoma. J Gastroenterol Hepatol 2006;21:1276-80.
140. Petitjean A, Cavard C, Shi H, et al. The expression of TA and DeltaNp63 are regulated by different mechanisms in liver cells. Oncogene 2005;24:512-9.
141. Lanza M, Marinari B, Papoutsaki M, et al. Cross-talks in the p53 family: deltaNp63 is an anti-apoptotic target for deltaNp73alpha and p53 gain-of-function mutants. Cell Cycle 2006;5:1996-2004.
142. Ruptier C, De Gaspéris A, Ansieau S, et al. TP63 P2 promoter functional analysis identifies β-catenin as a key regulator of ΔNp63 expression. Oncogene 2011;30:4656-65.
143. Petitjean A, Ruptier C, Tribollet V, et al. Properties of the six isoforms of p63: p53-like regulation in response to genotoxic stress and cross talk with ΔNp73. Carcinogenesis 2008;29:273-81.
144. Stiewe T, Tuve S, Peter M, et al. Quantitative TP73 transcript analysis in hepatocellular carcinomas. Clin Cancer Res 2004;10:626-33.
145. Castillo J, Goñi S, Latasa MU, et al. Amphiregulin induces the alternative splicing of p73 into its oncogenic isoform DeltaEx2p73 in human hepatocellular tumors. Gastroenterology 2009;137:1805-15.e1-4.
146. Müller M, Schilling T, Sayan AE, et al. TAp73/Delta Np73 influences apoptotic response, chemosensitivity and prognosis in hepatocellular carcinoma. Cell Death Differ 2005;12:1564-77.
147. Schuster A, Schilling T, De Laurenzi V, et al. ΔNp73β is oncogenic in hepatocellular carcinoma by blocking apoptosis signaling via death receptors and mitochondria. Cell Cycle 2010;9:2629-39.
148. Zhao L, Fu L, Xu Z, et al. The anticancer effects of cinobufagin on hepatocellular carcinoma Huh-7 cells are associated with activation of the p73 signaling pathway. Mol Med Rep 2019;19:4119-28.
149. Yoon MK, Ha JH, Lee MS, Chi SW. Structure and apoptotic function of p73. BMB Rep 2015;48:81-90.
150. González R, De la Rosa ÁJ, Rufini A, et al. Role of p63 and p73 isoforms on the cell death in patients with hepatocellular carcinoma submitted to orthotopic liver transplantation. PLoS One 2017;12:e0174326.
151. Gifu P, Pez F, Bian L, et al. DeltaNp73 isoforms are involved in the immature phenotype of liver cancer cells. J Hepatol 2017;66:S646.
152. Akita H, Marquardt JU, Durkin ME, et al. MYC activates stem-like cell potential in hepatocarcinoma by a p53-dependent mechanism. Cancer Res 2014;74:5903-13.
153. Li H, Wolfe A, Septer S, et al. Deregulation of Hippo kinase signalling in human hepatic malignancies. Liver Int 2012;32:38-47.
154. Tschaharganeh DF, Xue W, Calvisi DF, et al. p53-dependent Nestin regulation links tumor suppression to cellular plasticity in liver cancer. Cell 2014;158:579-92.
155. Brunt E, Aishima S, Clavien PA, et al. cHCC-CCA: consensus terminology for primary liver carcinomas with both hepatocytic and cholangiocytic differentation. Hepatology 2018;68:113-26.
156. Machida K. Existence of cancer stem cells in hepatocellular carcinoma: myth or reality? Hepatol Int 2017;11:143-7.
157. Mishra L, Banker T, Murray J, et al. Liver stem cells and hepatocellular carcinoma. Hepatology 2009;49:318-29.
158. Yamashita T, Wang XW. Cancer stem cells in the development of liver cancer. J Clin Invest 2013;123:1911-8.
159. Wu Y, Zhang J, Zhang X, et al. Cancer stem cells: a potential breakthrough in HCC-targeted therapy. Front Pharmacol 2020;11:198.
160. Li XF, Chen C, Xiang DM, et al. Chronic inflammation-elicited liver progenitor cell conversion to liver cancer stem cell with clinical significance. Hepatology 2017;66:1934-51.
161. Dubois-Pot-Schneider H, Fekir K, Coulouarn C, et al. Inflammatory cytokines promote the retrodifferentiation of tumor-derived hepatocyte-like cells to progenitor cells. Hepatology 2014;60:2077-90.
162. Belloni L, Di Cocco S, Guerrieri F, et al. Targeting a phospho-STAT3-miRNAs pathway improves vesicular hepatic steatosis in an in vitro and in vivo model. Sci Rep 2018;8:13638.
163. Pediconi N, Salerno D, Lupacchini L, et al. EZH2, JMJD3, and UTX epigenetically regulate hepatic plasticity inducing retro-differentiation and proliferation of liver cells. Cell Death Dis 2019;10:518.
164. Li M, He Y, Dubois W, et al. Distinct regulatory mechanisms and functions for p53-activated and p53-repressed DNA damage response genes in embryonic stem cells. Mol Cell 2012;46:30-42.
166. Yin X, Li YW, Zhang BH, et al. Coexpression of stemness factors Oct4 and Nanog predict liver resection. Ann Surg Oncol 2012;19:2877-87.
167. Zhao X, Sun B, Sun D, et al. Slug promotes hepatocellular cancer cell progression by increasing sox2 and nanog expression. Oncol Rep 2015;33:149-56.
168. Liang C, Zhang K, Ge H, et al. Prognostic and clinicopathological value of Nanog in hepatocellular carcinoma: a meta-analysis. Clin Chim Acta 2018;477:24-31.
169. Chen CL, Uthaya Kumar DB, Punj V, et al. NANOG metabolically reprograms tumor-initiating stem-like cells through tumorigenic changes in oxidative phosphorylation and fatty acid metabolism. Cell Metab 2016;23:206-19.
170. Shan J, Shen J, Liu L, et al. Nanog regulates self-renewal of cancer stem cells through the insulin-like growth factor pathway in human hepatocellular carcinoma. Hepatology 2012;56:1004-14.
171. Choi YJ, Lin CP, Ho JJ, et al. miR-34 miRNAs provide a barrier for somatic cell reprogramming. Nat Cell Biol 2011;13:1353-60.
172. He L, He X, Lim LP, et al. A microRNA component of the p53 tumour suppressor network. Nature 2007;447:1130-4.
173. Marchenko ND, Zaika A, Moll UM. Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J Biol Chem 2000;275:16202-12.
174. Blandino G, Valenti F, Sacconi A, Di Agostino S. Wild type- and mutant p53 proteins in mitochondrial dysfunction: emerging insights in cancer disease. Semin Cell Dev Biol 2020;98:105-17.
175. Comel A, Sorrentino G, Capaci V, Del Sal G. The cytoplasmic side of p53’s oncosuppressive activities. FEBS Lett 2014;588:2600-9.
176. Liu K, Lee J, Kim JY, et al. Mitophagy controls the activities of tumor suppressor p53 to regulate hepatic cancer stem cells. Mol Cell 2017;68:281-92.e5.
177. Liu K, Lee J, Ou JJ. Autophagy and mitophagy in hepatocarcinogenesis. Mol Cell Oncol 2018;5:e1405142.
178. Ungewitter E, Scrable H. Delta40p53 controls the switch from pluripotency to differentiation by regulating IGF signaling in ESCs. Genes Dev 2010;24:2408-19.
179. Voeltzel T, Flores-Violante M, Zylbersztejn F, et al. A new signaling cascade linking BMP4, BMPR1A, ΔNp73 and NANOG impacts on stem-like human cell properties and patient outcome. Cell Death Dis 2018;9:1011.
180. Siddique HR, Feldman DE, Chen CL, et al. NUMB phosphorylation destabilizes p53 and promotes self-renewal of tumor-initiating cells by a NANOG-dependent mechanism in liver cancer. Hepatology 2015;62:1466-79.
181. Shi H, Lambert JM, Hautefeuille A, et al. In vitro and in vivo cytotoxic effects of PRIMA-1 on hepatocellular carcinoma cells expressing mutant p53ser249. Carcinogenesis 2008;29:1428-34.
182. Bykov VJN, Eriksson SE, Bianchi J, Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer 2018;18:89-102.
183. Hientz K, Mohr A, Bhakta-Guha D, Efferth T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017;8:8921-46.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Caron de Fromentel C, Levrero M. p53 functional loss, stemness and hepatocellular carcinoma. Hepatoma Res 2020;6:80. http://dx.doi.org/10.20517/2394-5079.2020.77
AMA Style
Caron de Fromentel C, Levrero M. p53 functional loss, stemness and hepatocellular carcinoma. Hepatoma Research. 2020; 6: 80. http://dx.doi.org/10.20517/2394-5079.2020.77
Chicago/Turabian Style
Caron de Fromentel, Claude, Massimo Levrero. 2020. "p53 functional loss, stemness and hepatocellular carcinoma" Hepatoma Research. 6: 80. http://dx.doi.org/10.20517/2394-5079.2020.77
ACS Style
Caron de, Fromentel C.; Levrero M. p53 functional loss, stemness and hepatocellular carcinoma. Hepatoma. Res. 2020, 6, 80. http://dx.doi.org/10.20517/2394-5079.2020.77
About This Article
Copyright

Data & Comments
Data


 Cite This Article 42 clicks
Cite This Article 42 clicks

 Like This Article 8
likes
Like This Article 8
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.