
Targeted mutation-based therapy for intrahepatic cholangiocarcinoma
 0
0 , ...
, ... Abstract
Intrahepatic cholangiocarcinoma (iCCA) is a highly aggressive primary liver cancer with limited treatment options and poor prognosis. Although gemcitabine combined with cisplatin (GEMCIS) or newly GEMCIS plus durvalumab is the first-line systemic therapy for iCCA, several promising treatment targets have been identified in the past decade in both first- and subsequent-line settings, including neurotrophic tropomyosin-receptor tyrosine kinase (NTRK) fusions, RET fusions, high microsatellite instability (MSI-H), high tumor mutation burden (TMB-H), as well as fibroblast growth factor receptor 2 (FGFR2) fusions, BRAF V600E mutation, isocitrate dehydrogenase (IDH)-1 and IDH-2 mutations, and human epidermal growth factor receptor 2 [HER2 (ERBB2)] amplifications. Corresponding small molecule inhibitors and monoclonal antibodies have demonstrated improved efficacy and survival benefits in phase 2 or phase 3 studies, gained regulatory approvals or recommendations in guidelines, and reshaped the therapeutic management for advanced cholangiocarcinoma. Numerous novel targeted drugs and combination therapies have been developed and are under evaluation. Despite the progress made in targeted therapy, it still faces challenges such as acquired drug resistance, precise patient selection, and serious adverse events. Therefore, large-scale randomized phase 3 trials of novel targeted agents and innovative regimens are warranted to benefit this population. Herein, we present a comprehensive review of the literature of clinical significance on targeted therapy for iCCA in recent years, focusing on the advances in mutation-based targeted therapy.
Keywords
INTRODUCTION
iCCA is a highly aggressive primary liver cancer, representing approximately 20% of all malignant liver tumors[1]. Due to the distinctive biological characteristics, iCCA presents a hidden onset, elevated malignancy degree, restricted treatment options, and dismal prognosis. Radical surgical resection, currently the sole potential curative approach, is only executed in 30% of patients, and the median disease-specific survival was about 36 months in these resected patients[2]. Systemic therapy can delay progression for approximately 70% of patients with advanced or metastatic disease; however, the median overall survival (mOS) is still around one year[3]. Multiple potential therapeutic targets and genomic alterations have been discovered successively, with the advances of iCCA genomic profiling research and the progress of second-generation sequencing technology, raising the hope for patients with advanced iCCA to achieve improved oncologic outcomes[4].
Over the past decade, several promising therapeutic targets have been identified and will pave the way for a new era of iCCA management, such as fibroblast growth FGFR2 fusions and IDH-1 and IDH-2 mutations[5]. The FGFR2 inhibitor pemigatinib has been granted approval by the US Food and Drug Administration (FDA) as the first targeted therapy for locally advanced or metastatic iCCA with FGFR2 fusions or rearrangements[6]. Subsequently, a few FGFR inhibitors and IDH inhibitors have been approved, with more agents undergoing evaluation as subsequent-line therapy for advanced cholangiocarcinoma[7]. Furthermore, increasing interest has been shown in patients with cholangiocarcinoma who possess BRAF V600E mutation, amplifications or mutations of HER2 (ERBB2), fusions of NTRK, RET fusions, or KRAS mutations.
In this paper, we provide a comprehensive review of recent significant literature on targeted therapy for iCCA in recent years. Our focus is to summarize the latest advances in mutation-based targeted therapy and offer a valuable reference for both experimental and clinical research.
CURRENT GENETIC LANDSCAPE AND ACTIONABLE ABERRATIONS
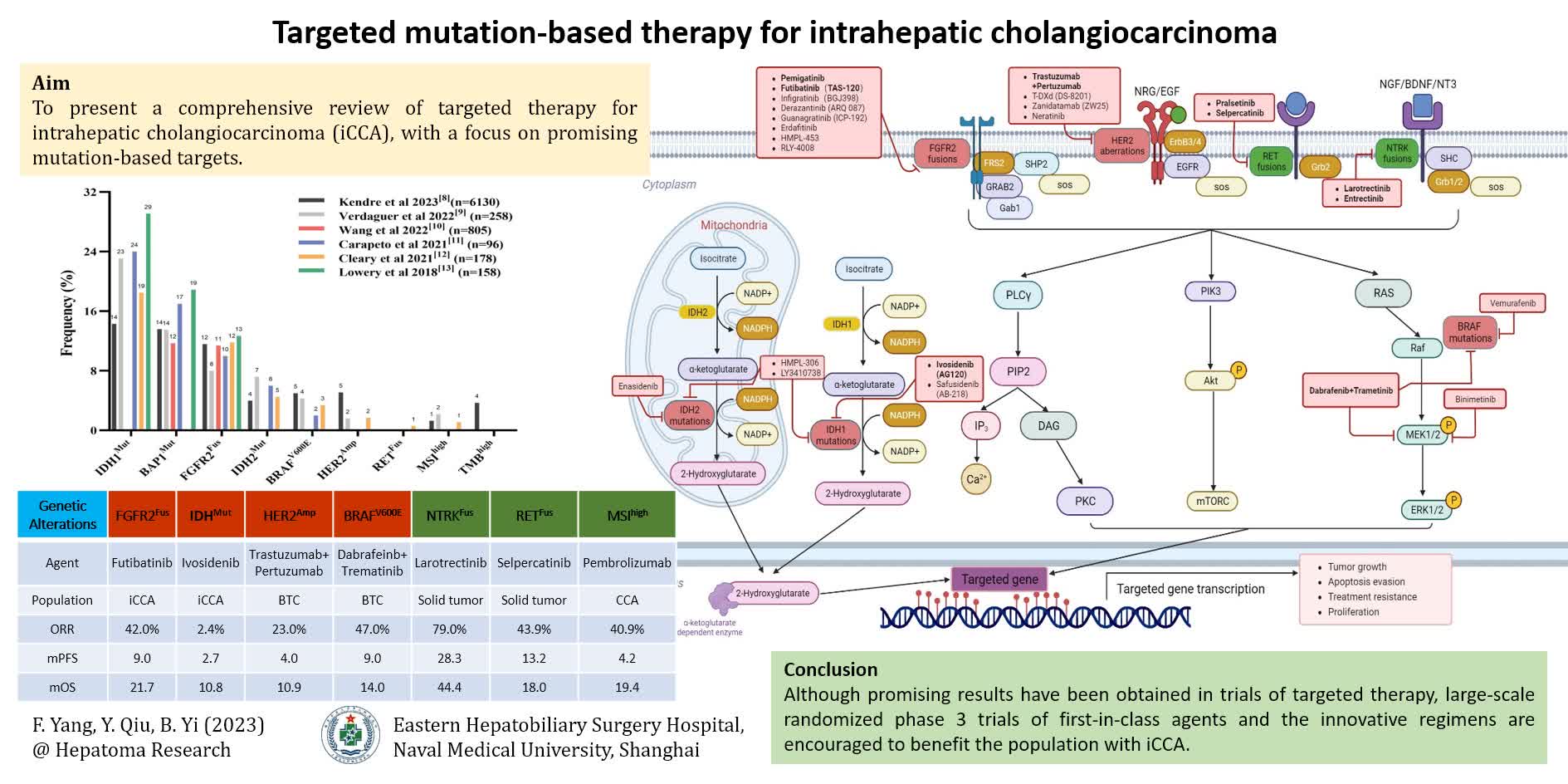
Given the genetic heterogeneity in iCCA, genomic profiling is crucial to inform targeted therapy. The advancement of next-generation sequencing (NGS) technology has enabled the identification of actionable alterations and facilitated the exploration of potential mechanisms underlying acquired resistance. Nearly 40% of iCCA patients carry targetable or potential actionable genetic alterations, including but not limited to tumor protein p53 (TP53), IDH1/2, AT-rich interaction domain 1A (ARID1A), BRCA1 associated protein 1 (BAP1), cyclin-dependent kinase inhibitor 2A (CDKN2A), KRAS, FGFR2, polybromo 1 (PBRM1), SMAD4, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA), MDM2, BRAF, BRCA1/2, ERBB2 (HER2), and MET[8-14]. IDH1 mutations (incidence of 14%-29%) and FGFR2 fusions or rearrangements (8%-13%) represent the most prevalent actionable alterations in iCCA. ERBB2 gene (encoding HER2 protein) amplification and/or overexpression, as well as BRAF V600E, have also been identified in 2%-5% of such patients [Figure 1 and 2 and Table 1]. Other alteration such as NRTK fusion, RET fusion, KRAS G12C mutation, or microsatellite instability (MSI) has been detected in less than 2% of iCCA patients. However, each of them has played an indispensable role in targeted therapy or immunotherapy[8,12,15].

Figure 1. Prevalence of actionable genomic alterations in intrahepatic cholangiocarcinoma. IDH1/2Mut: isocitrate dehydrogenase-1/2 mutation; BAP1Mut: BRCA1-Associated Protein 1 mutation; FGFR2Fus: fibroblast growth factor receptor 2 fusion; HER2Amp: human epidermal growth factor 2 amplification; BRAFV600E: BRAF V600E mutation; NTRKFus: NTRK fusion; RETFus: RET fusion; MSIhigh: high microsatellite instability; TMBhigh: high tumor mutation burden.

Figure 2. Schematic representation of main therapeutic targets, drugs and signaling pathways for intrahepatic cholangiocarcinoma in 2023. The next generation sequencing is recommended to define the genomic aberration status before systemic therapy for unresectable or metastatic patients. The altered gene textbox highlighted in green indicates that targeted monotherapy, such as anti-NTRKFus or anti-RETFus, is recommended by NCCN in first-line or subsequent-line settings. Accordingly, the gene textbox highlighted in red means that targeted therapy alone, such as anti-FGFR2Fus, anti-IDH1Mut, anti-HER2Amp, or anti-BRAFV600E, is recommended only after front-line systemic therapy. All the targeted agents recommended in the NCCN guideline of BTC (version 2. 2023) are displayed in bold for better individualized drug selection. IDH1/2Mut: isocitrate dehydrogenase-1/2 mutation; FGFR2Fus: fibroblast growth factor receptor 2 fusion; HER2Amp: human epidermal growth factor 2 amplification; BRAFV600E: BRAF V600E mutation; NTRKFus: NTRK fusion; RETFus: RET fusion.
Common genomic alterations and frequency (%) in intrahepatic cholangiocarcinoma
| Target | Kendre et al. 2023[8] (n = 6130) | Verdaguer et al. 2022[9] (n = 258) | Wang et al. 2022[10] (n = 805) | Carapeto et al. 2021[11] (n = 96) | Cleary et al. 2021[12] (n = 178) | Lowery et al. 2018[13] (n = 158) |
| ARID1A | 17.9 | 18.1 | 13.0 | 19.0 | NA | 23.0 |
| BAP1 | 13.6 | 13.5 | 11.7 | 17.0 | NA | 18.9 |
| BRAF V600E | 5.0 | 4.3 | NA | 2.0 | 3.4 | NA |
| BRCA1/2 | 3.4 | 3.1 | NA | NA | 1.7 | NA |
| CDKN2A | 30.2 | NA | 6.1 | 3.0 | NA | NA |
| HER2 | 5.1 | 1.6 | NA | NA | 1.7 | NA |
| FGFR2 | 11.6 | 8.0 | 11.4 | 10.0 | 11.8 | 12.7 |
| IDH1 | 14.3 | 23.1 | NA | 24.0 | 18.5 | 29.1 |
| IDH2 | 4.0 | 7.2 | NA | 6.0 | 4.5 | NA |
| IDH1/2 | 25.9 | 31.1 | 14.8 | 34.0 | 30.3 | NA |
| KRAS | 20.1 | 11.5 | NA | 6.0 | 17.3 | 7.0 |
| MDM2 | 4.3 | NA | NA | NA | NA | NA |
| MET | 2.2 | 2.4 | NA | NA | NA | NA |
| PBRM1 | 11.3 | NA | 8.3 | 7.0 | NA | NA |
| PIK3CA | 6.5 | 7.7 | 5.7 | 1.0 | NA | NA |
| RET | NA | NA | NA | NA | 0.6 | NA |
| SMAD4 | 7.1 | NA | NA | NA | NA | 5.1 |
| TP53 | 33.7 | NA | 22.0 | NA | NA | 17.8 |
| TMB-high | 3.7 | NA | NA | NA | NA | NA |
Genetic mutation-based clinical and histopathological characterization
The genomic diversity and heterogeneity of cholangiocarcinoma are closely related to tumor origin, as well as its epidemiological and clinicopathological features. In cholangiocarcinoma, iCCA is more likely to exhibit FGFR2 fusion, IDH1/2 mutation, and BRAF V600E mutation, while extrahepatic cholangiocarcinoma tends to have higher frequencies of TP53, KRAS, and BRCA1 mutations, and ERBB2 amplification/mutation is more commonly observed in gallbladder cancer[8,16,17]. Even in iCCA, the molecular characteristics of large duct types differ from those of small duct types. Small duct type of iCCA is more likely to have IDH1/2 mutations or FGFR2 fusions, and has a better prognosis than that of the opposite type[18,19]. Using proteomic profiling, Dong et al. identified four subtypes of distinct features in iCCA patients: inflammation (S1), interstitial (S2), metabolism (S3), and differentiation (S4)[20]. KRAS mutations exhibited significant enrichment in subtype S1, TP53 mutations predominated in S3, and the alterations of FGFR2, BAP1, and IDH1/2 enriched in S4. Compared to subtype S4, S1 presented higher CA19-9 levels, more metastasis, and worse prognosis. S3 was identified as a characteristic of HBV infection. Therefore, genetic mutation analysis could help recognize the clinicopathological characteristics and even prognosis of iCCA.
Genetic mutation-based tumor microenvironment and heterogeneity
ITH is a ubiquitous phenomenon in various types of cancer, as reported by Dentro et al., who showed that at least one subclonal expansion was found in over 75% of samples across all 38 cancer types except melanoma[21]. Recently, multi-region sequencing has revealed that iCCA exhibits extremely high ITH at both the genomic and immune microenvironment levels[22]. The differences in gene mutations and copy number changes exceed 40% between different regions of the same tumor, and more than half of iCCAs exhibit heterogeneous tumor-infiltrating immunity. iCCA driven by FGFR2 mutations has a lower neoantigen load and immune infiltration, suggesting that PD1 antibodies may need to be combined with FGRF2 inhibitors to achieve some efficacy in this situation. In addition, researchers screened the HLA binding ability and immunogenicity of immune peptides derived from TP53, KRAS, IDH1/2, and BAP1 mutations, and found that the peptide LVVVGADGV derived from KRAS G12D has strong immunogenicity and may serve as a new target for personalized immunotherapy. Actually, extensive ITH existed in iCCA at multiple levels, including genomics, transcriptomics, and the immune system. IDH-mutant iCCA displays increased ITH and colder tumor microenvironment (TME)[23]. Lin et al. employed multiomics analysis to categorize 255 iCCA patients into three immune subgroups: IG1 (suppressive in 25.1%), IG2 (exclusion in 42.7%), and IG3 (activated in 32.2%)[24]. These subgroups exhibited distinct genetic and molecular characteristics. Subgroup IG1 had the poorest prognosis, with the highest degree of bone marrow infiltration and abundant KRAS mutations. Evidently, TME and ITH have emerged as pivotal factors leading to resistance and failure of systemic therapies. Genetic mutation features may help not only guide targeted therapy but also provide further understanding of the TME and ITH of iCCA.
PROMISING THERAPEUTIC TARGETS IN ICCA
In the past two decades, a multitude of agents targeting VEGF or EGFR in unselected advanced biliary tract cancer (BTC) have not shown meaningful activity. In iCCA, only limited data of anti-VEGF suggest more activity in phase 2 second-line setting with a small sample size. To date, FGFR2 fusion or rearrangement, IDH1/2 mutation, BRAF V600E mutation, RET fusion, NTRK fusion, and ERBB2 amplification or overexpression have been well studied as therapeutic targets for advanced iCCA [Table 2], although these aberrant genes are less frequent except FGFR and IDH.
Clinical trials evaluating targeted therapies against most frequent actionable alterations in intrahepatic cholangiocarcinoma
| Phase | Trial | NO. of patients | Tumour type | Treatment | Objective response rate (%) | Duration of response (months) | Median progression free survival (months) | Median overall survival (months) | |
| FGFR | |||||||||
| Abou-Alfa et al. (2020)[28] | 2 | FIGHT-202/NCT02924376 | 107 (105) | CCA (FGFR2 fus) (iCCA) | Pemigatinib | 35.5 (95%CI, 26.5-45.4) | 7.5 (95%CI, 5.7-14.5) | 6.9 (95%CI, 6.2-9.6) | 21.1 (95%CI, 14.8-NE) |
| Shi et al. (2023)[29] | 2 | NCT04256980 | 31 (30) | CCA (FGFR2 fus) (iCCA) | Pemigatinib | 50.0 (95%CI 31.3-68.7) | NR (95%CI, 3.4-NE) | 6.3 (95%CI, 4.9-NE) | NR (95%CI, NE) |
| Goyal et al. (2023)[7] | 2 | FOENIX-CCA2/NCT02052778 | 103 | iCCA (FGFR2 fus) | Futibatinib | 42 (95%CI, 32-52) | 9.7 (95%CI, 7.6-17.0) | 9.0 (95%CI, 6.9-13.1) | 21.7 (95%CI, 14.5-NE) |
| Borad et al. (2022)[32] | 2 | FIDES-01/NCT03230318 | 103 40 | iCCA (FGFR2 fus) iCCA (FGFR2 mut/amp) | Derazantinib | 21.4 (95%CI, 13.9-30.5) 6.5 (95%CI, 0.8-21.4) | NA NA | 8.0 (95%CI, 5.5-8.3)8.3 (95%CI, 1.9-16.7) | 17.2 (95%CI, 12.5-22.4) 15.9 (95%CI, 8.4-NE) |
| Javle et al. (2021)[31] | 2 | NCT02150967 | 108 | CCA (FGFR2 fus) | Infigratinib | 23.1 (95%CI, 15.6-32.2) | 5.0 (IQR, 3.7-9.3) | 7.3 (95%CI, 5.6-7.6) | 12.2 (95%CI, 10.7-14.9) |
| Mazzaferro et al. (2019)[99] | 1/2 | NCT01752920 | 29 | iCCA (FGFR2 fus) | Derazantinib | 20.7 | 4.6 (95%CI: 2.3-8.9) | 5.7 (95%CI, 4.04-9.2) | NA |
| Bahleda et al. (2019)[100] | 1 | NCT01703481 | 11 | CCA (FGFR mut/fus) | Erdafitinib | 27.3 | 11.4 | NA | NA |
| Kim et al. (2019)[101] | 1 | NCT02368951 | 20 (4) | Solid Tumors (FGFR2-positive) (CCA) | Aprutumab ixadotin | 0 | NA | NA | NA |
| Ahn et al. (2022)[102] | pilot | NCT02265341 | 12 (10) | BTCs (FGFR2 fus) (iCCA) | Ponatinib | 8.3 | NA | 2.4 (95%CI, 1.9-9.2) | 15.7 (95%CI, 6.1-NE) |
| IDH | |||||||||
| Abou-Alfa et al. (2020)[48] | 3 | ClarIDHy/NCT02989857 | 185 | iCCA (IDH1 mut) | Ivosidenib vs. Placebo | 2.4 (95%CI, 0.5- 6.9)* | NA | 2.7 (95%CI, 1.6-3.6) vs. 1.4 (1.4-2.5), HR 0.47 (95%CI 0.33-0.68) | 10.8 (95%CI, 7.7-17.6) vs. 6.0 (3.6-6.3), HR 0.46 (95%CI 0.28-0.75), RPFST adjusted |
| BRAF | |||||||||
| Subbiah et al. (2020)[65] | 2 | ROAR/NCT02034110 | 43 (39) | BTCs (BRAF V600E) (iCCA) | Dabrafenib plus Trametinib | 47 (95%CI 31-62)* | 9 (95%CI, 6-14) | 9 (95%CI, 5-10) | 14 (95%CI, 10-33) |
| HER2 | |||||||||
| Lee et al. (2023)[103] | 2 | KCSG-HB19-14/NCT04722133 | 34 (6) | BTCs (HER2-positive) (iCCA) | Trastuzumab plus FOLFOX | 29.4 (95%CI, 16.7-46.3) | 4.9 (2.1-10.4) | 5.1 (95%CI, 3.6-6.7) | 10.7 (95%CI, 7.9-NR) |
| Javle et al. (2021)[54] | 2a | MyPathway/NCT02091141 | 39 (7) | BTCs (HER2-positive) (iCCA) | Pertuzumab plus Trastuzumab | 23 (95%CI, 11-39); iCCA: 0 | 10.8 (95%CI, 0.7-25.4); iCCA: NA | 4.0 (95%CI, 1.8-5.7); iCCA: 2.6 (95%CI, 1.0-5.3) | 10.9 (95%CI, 5.2-15.6); iCCA: 3.9 (95%CI, 1.2-8.1) |
| Harding et al. (2023)[56] | 2 | NCT01953926 | 25 (6) | BTCs (HER2 mut) (iCCA) | Neratinib | 16 (95%CI, 4.5-36.1) | Four PR: 3.0; 3.7 (censored); 3.8; 4.7. | 2.8 (95%CI, 1.1-3.7) | 5.4 (95%CI, 3.7-11.7) |
| Ohba et al. (2022)[55] | 2 | JMA-IIA00423 | 32 (24;8) (3) | BTCs (HER2-positive; -low) (iCCA) | Trastuzumab deruxtecan | HER2-positive: 36.4 (95%CI, 19.6-56.1); HER2-low: 12.5 (95%CI, 0.3-52.7) | NA | HER2-positive: 4.4 (95%CI, 2.8-8.3); HER2-low: 4.2 (95%CI, 1.3-6.2) | HER2-positive: 7.1 (95%CI, 4.7-14.6); HER2-low: 8.9 (95%CI, 3.0-12.8) |
| Meric-Bernstam et al. (2022)[57] | 1 | NCT02892123 | 20 (5) | BTCs (HER2-positive) (iCCA) | Zanidatamab | 47 (95%CI, 23-72) | 6.6 (95%CI, 3.2-NE) | NA | NA |
| RET | |||||||||
| Subbiah et al. (2022)[69] | 1/2 | ARROW/NCT03037385 | 29 (3) | solid tumors (RET fus)(CCA) | Pralsetinib | 57 (95%CI, 35-77) | 11.7 (95%CI, 5.5-19.0) | 7.4 (95%CI, 5.1-13.6) | 23.5 (95%CI, 19.8-23.9) |
| Cocco et al. (2018)[71] | 1/2 | LIBRETTO-001/NCT03157128 | 45 (1) | solid tumors (RET fus)(CCA) | Selpercatinib | 43·9 (95%CI, 28.5-60.3); CCA: 100.0 (95%CI, 2.5-100.0)* | 24.5 (95%CI, 9.2-NR); CCA: 5.6 (95%CI, NR-NR)* | 13.2 (95%CI, 7.4-26.2)* | 18.0 (95%CI, 10.0-NE)* |
| NTRK | |||||||||
| Hong et al. (2020)[75] | 1/2 | NCT02122913; NCT02637687; NCT02576431 | 159 (2) | solid tumors (TRK fus) (CCA) | Larotrectinib | 79 (95%CI, 72-85); CCA: 50 (95%CI, 1-99)* | 35.2 (95%CI, 22.8-NE); CCA: 7.5 (95%CI, NE-NE)* | 28·3 (95%CI 22-1-NE) | 44·4 (95%CI 36·5-NE) |
| Demetri et al. (2022)[76] | 2 | ALKA-372-001/ EudraCT 2012-000148-88; STARTRK-1/ NCT02097810; STARTRK-2/ NCT02568267 | 150 | solid tumors (NTRK fus) | Entrectinib | 61.3 (95%CI, 53.1-69.2) | 20.0 (95%CI, 13.2-31.1) | 13.8 (95%CI, 10.1-20.0) | 37.1 (95%CI, 27.2-NE) |
| MSI-H/dMMR | |||||||||
| Andre et al. (2021)[83] | 1 | GARNET/NCT02715284 | 106 (1) | solid tumors (dMMR) (CCA) | Dostarlimab | 38.7 (95%CI, 29.4-48.6); CCA: CR 100 | NR | NA | NA |
| Maio et al. (2022)[82] | 2 | KEYNOTE-158/ NCT02628067 | 351 (22) | solid tumors (dMMR) (CCA) | Pembrolizumab | 30.8 (95%CI, 25.8-36.2); CCA: 40.9 (95%CI, 20.7-63.6)* | 47.5 (95%CI, 2.1+-51.1+); CCA: 30.6 (95%CI, 6.2-40.5+)* | NA CCA: 4.2 (95%CI, 2.1-24.9)* | NA-CCA: 19.4 (95%CI, 6.5-NR)* |
FGFR alterations
FGFR1-5 belong to the transmembrane receptor tyrosine kinase (RTK) family and are high-affinity receptors for fibroblast growth factors (FGFs). The binding of FGF to inactive monomeric FGFRs triggers a conformational change in FGFRs, leading to the dimerization and activation of cytoplasmic tyrosine kinases by phosphorylating tyrosine residues within the cytoplasmic tails of FGFRs, which initiates a cascade of intracellular phosphorylation, signal transduction, and gene transcription[25]. FGFR1-4 are composed of extracellular regions, hydrophobic transmembrane regions, and intracellular tyrosine kinase domains. Their signaling axis relates to various physiological processes, including cell proliferation, differentiation, tissue repair, and angiogenesis[26]. The FGFR-activated signal transduction pathways, including JAK-STAT, RAS-BRAF-MEK-ERK, and PI3K-AKT-mTOR, are involved in cell proliferation, differentiation, and survival. The FGF/FGFR pathway is abnormally regulated through three mechanisms: (a) protein overexpression resulting from FGFR gene amplification; (b) activating mutations that typically enhance ligand affinity, receptor dimerization and activation (without ligand), or constitutive kinase domain activation; (c) gene fusion caused by chromosomal translocation.
Common FGFR mutations include gene amplification, activating mutations, gene fusions, and rearrangements, kinase domain duplications and autocrine activation. An NGS study of 4,853 cases across various solid tumors showed that FGFR dysfunction was present in 7.1% of tumor samples. FGFR1-4 amplification accounted for 3.5%, 1.5%, 2.0%, and 0.5% of the patients, respectively[27]. FGFR2 rearrangements or fusions have been reported in iCCA with an incidence of 8%-13%[8-13]. A retrospective sequencing analysis of 6130 cases of iCCA showed that the incidence of FGFR2 aberrations was 11.2%, including rearrangements (9.4%), short variants (2.2%), and copy number variations (0.46%). The most common fusion partners were BiCCA1, FGFR2-N/A, SORBS1, AHCYL1, and CCDC6[8].
The emergence of oncogenic FGFR2 fusion protein presents a promising opportunity for the development of targeted therapy. Pemigatinib, futibatinib, infigratinib, derazantinib, gunagratinib, aprutumab ixadotin, and ponatinib have emerged as promising candidates in phase 1 or 2 clinical trials for cholangiocarcinoma harboring FGFR2 fusion/rearrangement. Phase 3 trials evaluating the efficacy of pemigatinib (NCT03656536) and futibatinib (NCT04093362) are poised to challenge GEMCIS in the first-line treatment of cholangiocarcinoma [Supplementary Table 1].
Pemigatinib
Pemigatinib, a small molecule reversible inhibitor of FGFR1-3, has gained approval as the first targeted therapy for patients with cholangiocarcinoma carrying FGFR2 fusions or rearrangements in subsequent-line settings due to the studies of FIGHT-202 and CIBI375A201[6]. FIGHT-202, a phase 2, open-label and single-arm study, enrolled 146 patients with metastatic or advanced cholangiocarcinoma who experienced progression following first-line or subsequent treatments[28] and were relocated into three cohorts: cohort A, FGFR2 fusion or rearrangement; cohort B, other mutations on FGFR; and cohort C, no mutations on FGFR. In cohort A (n = 107), the objective response rate (ORR) and median duration of response (mDOR) achieved 35.5% and 7.5 months, respectively, with median progression-free survival (mPFS) of 6.9 months (95%CI 6.2-9.6) and mOS of 21.1 months (95%CI 14.8- not estimable). Hyperphosphatemia constituted the most frequent adverse events (AEs) (all-grade 60% and ≥ grade 3 12%)[28]. The outcomes of cohort A were found to be better than those of cohort B with other mutations or cohort C with no mutations. The ORR, mPFS, and mOS were 0, 4.0 months (95%CI 2.3-6.5), and 6.7 months (95%CI 2.1-10.6) in cohort B, respectively; in cohort C, they were 0, 1.7 months (95%CI 1.3-1.8) and 2.1 months (95%CI 1.2-4.9), respectively. In the bridging study (CIBI375A201) conducted in China, comparable results were observed among 30 evaluable subjects harboring FGFR2 fusion/rearrangement with ORR of 50% and mPFS of 6.3 months[29].
Futibatinib (TAS-120)
Futibatinib (TAS-120), an irreversible inhibitor of FGFR1-4 with a unique capability of forming covalent bonding in the ATP pocket of the tyrosine kinase domain, has been recommended as a second-line treatment for FGFR2-rearranged iCCA in the NCCN Guidelines (2022)[30]. FOENIX-CCA2, a phase 2 single-arm study, included 103 patients with unresectable or metastatic iCCA of FGFR2 fusions/rearrangements after previous therapy (excluding FGFR inhibitors). The ORR achieved 41.7%, with mDOR of 9.7 months (95%CI, 7.6-17.0), mPFS of 9.0 months (95%CI 6.9-13.1), and mOS of 21.7 months (95%CI 14.5- not estimable). Hyperphosphatemia remained the most common AE (all-grade 85% and ≥ grade 3 30%)[7].
Infigratinib (BGJ398)
Infigratinib, a selective ATP-competitive inhibitor targeting FGFR1-3, was assessed in a phase 2 single-arm trial for advanced iCCA with FGFR fusions/rearrangements after first-line gemcitabine-based treatment[31]. In cohort one (n = 108), the ORR, mDOR, mPFS, and mOS were 23.1%, 5.0 months, 7.3 months (95%CI 5.6-7.6), and 12.2 months (95%CI 10.7-14.9), respectively. The most frequent treatment-related adverse events (TRAEs) of any grade were hyperphosphatemia (76.9%). Unfortunately, the phase 3 trial of infigratinib (NCT03773302) was closed due to the withdrawal of New Drug Application.
Derazantinib (ARQ 087)
Derazantinib, an ATP-competitive inhibitor of FGFR1-3, was evaluated in a phase 2 single-arm trial (FIDES-01) for advanced iCCA with FGFR2 fusions (FGFR2F) or mutations/amplifications (FGFR2MA) following one or more lines of chemotherapy[32]. FIDES-01 study included 143 patients (103 with FGFR2F and 40 with FGFR2MA). In FGFR2F cohort, the ORR was 21.4%, with mPFS of 8.0 months (95%CI 5.5 - 8.3) and mOS of 17.2 months (95%CI 12.5-22.4). By contrast, in FGFR2MA cohort, ORR was only 6.5%, with mPFS of 8.3 months (95%CI 1.9-16.7) and mOS of 15.9 months (95%CI 8 .4- not estimable). The most frequent AEs (any grade/ ≥ grade-3) were hyperphosphatemia (37%/3%). Derazantinib demonstrated significant clinical benefits for iCCA with FGFR2 alterations, including fusions, mutations, and amplifications.
IDH1/2 mutations
In the citric acid cycle, IDH is one of the crucial rate-limiting enzymes, which catalyzes the oxidative decarboxylation of isocitrate to generate alpha-ketoglutarate (α-KG), simultaneously producing carbon dioxide and NADPH/NADH. The IDH family can be divided into two categories according to their catalytic characteristics. One is NAD+-dependent IDH3, which includes α, β, and γ subtypes. The other comprises NADP+-dependent IDH1 and IDH2, which category generates NADPH in peroxisomes and cytoplasm (IDH1) or in mitochondria (IDH2). IDH3 mutations are infrequent in iCCA, but IDH1 and IDH2 mutations can lead to a conversion of α-KG to 2-hydroxyglutarate (2-HG)[33]. Excessive accumulation of this metabolite can competitively inhibit the activity of α-KG-dependent dioxygenases[34]. Additionally, IDH1/2 mutations not only decrease the ability of NADP+ to be reduced to NADPH, but also promote the reverse conversion of NADPH to NADP+[35]. Excessive accumulation of 2-HG and impaired NADPH production jointly result in epigenetic changes, DNA repair damage, and aberrant cell metabolism, thereby promoting tumorigenesis[36].
The oncogenic transformation of IDH1/2 is due to arginine 132 (R132) in IDH1, and either R140 or R172 in IDH2. Notably, mutations in IDH1 and IDH2 are mutually exclusive events in iCCA. The oncogenic metabolite 2-HG, produced as a result of IDH1 R132 mutation, directly binds to DNMT1 and promotes its association with the promoter region of RIP3. This leads to an increase in DNA methylation levels on the RIP3 promoter and a decrease in RIP3 expression, ultimately hindering cellular necrosis[37]. IDH1 mutations induce metabolic reprogramming and iCCA tumorigenesis through PFKP-induced aerobic glycolysis and AMPK activation[38], and inhibit interferon-TET2 signaling to promote immune evasion and tumor maintenance[39]. IDH2 mutations increase the dependence of cells on mitochondrial oxidative phosphorylation and reduce glycolysis under hypoxia[40]. Xiang et al. characterized the intratumor heterogeneity (ITH) landscape of iCCA patients by utilizing exome sequencing and bulk RNA sequencing techniques[23]. The subgroup of IDH mutation (IDH-SG) is less associated with TME, and presents reduced T-cell infiltration and decreased T-cell toxicity. This result was also confirmed in single-cell sequencing. In all types of BTC, IDH1/2 mutations only occur in iCCA, and IDH1 mutations are more common, with an incidence of 5%-24%. The prognostic impact of IDH mutations in patients with BTC remains unclear. Some reports showed that IDH mutations are associated with better prognosis in iCCA patients, with longer mOS, smaller tumor size, and less tumor recurrence [41-44]. However, others suggested that IDH mutations have no prognostic impact on iCCA. Goyal et al. analyzed 104 iCCA patients (25.0% IDH1 mutation, 3.8% IDH2 mutation) and found no significant difference in mOS between IDH-mutant and IDH-wild type patients (15.0 vs. 20.1 months, P = 0.17)[45]. In lymph node-negative patients, IDH mutations did not impact prognosis[46]. In addition, a propensity score matching analysis showed that IDH1/2 mutations did not exhibit higher homologous recombination defects and were not associated with increased sensitivity to platinum-based chemotherapy[47].
Ivosidenib
To date, IDH1 inhibition has shown promising application prospects in iCCA patients with IDH1 mutations. Based on the findings of the ClarIDHy trial, ivosidenib was granted FDA approval as a second-line therapy for cholangiocarcinoma in 2021. ClarIDHy, a phase 3 randomized (2:1) trial, enrolled 185 previously treated cholangiocarcinoma patients carrying IDH1 mutations, and assessed the safety and efficacy of ivosidenib in such patients (124 receiving ivosidenib vs. 61 receiving placebo)[48]. The ORR of ivosidenib arm was 2.4%. However, compared to the placebo arm, the ivosidenib arm had significantly longer mPFS [2.7 vs. 1.4 months; HR 0.47 (95%CI 0.33- 0.68)]. After adjustment of the crossover of 70% placebo patients to ivosidenib, mOS was also better with ivosidenib [10.8 vs. 6.0 months; HR:0.46 (95%CI 0.28-0.75)]. The most common TRAEs of ivosidenib were nausea (38%), diarrhea (32%), as well as fatigue (28%).
Other IDH inhibitors
Apart from ivosidenib, other inhibitors targeting IDH1 (AB-218), IDH2 (Enasidenib), as well as dual IDH1/2 inhibitors (HMPL-306; LY3410738) are currently under investigation. PARP inhibitors (Olaparib; AZD6738) and PARP inhibitors combined with immune checkpoint inhibitors (Olaparib plus Durvalumab) are being evaluated for IDH1-mutant iCCA with impaired homologous recombination repair through an alternative approach of synthetic lethality [Supplementary Table 1].
EGFR pathway and HER2 aberrations
The epidermal growth factor receptor (EGFR) is a member of the ERBB family (including ERBB1/EGFR/HER1, ERBB2/HER2/Neu, ERBB3/HER3, and ERBB4/HER4) of receptor tyrosine kinases. The activation of downstream signaling pathways, including RAS/RAF/MEK/ERK, PI3K/AKT/mTOR, PLCγ/PKC, and STAT, is initiated by the binding of epidermal growth factor (EGF), transforming growth factor α (TGF-α), or nerve growth factor (NGF), and results in persistent pathway activation that promotes tumor cell proliferation, invasion and metastasis[49,50]. Targeted therapy against the ERBB family is currently considered a pivotal approach in tumor treatment, commonly using two categorical drugs: monoclonal antibodies and small molecule tyrosine kinase inhibitors (TKI)[51]. Monoclonal antibodies primarily impede signal transduction by obstructing ligand binding or interfering with receptor dimerization. For instance, cetuximab and panitumumab specifically target EGFR, while trastuzumab and pertuzumab effectively target HER2. On the other hand, small molecule TKIs competitively bind to the receptor kinase domain to inhibit signal transduction. Notable examples include selectively targeting EGFR with gefitinib, erlotinib, or osimertinib, and targeting HER2 with lapatinib, neratinib, or tucatinib. Although EGFR/ERBB1 mutations and amplifications have been reported in CCA, none of the Phase 2/3 trials targeting EGFR in CCA have shown substantial improvement in survival over the past few decades. HER2 gene (also known as Neu/ERBB2) encodes product HER2 protein, which enhances invasion, motility, and proliferation of cholangiocarcinoma cells and exerts carcinogenic effects by activating the ERBB receptor family[52]. ERBB2 gene amplification is observed in approximately 5%-15% of BTC and about 1.6%-4% of iCCA[8,9,11]. ERBB2 mutation accounts for around 1.6% of iCCA[8]. Interestingly, targeted treatment against HER2 amplification, together with overexpression in BTC, has made more progress.
Trastuzumab plus pertuzumab
Trastuzumab, the monoclonal antibody targeting the subdomain IV of the extracellular domain of HER2 receptor, inhibits downstream PI3K/AKT and Ras/MEK signaling pathways to exert anti-tumor effects[53]. Pertuzumab is a HER-2 inhibitor targeting the subdomain II of the extracellular domain. MyPathway study, a phase 2a multi-basket trial of trastuzumab combined with pertuzumab, enrolled 39 advanced BTC patients (18% with iCCA) who had HER2-amplified, overexpressed or both[54]. In BTC cohort, the ORR was 23% and mDOR was 10.8 months, with mPFS of 4.0 months (95%CI 1.8-5.7) and mOS of 10.9 months (95%CI 5.2-15.6). This regimen is thus recommended as subsequent therapy for HER2-positive BTC in NCCN guidelines (2022). However, in iCCA subset (n = 7), the ORR was 0% and mDOR was not applicable, with mPFS of 2.6 months (95%CI 1.0-5.3) and mOS of 3.9 months (95%CI 1.2-8.1). Grade 3-4 AEs were reported in 46% of BTC patients, and the most frequent AEs were elevated alanine aminotransferase and aspartate aminotransferase (each 13%).
Other ERBB2 alterations inhibitors
Trastuzumab deruxtecan (DS-8201/T-DXd), a new drug of antibody-drug conjugates consisting of a HER2-monoclonal antibody, a chemical linker, and a topoisomerase I inhibitor, was investigated in a phase 2 single-arm study (HERB trail) among patients with HER2-expressing BTC[55]. Among 22 HER2-positive BTC patients (3 with iCCA) identified for analysis, the ORR was 36.4% (2 CR and 6 PR), with mPFS of 4.4 months (95%CI 2.8-8.3) and mOS of 7.1 months (95%CI 4.7-14.6).
Neratinib, an irreversible inhibitor targeting pan-HER tyrosine kinase, exhibits efficacy against HER2-mutant tumors. SUMMIT trial, its phase 2, single-arm, basket study in solid tumors carrying HER2 mutations, enrolled 25 BTC patients (11 cholangiocarcinoma including 6 iCCA, 10 gallbladder and 4 ampullary cancer)[56]. In BTC cohort, the ORR was 16%, and DORs for the 4 PR patients were 3.0, 3.6 (censored), 3.7, and 4.7 months, respectively, with mPFS of 2.8 months (95%CI 1.1-3.7) and mOS of 5.4 months (95%CI 3.7-11.7).
Zanidatamab (ZW25), a novel bispecific antibody, targets extracellular subdomains IV and II, which are targeted by trastuzumab and pertuzumab, respectively. HERIZON-BTC-01, its phase 2 single-arm trial in BTC patients with HER2-amplification after prior gemcitabine-base treatment, enrolled 80 patients with HER2 immunohistochemistry (IHC) 2+ or 3+[57]. In these patients (23 with iCCA), the ORR was 41.3% (30.4% in iCCA) and mDOR was 12.9 months (95%CI 6.0- not estimable), with mPFS of 5.5 months (95%CI 3.7-7.2) and immature mOS. The grade 3 TRAE rate was 18%, with the most frequent TRAEs of diarrhea (5%) and decreased ejection fraction (3%). No grade 4/5 TRAEs were observed.
BRAF and KRAS mutations
The BRAF and KRAS genes play crucial roles in signaling pathways that regulate cell growth and differentiation. BRAF, also known as proto-oncogene B-Raf and v-Raf murine sarcoma viral oncogene homolog B, encodes serine/threonine-protein kinase B-Raf[58]. B-Raf belongs to the Raf kinase family of proteins involved in growth signal transduction, and functions to regulate the MAP kinase/ERKs pathway, which impacts cell division, differentiation, and secretion. B-Raf mutation was discovered in certain human tumors[59]. About 5% of iCCAs harbor BRAF mutations, with the most prevalent type of BRAF V600E. iCCA patients carrying BRAF V600E exhibit higher cancer stage, greater lymph node involvement, and poorer overall survival, compared to those without such mutation[60]. Recently, combined inhibition along the RAS-RAF-MEK-ERK pathway targeting BRAF V600E has demonstrated significant clinical activities.
KRAS proto-oncogene, a member of the RAS superfamily that also includes H-RAS and N-RAS, is frequently mutated in human cancers. The KRAS-encoded protein is a small GTPase that plays a crucial role in regulating multiple signaling pathways, including MAPK, PI3K, and Ral-GEFs, thereby impacting cellular growth, differentiation, survival, and migration[61]. KRAS mutations occur in 6% to 20% of iCCA patients[8,9,12,13], predominantly at codon 12 or 13 on exon 2. The most prevalent mutations are G12D and G12E[62], which result in the impaired ability of KRAS protein to hydrolyze GTP to GDP after binding to GTP, thereby leading to a persistent activation. Consequently, this sustained activation excessively stimulates downstream signaling pathways and facilitates the initiation and progression of iCCA. Currently, there are two main treatment strategies for KRAS mutations: one involves direct inhibition of KRAS protein or its GTP binding capacity, exemplified by sotorasib (AMG 510) and adagrasib (MRTX849) targeting G12C in non-small cell lung cancer (NSCLC); the other entails indirect inhibition of downstream signaling pathways or upstream regulatory factors associated with KRAS, such as MEK inhibitors and SOS1 inhibitors[62]. For iCCA, further research is currently needed[63,64].
Darafenib plus trametinib
Dual inhibition of BRAF and MEK (dabrafenib plus trametinib) was evaluated and approved as the first tissue-agnostic treatment for patients with solid tumors harboring BRAF V600E mutation, on the basis of phase 2 Rare Oncology Agnostic Research (ROAR)[65] and NCI-MATCH[66] studies. Concerning iCCA, ROAR study, a single-arm and basket trial, enrolled 43 BTC patients (iCCA accounted for 91%) after previous systemic treatment[65]. In BTC cohort, the independent review ORR reached 47%, and mDOR was 9 months. Among responded BTC patients, 59% of them had a DOR > 6 months, and 7 patients had a DOR > 12 months. The mPFS was 9 months (95%CI 5-10), and mOS achieved 14 months (95%CI 10-33). This cohort experienced serious AEs (40%) and TRAEs (21%), with pyrexia being the most frequent AE (19%). No deaths of TRAE were reported.
In EAY131-H study (the subprotocol H of NCI-MATCH trial), this dual inhibition was evaluated in patients with BRAF V600-mutant refractory solid tumors (including 4 iCCA patients), lymphoma or multiple myeloma[66]. In 29 patients for analysis, the ORR was 37.9%, with a mDOR of 25.1 months. The mPFS was 11.4 months (95%CI 8.4-16.3), while mOS achieved 28.6 months. Three of 4 patients with iCCA exhibited a PR, with individual PFS of 12.8, 9.1, and 29.4 months, respectively. The most frequent AEs were fatigue (74%), nausea (57%), fever (51%) and chills (54%). In 2021, this regimen was recommended as a subsequent-line therapy for BTCs with BRAF V600E mutation in NCCN guidelines.
Other RAS/RAF/MEK/ERK pathway inhibitors
VE-BASKET, a phase 2, single-arm, basket trial of vemurafenib in non-melanoma cancers with BRAF V600 mutations, enrolled 9 patients with cholangiocarcinoma (8 with BRAF V600E; 1 with V600X)[67]. Two of the 9 patients achieved PR with a DOR of 3.6 and 22.1 months, respectively. The mPFS was 3.0 months, while the mOS was 11.2 months.
Binimetinib, a selective MEK1/2 inhibitor, was assessed in combination with capecitabine in a phase 1b trial. A total of 34 patients (26 detected for mutations using NGS) with advanced BTC were enrolled after prior gemcitabine-base treatment[68]. The ORR, mPFS, and mOS were 20.6%, 4.1 months (95%CI 2.8-5.7), and 7.8 months (95%CI 5.9-12.2), respectively. The patients with mutant-type (n = 10) on RAS/RAF/MEK/ERK pathway (7 on KRAS; 2 on MAP2K1; 1 on NRAS) exhibited superior outcomes than those with wild-type (n = 16), in terms of ORR (40.0% vs. 12.5%, P = 0.028), mPFS (5.4 vs. 3.5 months, P = 0.010), and mOS (10.8 vs. 5.9 months, P = 0.160).
RET fusions
The RET gene is an oncogene that encodes the tyrosine kinase receptor superfamily RET protein. The tumorigenic activation of RET can take place through two mechanisms: one involves chromosomal rearrangements, by which RET kinase domain is fused with a partner protein containing a dimerization domain; the other involves RET mutations, which results in direct or indirect kinase activation. The fusion of the RET gene can cause ligand-independent dimerization and constitutive activation of RET kinase, thereby inducing cell overproliferation and tumor formation by activating downstream pathways such as RAS-MARK, PI3K-AKT, JAK-STAT, PLCγ, etc. RET fusion genes are widely distributed across various types of cancer. Despite the low incidence in iCCA population, many individuals carrying RET fusions may still benefit from targeted therapies. Although previous multi-kinase inhibitors like cabozantinib and vandetanib have shown moderate activity against RET-driven malignancies, recent selective inhibitors such as selpercatinib and pralsetinib have demonstrated significant efficacy against both mutations and fusions.
Pralsetinib
Pralsetinib, a selective inhibitor targeting RET receptor tyrosine kinase, was investigated in phase 1/2 trial ARROW in patients with advanced solid tumors harboring RET fusion[69]. Twenty-nine patients with 12 types of tumors, excluding NSCLC and thyroid cancer, who had previously received or were not eligible for standard therapies, were enrolled. In 23 evaluable patients (3 with cholangiocarcinoma), the ORR achieved 57% and mDOR was 11.7 months (95%CI 5.5-19.0). Notably, PR was achieved in two cholangiocarcinoma patients, including one iCCA patient, who was enrolled with the best response of PD on three lines of previous therapy and received pralsetinib for more than 20 months. The mPFS was 7.4 months (95%CI 5.1-13.6), with mOS of 13.6 months (95%CI 7.5- not reached). The most frequent grade ≥ 3 TRAEs were neutropenia (31%), anemia (14%), and increased aspartate aminotransferase (AST) (10%).
Selpercatinib
Selpercatinib, a highly selective inhibitor of the RET kinase with CNS activity, was assessed in phase 1/2 trial IBRETTO-001 for patients with advanced solid tumors harboring RET fusion[70]. A total of 45 patients with 14 kinds of tumors, excluding lung and thyroid cancer, who had experienced progression on prior systemic therapies or lacked satisfactory treatment options, were enrolled in the study. Among the 41 evaluable patients (one with cholangiocarcinoma), the ORR was 43.9%, with mDOR of 24.5 months (95%CI, 9.2 to not evaluable). The patient with cholangiocarcinoma achieved PR with a DOR of 5.6 months. The mPFS was 13.2 months (95%CI, 7.4 to 26.2), with an mOS of 18.0 months (95%CI,10.7 to not evaluable). The most prevalent grade ≥ 3 TRAEs were elevated alanine aminotransferase (ALT) (16%), elevated AST (11%), and hypertension (13%). In 2022, selpercatinib was approved as the first RET inhibitor for adults with advanced or metastatic solid tumors harboring RET fusions, irrespective of tumor type.
Although the NCCN guideline recommends both the inhibitors in primary treatment and subsequent-line therapy for BTCs with RET fusions, further studies are needed to determine their anti-cancer effects in iCCA due to the very limited sample size.
NTRK fusions
The NTRK gene family comprises NTRK1, NTRK2, and NTRK3, which encode the receptors of the tropomyosin receptor kinase (TRK) family as TRKA, TRKB, and TRKC, respectively. Changes in the TRK signaling pathway primarily involve gene fusion, protein overexpression, single nucleotide variations, and splicing alterations. Among these changes, NTRK gene fusion is the most definitive oncogenic driver[71]. Current research has revealed that NTRK gene fusion is mutually exclusive with other oncogenic drivers, such as EGFR, ALK, ROS1, KRAS, etc. However, variations in passenger genes related to NTRK intracellular pathways may occur concurrently[72]. NTRK gene fusion has been identified in numerous tumor types, albeit its incidence is below 5% in certain solid tumors such as lung, colorectal, and cholangiocarcinoma[71]. With the emergence of NTRK gene fusion, receptor conformational activation ensues, leading to subsequent activation of downstream signal pathways such as MAPK, PI3K, and PKC, thereby promoting tumorigenesis[73]. NTRK gene fusion has emerged as a pivotal and prevalent molecular marker for pan-cancer targeted therapy. NTRK inhibitors have exhibited remarkable response rates and durable responses in advanced solid tumors with NTRK fusions, including cholangiocarcinoma.
Larotrectinib
Larotrectinib, a highly selective TRK inhibitor, was granted FDA approval in 2018 for solid tumors carrying NTRK gene fusions, for the second time based on a common biomarker rather than the location[74]. In an updated pooled analysis of three phase 1/2 trials (NAVIGATE, SCOUT, and LOXO-TRK-14001), 159 patients with NTRK fusion-positive cancers (including 2 cholangiocarcinomas) were enrolled and treated with larotrectinib[75]. Among the 153 evaluable patients (including 2 cholangiocarcinomas), the ORR was 79.1% (CR 15.7%; PR 63.4%), with an mDOR of 35.2 months (95%CI 22.8- not estimable). One responded patient with cholangiocarcinoma had a DOR of 7.3 months. The mPFS achieved 28.3 months (95%CI 22.1- not estimable), with mOS of 44.4 months (95%CI 36.5- not estimable). The most frequent grade 3/4 TRAEs were elevated ALT in 3%, anemia in 2%, and neutropenia in 2%.
Entrectinib
Entrectinib is an effective inhibitor of TRK with CNS activity. In an updated integrated analysis of three phase 1/2 trials (ALKA-372-001, STARTRK-1, and STARTRK-2), 121 patients with solid tumors (1 with cholangiocarcinoma) harboring NTRK fusions treated with entrectinib were included for efficacy evaluation. The ORR was 61.2% (CR 15.7%; PR 44.5%), and the mDOR was 20.0 months (95%CI 13.0-38.2). The one patient with cholangiocarcinoma achieved a PR, with DOR of 9.3 months, PFS of 12.0 months, and OS of 23.4 months[76]. The mPFS was 13.8 months (95%CI 10.1-19.9), with mOS of 33.8 months (95%CI 23.4-46.4). The majority of TRAEs were grade 1/2 and manageable/reversible with dose modifications.
In 2020, NCCN guidelines recommended both larotrectinib and entrectinib as a front or subsequent-line treatment for patients with BTCs carrying NTRK fusions.
Targeted immunotherapy
In 2017, the PD-1 inhibitor pembrolizumab was granted FDA approval as the first cancer therapy for any solid tumor harboring a specific genetic feature, MSI-H or mismatch repair deficiency (dMMR), marking the dawn of the era of tumor immunotherapy[77]. The mechanism of mismatch repair is highly conserved, and regulated by four genes: MLH1, MSH2, MSH6, and PMS2. Biallelic inactivation of any one of these genes caused by mutations or epigenetic silencing results in dMMR and increased mutations[78]. The accumulated mutations, detectable through MSI testing, can further alter the status of tumor mutation burden (TMB) by increasing the neoantigen rate, which is associated with the tumor microenvironment and increased anti-cancer T cells in the periphery[79]. The frequencies of MSI-H and TMB-H in iCCA were approximately 1.2% and 3.7%, respectively[8]. Accumulating evidence suggests that TMB-H (≥ 10 mut/Mb) is more likely to benefit from PD-1 blockade immunotherapy, as is MSI-H[80-82].
MSI-H/dMMR
The activity of immune monotherapy utilizing monoclonal antibody of PD-1 has been evaluated in solid tumors of MSI-H/dMMR in substantial trials. KEYNOTE-158, a phase 2 basket trial, was designed to investigate predictive biomarkers for immunotherapy using pembrolizumab in patients with advanced solid tumors after previous treatment[82]. In this trial, 321 patients with advanced non-colorectal cancer (22 with cholangiocarcinoma) of MSI-H/dMMR were enrolled and evaluated for efficacy. The ORR was 30.8% (CR 8.4%; PR 22.4%), with mDOR of 47.5 months (95%CI 2.1+-51.1+). Furthermore, the percentages of patients who achieved DOR ≥ 1 year, ≥ 2 years, and ≥ 3 years were 88.0%, 74.1%, and 70.1%, respectively. In cholangiocarcinoma cohort, the ORR was 40.9% (CR 13.6%; PR 23.7%), and mDOR was 30.6 months (95%CI, 6.2 to 40.5+), with mPFS of 4.2 months (95%CI 2.1-24.9) and mOS of 19.4 months (95%CI 6.5- not reached). Thereby, the NCCN guideline of BTC also recommends the primary treatment of pembrolizumab for patients with MSI-H/dMMR, but with limited trial data in the upfront setting. Additionally, the ongoing phase 1 GARNET is currently assessing dostarlimab in patients with advanced solid tumors. In a presentation of Cohort F, 106 evaluable patients with dMMR were enrolled, of which 99 had GI tumors (including 2 with BTC)[83]. The ORR was 38.7% (CR 7.5%; PR 31.2%), and mDOR was not reached. The patient, either with biliary neoplasm or with gallbladder cancer, was confirmed to achieve CR. TRAEs were reported in 68.8% of all patients, and 8.3% experienced grade ≥ 3 TRAEs.
TMB-H
Nivolumab combined with ipilimumab or nivolumab alone was assessed in CheckMate 848, a phase 2 randomized trial in patients with metastatic or advanced solid tumors of high tTMB or bTMB who were immunotherapy-naïve and refractory to standard local therapies. In an oral presentation[84], the combination arm exhibited improved ORR and survival in patients of tTMB-H. In tTMB-H cohort (n = 68) of dual checkpoint inhibition, the ORR was 35.3%, with mPFS of 4.1 months (95%CI 2.8-11.3) and mOS of 14.5 months (95%CI 7.7- not evaluable).
The PD-L1 status was investigated for advanced BTC in a phase 2 trial of nivolumab, which enrolled 54 patients[85]. The ORR in all patients was 22% by investigator assessment, but was merely 11% by independent central review. The mPFS was 3.7 months (95%CI 2.3-5.7) with mOS of 14.2 months (95%CI 6.0 - not reached). In the subgroup (n = 18) of PD-L1 positive (≥ 1% of tumor cells), the investigator-assessed and centrally assessed ORRs were 50% and 28%, respectively, with mPFS of 10.4 months and mOS not reached.
Of late, for unselected BTC patients, non-targeted immunotherapy of immuno-checkpoint inhibition combined with chemotherapy has demonstrated improved survival results and represented a new standard of care as first-line treatment, which may imply the increasing role of immunotherapy combination for unselected patients. TOPAZ1, a phase 3 randomized trial of GEMCIS plus durvalumab (PD-L1 inhibitor) compared with GEMCIS alone in patients with BTC, has yielded positive results of mOS and mPFS in not only the full analysis set but also iCCA subgroup: Arm durvalumab n = 341, ORR 26.7%, mDOR 6.4 months, mPFS 7.2 months with HR 0.75 [95%CI 0.63-0.890] and P value of 0.001, and mOS 12.8 months with HR 0.80 [95%CI 0.66-0.97] and P value of 0.021); Arm placebo (n = 344, ORR 18.7%, mDOR 6.2 months, mPFS 5.7 months, and mOS 11.3 months)[86].
More recently, KEYNOTE966, a phase 3 randomized trial of GEMCIS plus pembrolizumab compared with GEMCIS enrolling a greater proportion of non-Asian participants with BTC, has published improved results in intention-to-treat population and iCCA subset: Arm pembrolizumab (n = 533, ORR 29%, mDOR 9.7 months, mPFS 6.5 months with HR 0.86 [95%CI 0.75-1.00] and P value of 0.023, and mOS 12.7 months with HR 0.83 [95%CI 0.72-0.95] and P value of 0.003; Arm placebo (n = 536, ORR 28%, mDOR 6.9 months, mPFS 5.6 months, and mOS 10.9 months)[87].
MOLECULAR TARGET DETECTION AND LIQUID BIOPSY
Multiple molecular target detection modalities, which involve DNA, RNA and protein detection through tumor tissue or body fluid, are required to identify the status of actionable genomic alterations to determine whether the patients with iCCA can respond. These modalities include NGS, polymerase chain reaction (PCR), fluorescence in situ hybridization (FISH), and IHC in daily clinical practice. NGS allows the sequencing of a huge number of nucleotides in a single analysis and plays a significant role in cancer-targeted therapy, for precise candidate selection, treatment monitoring, and resistant mutation identification. An ideal NGS panel test in clinical laboratories should be limited to a certain tumor type, iCCA/BTC, based on the tumor-specific somatic and/or germline aberrations and the available targeted agents. For all patients with unresectable or metastatic CCA, NGS has been recommended by the NCCN guideline of BTC and ESMO Precision Medicine Working Group[88].
Generally, for NTRK fusion screening, RNA-based NGS panel testing or IHC is preferable[89]. RET fusion testing is recommended using a comprehensive NGS panel, which may identify coexisting aberrations. As to FGFR2 fusions, NGS gene panels and break-apart FISH can be used for the identification of fusions and preferentially on tumor tissue[90]. In terms of IDH1/2 mutations, NGS gene panels or PCR for hotspot mutations can be applied to detect common mutations, such as IDH1 R132X. For BRAF V600E, NGS or PCR can be performed to detect the mutation. Concerning HER2 alterations, HER2 amplification can be tested by IHC, FISH, or NGS approach, protein overexpression identified by IHC has significant heterogeneity, and HER2 activating mutations should be detected using the NGS approach. For dMMR/MSI, IHC staining is more frequently used to assess MLH1, MSH2, MSH6, and PMS2 proteins, and NGS analysis is more powerful to identify inactivating mutations. TMB-H can only be detected by validated NGS approaches, which may vary by platform.
Obtaining tumor components and genetic information through traditional tissue sampling is crucial for diagnosis, management, and targeted therapy selection. Liquid biopsy, which utilizes biomarkers such as circulating tumor DNA (ctDNA)/cell-free DNA (cfDNA), circulating tumor cells (CTC), or exosomes in blood or other bodily fluids, offers a non-invasive and dynamic solution for the detection and monitoring of tumors. This platform possesses the capability to accurately detect driver gene alterations in cholangiocarcinoma, encompassing FGFR2 fusion/rearrangement, IDH1/2 mutation, KRAS mutation, and more[91].
To date, the most comprehensive study on ctDNA involved 2068 samples from 1671 BTC patients[92]. Genetic alterations were detected in 84% of patients, with targetable alterations identified in 44%. The concordance of mutation detection between ctDNA and tissue DNA was high for IDH1 mutations (87%) and BRAF V600E (100%), but low for FGFR2 fusions (18%). Additionally, ctDNA analysis did reveal novel potential mechanisms of resistance to targeted therapies, including the mutation in cysteine residue (FGFR2 C492F) that interacts with covalent FGFR inhibitors. A high pre-treatment ctDNA variant allele fraction was associated with unfavorable prognosis and reduced response duration to both chemotherapy and targeted therapy.
However, liquid biopsy cannot serve as a substitute for tissue biopsy since the latter remains necessary for tumor histological evaluation, staging, and diagnosis. Particularly in early lesions or low-burden tumors with limited concentrations of ctDNA, crucial gene alterations may go undetected.
DILEMMA AND OUTLOOK
Targeted mutation-based therapy has shown higher response rates and more survival benefits in the treatment of BTC/iCCA compared to contemporaneous standard therapy in subsequent-line settings. FGFR inhibitors (pemigatinib and futibatinib), IDH inhibitors (ivosidenib), BRAF and MEK tyrosine kinase inhibitors (dabrafenib and trametinib), RET inhibitors (selpercatinib and pralsetinib), NTRK inhibitors (larotrectinib and entrectinib), and anti-HER2 monoclonal antibodies (trastuzumab plus pertuzumab) have been approved by the FDA or recommended in Guidelines for the treatment of iCCA patients. Although targeted therapies for CCA have achieved ORR usually more than 30% and mPFS commonly beyond 6 months, the use of targeted agents still faces challenges such as acquired drug resistance, precise patient selection, and serious adverse events.
Acquired resistance to FGFR inhibitors (infigratinib and Debio1347) has been observed in small sample studies. After disease progression, subsequent biopsy and ctDNA sequencing results showed the emergence of secondary mutations within the FGFR2 kinase domain[93]. In preclinical models of iCCA, various FGFR inhibitors exhibited different sensitivities to FGFR2 kinase domain mutations. Planned adjustment of the use order of different FGFR inhibitors may play an important role in overcoming resistance to themselves[94]. There are two potential solutions to acquired resistance: one is to develop covalent inhibitors, anti-FGFR monoclonal antibodies, and FGF ligand traps; the other is therapy combination or the development of biparatopic compounds or antibodies.
Resistance to targeted mutation-based therapy has also been reported in various other malignancies. For instance, resistance to IDH inhibitors is associated with the accumulation of 2-HG and has been observed in acute myeloid leukemia and glioma[95]. One study suggests that certain mutations occur at the interface of IDH heterodimers and thus affect the binding of inhibitors[96]. “Isoform switching” between IDH1 and IDH2 subtypes has been reported as another mechanism to restore the activity of mutated IDH and to produce 2-HG[97]. Due to the lack of clinically relevant IDH mutation cell or animal models, the resistance mechanisms caused by 2-HG restoration remain incompletely understood. A study on BRAF inhibitor resistance shows that the dominated resistance mechanism results in ERK signaling not sensitive to RAF inhibitors by upregulating RAF dimers in melanoma, while other mechanisms detour the tumor's dependence on BRAF mutation[98]. Therefore, the combined use with other targeted or immunotherapy regimens may help break through the limitations of targeted monotherapy.
Targeted therapy is a type of and the foundation of precision medicine. The latter needs to identify biological information of genes, RNA, and proteins to stratify patients, selecting patients most likely to benefit from a specific therapy. Therefore, it is crucial to determine the precise targets and select the appropriate agents for the optimized efficacy of treatment. Although high-throughput sequencing of tumor tissue or peripheral blood can provide more comprehensive genetic profiling, the molecular heterogeneity and biological regulation of cancer as iCCA never occur in genomics alone. In the near future, new advances in epigenomics, transcriptomics, proteomics, and metabolomics will be indispensable for the improvement of tailored treatments for patients with iCCA.
CONCLUSION
In the past decade, the recognition of the molecular and genomic characteristics of iCCA has led to a paradigm shift in treatment approaches and strategies. Next-generation sequencing revealed that the majority of iCCA patients harbor at least one actionable mutation, prompting numerous clinical trials to evaluate the efficacy and safety of novel therapies targeting tumor-specific molecules or pathways. Although promising results have been obtained in extensive trials, large-scale randomized phase 3 trials of first-in-class agents and innovative regimens are encouraged to benefit the population with iCCA.
DECLARATIONS
Authors’ contributions
Contribute equally to this work, and were major contributors in collecting literature and drafting the manuscript: Yang F, Qiu Y
in charge of the methodology and administrated the project and reviewed the manuscript: Yi B
Availability of data and materials
All data generated or analysed during this study are included in this published article and its additional information files.
Financial support and sponsorship
This study was financially supported by the National Natural Science Foundation of China (Dr Bin Yi, No. 82173213) and by the Shanghai Natural Science Foundation (Dr Bin Yi, No.21Y11912500).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2023.
Supplementary Materials
REFERENCES
2. Endo I, Gonen M, Yopp AC, et al. Intrahepatic cholangiocarcinoma: rising frequency, improved survival, and determinants of outcome after resection. Ann Surg 2008;248:84-96.
3. Moris D, Palta M, Kim C, Allen PJ, Morse MA, Lidsky ME. Advances in the treatment of intrahepatic cholangiocarcinoma: an overview of the current and future therapeutic landscape for clinicians. CA Cancer J Clin 2023;73:198-222.
4. Bekaii-saab T, Bridgewater J, Normanno N. Practical considerations in screening for genetic alterations in cholangiocarcinoma. Ann Oncol 2021;32:1111-26.
5. Lamarca A, Barriuso J, McNamara MG, Valle JW. Molecular targeted therapies: ready for "prime time" in biliary tract cancer. J Hepatol 2020;73:170-85.
7. Goyal L, Meric-Bernstam F, Hollebecque A, et al. FOENIX-CCA2 Study Investigators. Futibatinib for FGFR2-rearranged intrahepatic cholangiocarcinoma. N Engl J Med 2023;388:228-39.
8. Kendre G, Murugesan K, Brummer T, Segatto O, Saborowski A, Vogel A. Charting co-mutation patterns associated with actionable drivers in intrahepatic cholangiocarcinoma. J Hepatol 2023;78:614-26.
9. Verdaguer H, Saurí T, Acosta DA, et al. ESMO scale for clinical actionability of molecular targets driving targeted treatment in patients with cholangiocarcinoma. Clin Cancer Res 2022;28:1662-71.
10. Wang XY, Zhu WW, Wang Z, et al. Driver mutations of intrahepatic cholangiocarcinoma shape clinically relevant genomic clusters with distinct molecular features and therapeutic vulnerabilities. Theranostics 2022;12:260-76.
11. Carapeto F, Bozorgui B, Shroff RT, et al. The immunogenomic landscape of resected intrahepatic cholangiocarcinoma. Hepatology 2022;75:297-308.
12. Cleary JM, Raghavan S, Wu Q, et al. FGFR2 extracellular domain in-frame deletions are therapeutically targetable genomic alterations that function as oncogenic drivers in cholangiocarcinoma. Cancer Discov 2021;11:2488-505.
13. Lowery MA, Ptashkin R, Jordan E, et al. Comprehensive molecular profiling of intrahepatic and extrahepatic cholangiocarcinomas: potential targets for intervention. Clin Cancer Res 2018;24:4154-61.
14. Jusakul A, Cutcutache I, Yong CH, et al. Whole-genome and epigenomic landscapes of etiologically distinct subtypes of cholangiocarcinoma. Cancer Discov 2017;7:1116-35.
15. Verlingue L, Hollebecque A, Boige V, Ducreux M, Malka D, Ferté C. Matching genomic molecular aberrations with molecular targeted agents: are biliary tract cancers an ideal playground? Eur J Cancer 2017;81:161-73.
16. Weinberg BA, Xiu J, Lindberg MR, et al. Molecular profiling of biliary cancers reveals distinct molecular alterations and potential therapeutic targets. J Gastrointest Oncol 2019;10:652-62.
17. Zhang Y, Ma Z, Li C, et al. The genomic landscape of cholangiocarcinoma reveals the disruption of post-transcriptional modifiers. Nat Commun 2022;13:3061.
18. Komuta M. Intrahepatic cholangiocarcinoma: tumour heterogeneity and its clinical relevance. Clin Mol Hepatol 2022;28:396-407.
19. Song G, Shi Y, Meng L, et al. Publisher correction: single-cell transcriptomic analysis suggests two molecularly distinct subtypes of intrahepatic cholangiocarcinoma. Nat Commun 2022;13:2848.
20. Dong L, Lu D, Chen R, et al. Proteogenomic characterization identifies clinically relevant subgroups of intrahepatic cholangiocarcinoma. Cancer Cell 2022;40:70-87.e15.
21. Dentro SC, Leshchiner I, Haase K, et al. PCAWG Evolution and Heterogeneity Working Group and the PCAWG Consortium. Characterizing genetic intra-tumor heterogeneity across 2,658 human cancer genomes. Cell 2021;184:2239-54.e39.
22. Lin Y, Peng L, Dong L, et al. Geospatial immune heterogeneity reflects the diverse tumor-immune interactions in intrahepatic cholangiocarcinoma. Cancer Discov 2022;12:2350-71.
23. Xiang X, Liu Z, Zhang C, et al. IDH mutation subgroup status associates with intratumor heterogeneity and the tumor microenvironment in intrahepatic cholangiocarcinoma. Adv Sci 2021;8:e2101230.
24. Lin J, Dai Y, Sang C, et al. Multimodule characterization of immune subgroups in intrahepatic cholangiocarcinoma reveals distinct therapeutic vulnerabilities. J Immunother Cancer 2022;10:e004892.
25. Farrell B, Breeze AL. Structure, activation and dysregulation of fibroblast growth factor receptor kinases: perspectives for clinical targeting. Biochem Soc Trans 2018;46:1753-70.
26. Wiedemann M, Trueb B. Characterization of a novel protein (FGFRL1) from human cartilage related to FGF receptors. Genomics 2000;69:275-9.
27. Helsten T, Elkin S, Arthur E, Tomson BN, Carter J, Kurzrock R. The FGFR landscape in cancer: analysis of 4,853 tumors by next-generation sequencing. Clin Cancer Res 2016;22:259-67.
28. Abou-Alfa GK, Sahai V, Hollebecque A, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol 2020;21:671-84.
29. Shi GM, Huang XY, Wen TF, et al. Pemigatinib in previously treated Chinese patients with locally advanced or metastatic cholangiocarcinoma carrying FGFR2 fusions or rearrangements: a phase II study. Cancer Med 2023;12:4137-46.
30. NCCN. NCCN Guidelines. Available from: https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1517 [Last accessed on 14 Nov 2023].
31. Javle M, Roychowdhury S, Kelley RK, et al. Infigratinib (BGJ398) in previously treated patients with advanced or metastatic cholangiocarcinoma with FGFR2 fusions or rearrangements: mature results from a multicentre, open-label, single-arm, phase 2 study. Lancet Gastroenterol Hepatol 2021;6:803-15.
32. Borad M, Javle M, Shaib W, et al. 59P efficacy of derazantinib in intrahepatic cholangiocarcinoma (iCCA) patients with FGFR2 fusions, mutations or amplifications. Ann Oncol 2022;33:S567-8.
33. Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010;17:225-34.
34. Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009;462:739-44.
35. Bleeker FE, Atai NA, Lamba S, et al. The prognostic IDH1(R132) mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol 2010;119:487-94.
36. Pirozzi CJ, Yan H. The implications of IDH mutations for cancer development and therapy. Nat Rev Clin Oncol 2021;18:645-61.
37. Yang Z, Jiang B, Wang Y, et al. 2-HG inhibits necroptosis by stimulating DNMT1-dependent hypermethylation of the RIP3 promoter. Cell Rep 2017;19:1846-57.
38. Fujiwara H, Tateishi K, Misumi K, et al. Mutant IDH1 confers resistance to energy stress in normal biliary cells through PFKP-induced aerobic glycolysis and AMPK activation. Sci Rep 2019;9:18859.
39. Wu MJ, Shi L, Dubrot J, et al. Mutant IDH Inhibits IFNγ-TET2 signaling to promote immunoevasion and tumor maintenance in cholangiocarcinoma. Cancer Discov 2022;12:812-35.
40. Chen X, Yang P, Qiao Y, et al. Effects of cancer-associated point mutations on the structure, function, and stability of isocitrate dehydrogenase 2. Sci Rep 2022;12:18830.
41. Wintheiser G, Zemla T, Shi Q, et al. Isocitrate dehydrogenase-mutated cholangiocarcinoma: natural history and clinical outcomes. JCO Precis Oncol 2022;6:e2100156.
42. Wang J, Zhang ZG, Ding ZY, et al. IDH1 mutation correlates with a beneficial prognosis and suppresses tumor growth in IHCC. J Surg Res 2018;231:116-25.
43. Jiao Y, Pawlik TM, Anders RA, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat Genet 2013;45:1470-3.
44. Wang P, Dong Q, Zhang C, et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene 2013;32:3091-100.
45. Goyal L, Govindan A, Sheth RA, et al. Prognosis and clinicopathologic features of patients with advanced stage isocitrate dehydrogenase (IDH) mutant and IDH wild-type intrahepatic cholangiocarcinoma. Oncologist 2015;20:1019-27.
46. Jolissaint JS, Soares KC, Seier KP, et al. Intrahepatic cholangiocarcinoma with lymph node metastasis: treatment-related outcomes and the role of tumor genomics in patient selection. Clin Cancer Res 2021;27:4101-8.
47. Niger M, Nichetti F, Casadei-Gardini A, et al. Platinum sensitivity in patients with IDH1/2 mutated vs wild-type intrahepatic cholangiocarcinoma: a propensity score-based study. Int J Cancer 2022;151:1310-20.
48. Abou-Alfa GK, Macarulla T, Javle MM, et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): a multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol 2020;21:796-807.
49. Levantini E, Maroni G, Del Re M, Tenen DG. EGFR signaling pathway as therapeutic target in human cancers. Semin Cancer Biol 2022;85:253-75.
50. Hsu JL, Hung MC. The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer Metastasis Rev 2016;35:575-88.
51. Kumagai S, Koyama S, Nishikawa H. Antitumour immunity regulated by aberrant ERBB family signalling. Nat Rev Cancer 2021;21:181-97.
52. Treekitkarnmongkol W, Suthiphongchai T. High expression of ErbB2 contributes to cholangiocarcinoma cell invasion and proliferation through AKT/p70S6K. World J Gastroenterol 2010;16:4047-54.
53. Swain SM, Shastry M, Hamilton E. Targeting HER2-positive breast cancer: advances and future directions. Nat Rev Drug Discov 2023;22:101-26.
54. Javle M, Borad MJ, Azad NS, et al. Pertuzumab and trastuzumab for HER2-positive, metastatic biliary tract cancer (MyPathway): a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol 2021;22:1290-300.
55. Ohba A, Morizane C, Kawamoto Y, et al. Trastuzumab deruxtecan (T-DXd; DS-8201) in patients (pts) with HER2-expressing unresectable or recurrent biliary tract cancer (BTC): An investigator-initiated multicenter phase 2 study (HERB trial). JCO 2022;40:4006.
56. Harding JJ, Piha-Paul SA, Shah RH, et al. Antitumour activity of neratinib in patients with HER2-mutant advanced biliary tract cancers. Nat Commun 2023;14:630.
57. Meric-Bernstam F, Beeram M, Hamilton E, et al. Zanidatamab, a novel bispecific antibody, for the treatment of locally advanced or metastatic HER2-expressing or HER2-amplified cancers: a phase 1, dose-escalation and expansion study. Lancet Oncol 2022;23:1558-70.
58. Sithanandam G, Druck T, Cannizzaro LA, Leuzzi G, Huebner K, Rapp UR. B-raf and a B-raf pseudogene are located on 7q in man. Oncogene 1992;7:795-9.
59. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949-54.
60. Robertson S, Hyder O, Dodson R, et al. The frequency of KRAS and BRAF mutations in intrahepatic cholangiocarcinomas and their correlation with clinical outcome. Hum Pathol 2013;44:2768-73.
61. Iversen L, Tu HL, Lin WC, et al. Molecular kinetics. Ras activation by SOS: allosteric regulation by altered fluctuation dynamics. Science 2014;345:50-4.
62. Huang L, Guo Z, Wang F, Fu L. KRAS mutation: from undruggable to druggable in cancer. Signal Transduct Target Ther 2021;6:386.
63. Zhu C, Guan X, Zhang X, et al. Targeting KRAS mutant cancers: from druggable therapy to drug resistance. Mol Cancer 2022;21:159.
64. Parikh K, Banna G, Liu SV, et al. Drugging KRAS: current perspectives and state-of-art review. J Hematol Oncol 2022;15:152.
65. Subbiah V, Lassen U, Élez E, et al. Dabrafenib plus trametinib in patients with BRAFV600E-mutated biliary tract cancer (ROAR): a phase 2, open-label, single-arm, multicentre basket trial. Lancet Oncol 2020;21:1234-43.
66. Salama AKS, Li S, Macrae ER, et al. Dabrafenib and trametinib in patients with tumors with BRAFV600E mutations: results of the NCI-MATCH trial subprotocol H. J Clin Oncol 2020;38:3895-904.
67. Subbiah V, Puzanov I, Blay JY, et al. Pan-cancer efficacy of vemurafenib in BRAFV600-mutant non-melanoma cancers. Cancer Discov 2020;10:657-63.
68. Kim JW, Lee KH, Kim JW, et al. Enhanced antitumor effect of binimetinib in combination with capecitabine for biliary tract cancer patients with mutations in the RAS/RAF/MEK/ERK pathway: phase Ib study. Br J Cancer 2019;121:332-9.
69. Subbiah V, Cassier PA, Siena S, et al. Pan-cancer efficacy of pralsetinib in patients with RET fusion-positive solid tumors from the phase 1/2 ARROW trial. Nat Med 2022;28:1640-5.
70. Subbiah V, Wolf J, Konda B, et al. Tumour-agnostic efficacy and safety of selpercatinib in patients with RET fusion-positive solid tumours other than lung or thyroid tumours (LIBRETTO-001): a phase 1/2, open-label, basket trial. Lancet Oncol 2022;23:1261-73.
71. Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol 2018;15:731-47.
72. Yoshino T, Pentheroudakis G, Mishima S, et al. JSCO-ESMO-ASCO-JSMO-TOS: international expert consensus recommendations for tumour-agnostic treatments in patients with solid tumours with microsatellite instability or NTRK fusions. Ann Oncol 2020;31:861-72.
73. Vaishnavi A, Le AT, Doebele RC. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov 2015;5:25-34.
75. Hong DS, DuBois SG, Kummar S, et al. Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol 2020;21:531-40.
76. Demetri GD, De Braud F, Drilon A, et al. Updated integrated analysis of the efficacy and safety of entrectinib in patients with NTRK fusion-positive solid tumors. Clin Cancer Res 2022;28:1302-12.
77. Marcus L, Lemery SJ, Keegan P, Pazdur R. FDA approval summary: pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clin Cancer Res 2019;25:3753-8.
79. Jung J, Heo YJ, Park S. High tumor mutational burden predicts favorable response to anti-PD-(L)1 therapy in patients with solid tumor: a real-world pan-tumor analysis. J Immunother Cancer 2023;11:e006454.
80. Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409-13.
81. Borghaei H, Ciuleanu TE, Lee JS, et al. Long-term survival with first-line nivolumab plus ipilimumab in patients with advanced non-small-cell lung cancer: a pooled analysis. Ann Oncol 2023;34:173-85.
82. Maio M, Ascierto PA, Manzyuk L, et al. Pembrolizumab in microsatellite instability high or mismatch repair deficient cancers: updated analysis from the phase II KEYNOTE-158 study. Ann Oncol 2022;33:929-38.
83. Andre T, Berton D, Curigliano G, et al. Safety and efficacy of anti-PD-1 antibody dostarlimab in patients (pts) with mismatch repair-deficient (dMMR) solid cancers: results from GARNET study. JCO 2021;39:9.
84. Schenker M, Burotto M, Richardet M, et al. Abstract CT022: checkMate 848: a randomized, open-label, phase 2 study of nivolumab in combination with ipilimumab or nivolumab monotherapy in patients with advanced or metastatic solid tumors of high tumor mutational burden. Cancer Res 2022;82:CT022.
85. Kim RD, Chung V, Alese OB, et al. A Phase 2 multi-institutional study of nivolumab for patients with advanced refractory biliary tract cancer. JAMA Oncol 2020;6:888-94.
86. Oh DY, He AR, Qin S, et al. Durvalumab plus gemcitabine and cisplatin in advanced biliary tract cancer. NEJM Evid 2022;1:EVIDoa2200015.
87. Kelley RK, Ueno M, Yoo C, et al. KEYNOTE-966 Investigators. Pembrolizumab in combination with gemcitabine and cisplatin compared with gemcitabine and cisplatin alone for patients with advanced biliary tract cancer (KEYNOTE-966): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2023;401:1853-65.
88. Mosele F, Remon J, Mateo J, et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: a report from the ESMO precision medicine working group. Ann Oncol 2020;31:1491-505.
89. Marchiò C, Scaltriti M, Ladanyi M, et al. ESMO recommendations on the standard methods to detect NTRK fusions in daily practice and clinical research. Ann Oncol 2019;30:1417-27.
90. Graham RP, Barr Fritcher EG, Pestova E, et al. Fibroblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma. Hum Pathol 2014;45:1630-8.
91. Rompianesi G, Di Martino M, Gordon-Weeks A, Montalti R, Troisi R. Liquid biopsy in cholangiocarcinoma: Current status and future perspectives. World J Gastrointest Oncol 2021;13:332-50.
92. Berchuck JE, Facchinetti F, DiToro DF, et al. The clinical landscape of cell-free DNA alterations in 1671 patients with advanced biliary tract cancer. Ann Oncol 2022;33:1269-83.
93. Goyal L, Shi L, Liu LY, et al. TAS-120 overcomes resistance to ATP-competitive FGFR inhibitors in patients with FGFR2 fusion-positive intrahepatic cholangiocarcinoma. Cancer Discov 2019;9:1064-79.
94. Sootome H, Fujita H, Ito K, et al. Futibatinib is a novel irreversible FGFR 1-4 inhibitor that shows selective antitumor activity against FGFR-deregulated tumors. Cancer Res 2020;80:4986-97.
95. Calvert AE, Chalastanis A, Wu Y, et al. Cancer-associated IDH1 promotes growth and resistance to targeted therapies in the absence of mutation. Cell Rep 2017;19:1858-73.
96. Intlekofer AM, Shih AH, Wang B, et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature 2018;559:125-9.
97. Harding JJ, Lowery MA, Shih AH, et al. Isoform switching as a mechanism of acquired resistance to mutant isocitrate dehydrogenase inhibition. Cancer Discov 2018;8:1540-7.
98. Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med 2013;19:1401-9.
99. Mazzaferro V, El-Rayes BF, Droz Dit Busset M, et al. Derazantinib (ARQ 087) in advanced or inoperable FGFR2 gene fusion-positive intrahepatic cholangiocarcinoma. Br J Cancer 2019;120:165-71.
100. Bahleda R, Italiano A, Hierro C, et al. Multicenter phase I study of erdafitinib (JNJ-42756493), oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced or refractory solid tumors. Clin Cancer Res 2019;25:4888-97.
101. Kim SB, Meric-Bernstam F, Kalyan A, et al. First-in-human phase I study of aprutumab ixadotin, a fibroblast growth factor receptor 2 antibody-drug conjugate (BAY 1187982) in patients with advanced cancer. Target Oncol 2019;14:591-601.
102. Ahn DH, Uson Junior PLS, Masci P, et al. A pilot study of Pan-FGFR inhibitor ponatinib in patients with FGFR-altered advanced cholangiocarcinoma. Invest New Drugs 2022;40:134-41.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Yang F, Qiu Y, Yi B. Targeted mutation-based therapy for intrahepatic cholangiocarcinoma. Hepatoma Res 2023;9:48. http://dx.doi.org/10.20517/2394-5079.2023.68
AMA Style
Yang F, Qiu Y, Yi B. Targeted mutation-based therapy for intrahepatic cholangiocarcinoma. Hepatoma Research. 2023; 9: 48. http://dx.doi.org/10.20517/2394-5079.2023.68
Chicago/Turabian Style
Yang, Facai, Yinghe Qiu, Bin Yi. 2023. "Targeted mutation-based therapy for intrahepatic cholangiocarcinoma" Hepatoma Research. 9: 48. http://dx.doi.org/10.20517/2394-5079.2023.68
ACS Style
Yang, F.; Qiu Y.; Yi B. Targeted mutation-based therapy for intrahepatic cholangiocarcinoma. Hepatoma. Res. 2023, 9, 48. http://dx.doi.org/10.20517/2394-5079.2023.68
About This Article
Special Issue
Copyright

Data & Comments
Data
0


 Cite This Article 7 clicks
Cite This Article 7 clicks

 Like This Article 8
likes
Like This Article 8
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.