
Genomic alterations in intrahepatic cholangiocarcinoma
 0
0Abstract
Intrahepatic cholangiocarcinoma (iCCA) is an aggressive and heterogeneous biliary cancer with a poor prognosis and limited treatment options. The molecular pathogenesis of iCCA involves a highly complex process entailing multiple genetic alterations and dysregulation of signaling pathways. Recent advancements in our understanding of the genetic landscape of iCCA have opened new opportunities for therapeutic interventions. Technologies such as next-generation sequencing (NGS) have contributed to elucidating the genetic heterogeneity of iCCA, leading to the identification of numerous potentially actionable genetic alterations. Despite these advances, the prognosis of iCCA patients remains dismal. In this review, we provide an extensive summary of the current knowledge on genetic alterations in iCCA, their biological impact on patients, potential therapeutic targets, approved targeted therapies, and ongoing clinical trials with targeted agents. Furthermore, we discuss the main technologies available for studying genetic alterations and their advantages and limitations. Finally, we highlight future directions in studying genetic alterations and the development of new targeted therapies and personalized medicine approaches.
Keywords
INTRODUCTION
Cholangiocarcinoma (CCA) is a rare but fatal malignancy arising in the epithelial cells of the biliary tract[1,2]. Second only to hepatocellular carcinoma (HCC), CCA is one of the most prevalent primary liver cancers, occurring in 15%-20% of patients[1]. Based on anatomical origin, CCAs are classified into intrahepatic (iCCA) and extrahepatic cholangiocarcinoma (eCCA), with the latter being further subtyped into perihilar (pCCA) and distal (dCCA) CCA. These subtypes not only differ in the anatomical location and cell of origin (as extensively reviewed by Moeini et al. 2021[2]), but also present with distinct risk factors, clinical presentations and management, as well as different molecular pathogenesis[1,2]. In this review, we will focus on iCCA.
Risk factors for iCCA vary and include obesity, liver fluke infestation, primary sclerosing cholangitis, and chronic liver diseases, such as viral hepatitis and non-alcoholic fatty liver disease[1,3]. However, most of iCCA cases are sporadic and do not have identifiable risk factors[1]. Age-standardized incidence rates have been steadily increasing in both males and females[3]. The highest mortality rates are still in Asian countries, but mortality rates in European and other Western nations have been on the rise over the last decade[1,3,4].
Diagnosis of iCCA is typically made at advanced stages because of the lack of symptoms and effective biomarkers[3]. Over 70% of patients diagnosed with iCCA have unresectable tumors or extrahepatic spread, lowering the median overall survival to less than 12 months[1]. Traditionally, the diagnosis of CCA is based on the anatomical location of tumors and the evaluation of histological features obtained from tumor biopsies[5,6]. With an examination of gross morphology, iCCA is categorized into either mass-forming, periductal-infiltrating, or mixed-type iCCA[7,8]. At the microscopic level, iCCAs can be divided into
For over a decade, the standard of care treatment for iCCA and other biliary tract cancers has been a regimen of gemcitabine plus cisplatin (GemCis)[10-14]. Since results with GemCis are only modest (median overall survival: 11.7 months), it has been challenging to improve patient survival in iCCA patients[10-14], leaving an unmet need for more effective first- and second-line therapies[1]. In an effort to meet this need, clinical trials have been designed to explore adding a third agent or using immune checkpoint inhibitors to enhance the effectiveness of the standard of care therapy, as extensively reviewed by Rizzo and Brandi[15]. Notably, while immune checkpoint inhibitors as single agents have been unsuccessful, the recent TOPAZ 1 trial has shown that the combination of cisplatin and gemcitabine with durvalumab, an immune checkpoint inhibitor (ICI) targeting PD-L1, has led to greater overall survival (median overall survival:
In addition, over the past few years, next-generation sequencing (NGS) technologies have shed light on the molecular landscape of iCCA[18-21], and unveiled various oncogenic drivers and potential therapeutic targets[18,22-24]. These discoveries have spurred the development of targeted therapies tailored to iCCA patients with specific molecular profiles. For instance, the first FGFR inhibitors have received FDA approval for patients harboring FGFR2 fusions[25,26]. Moreover, comprehensive genomic profiling of iCCA patients has identified the presence of biomarkers such as tumor mutational burden and microsatellite instability[27]. These results suggest that deepening our understanding of genetic alterations (GAs) and molecular profiling in iCCA is crucial for advancing personalized therapeutic strategies for iCCA.
In this review, we first summarize current knowledge on GAs in iCCA and their biological impact. Next, we discuss the various molecular classification systems used to stratify patients for treatment strategies and the clinical application of different testing procedures to identify GAs harbored by each iCCA patient. Finally, we discuss the knowledge gaps and the approaches being used to close those gaps and develop more effective therapeutics and treatment strategies for patients with strategies for this disease.
GENETIC ALTERATIONS IN iCCA
In recent years, thanks to the identification of recurrent and “druggable” GAs, iCCA has emerged as a model for precision cancer treatment within gastrointestinal malignancies, with more than 45% of patients harboring at least one clinically actionable genetic alteration according to the results of the FIGHT-202 trial[28]. Among the identified GAs, those in isocitrate dehydrogenase (IDH) and fibroblast growth factor receptor 2 (FGFR2) genes account for the most frequent and clinically-relevant alterations. In addition, despite occurring at lower frequencies in patients, GA’s in B-raf proto-oncogene (BRAF), erb-b2 receptor tyrosine kinase 2 (ERBB2), neurotrophic tyrosine receptor kinase (NRTK), BReast CAncer gene 1 (BRCA1), and Kirsten rat sarcoma virus (KRAS), among others, are prime targets for new and developing therapeutic options. In the following paragraphs, we will provide an overview of the GAs occurring in iCCA [Figure 1] with particular emphasis on the most clinically relevant alterations. Ongoing clinical trials investigating therapeutic agents targeting the identified GAs are summarized in Table 1.
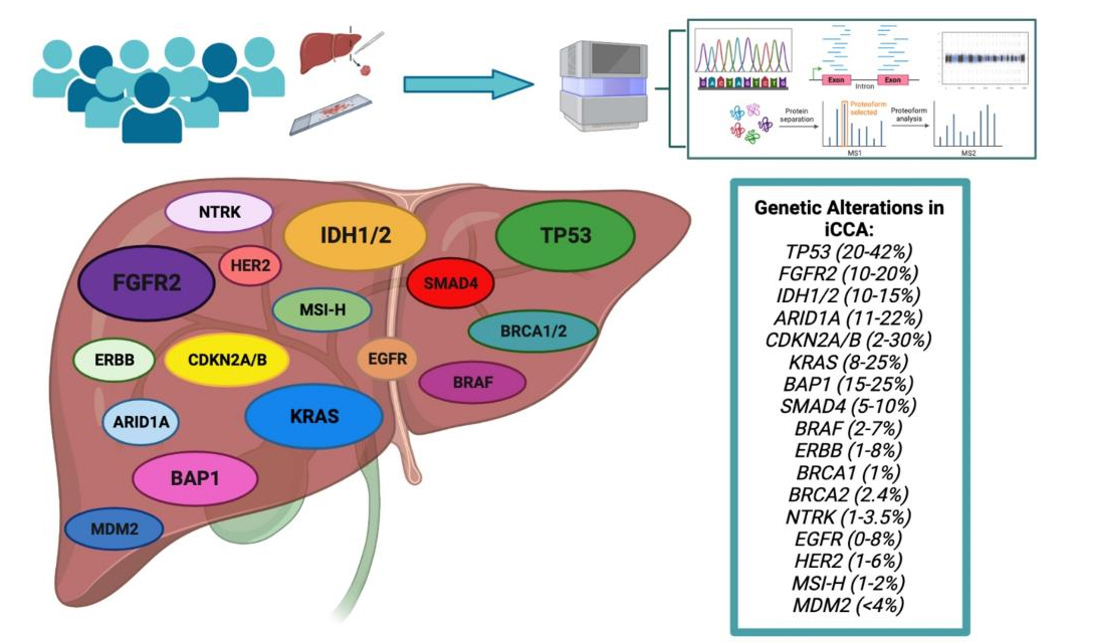
Figure 1. Genetic alterations in iCCA. Multi-omic analyses of large patient cohorts have revealed the most prevalent alterations in iCCA. FGFR2 fusions and mutations in IDH1/2 are the most prevalent, while alterations in KRAS, ARID1A, CDKN2A/B, and BAP1 occur at slightly lower frequencies. Other alterations occur in iCCA at low frequencies but are still considered actionable therapeutic targets are herein depicted. Created with https://www.biorender.com/ .
Current clinical trials investigating target therapies for actionable driver mutations in iCCA
| Target gene | Drug | Combination therapies included | Drug class | FDA approval | Ongoing clinical trials | Conditions | Status |
| IDH1 mutations (20%-25%) | Ivosidenib | - | IDH1 inhibitor | For advanced cholangiocarcinoma with IDH mutations | NCT02989857 | Advanced, metastatic cholangiocarcinoma | Completed |
| Ivosidenib | In combination with Gemcitabine and Cisplatin | IDH1 inhibitor | For advanced cholangiocarcinoma with IDH mutations | NCT04088188 | Unresectable or metastatic cholangiocarcinoma | Recruiting | |
| Olaparib | - | PARP inhibitor | For breast, ovarian, and pancreatic cancers | NCT03212274 | Advanced Glioma, Cholangiocarcinoma, or Solid Tumors with IDH1 or IDH2 Mutations | Recruiting | |
| Olaparib | In combination with Ceralasertib (AZD6738) | PARP inhibitor | For breast, ovarian, and pancreatic cancers | NCT03878095 | IDH1 and IDH2 mutant cholangiocarcinoma or solid tumors | Recruiting | |
| Olaparib | In combination with Durvalumab | PARP inhibitor | For breast, ovarian, and pancreatic cancers | NCT03991832 | IDH1-Mutated solid tumors | Recruiting | |
| AB-218 | - | IDH1 inhibitor | No | NCT05814536 | Advanced IDH1 mutant cholangiocarcinoma | Not yet recruiting | |
| LY3410738 | In combination with Gemcitabine, Cisplatin, and Durvalumab | covalent mutant IDH inhibitor | No | NCT04521686 | Advanced solid tumors with IDH1 or IDH2 mutations | Recruiting | |
| HMPL-306 | - | dual IDH1/2 inhibitor | No | NCT04762602 | Advanced solid tumors with IDH mutations | Recruiting | |
| IDH305 | - | mutant-selective IDH1 inhibitor | No | NCT02381886 | Advanced Malignancies that Harbor IDH1R132 Mutations | Active, not recruiting | |
| FGFR2 fusions/alterations (10%-20%) | Infigratinib | - | selective ATP-competitive inhibitor | For metastatic cholangiocarcinoma with FGFR2 fusion/rearrangement | NCT04233567 | Advanced or metastatic solid tumors with FGFR Gene Mutations | Recruiting |
| Infigratinib | In combination with Nab-paclitaxel, Gemcitabine, and Cisplatin | selective ATP-competitive inhibitor | For metastatic cholangiocarcinoma with FGFR2 fusion/rearrangement | NCT05514912 | Resectable Intrahepatic Cholangiocarcinoma | Not yet recruiting | |
| Infigratinib | In combination with Atezolizumab and Bevacizumab | selective ATP-competitive inhibitor | For metastatic cholangiocarcinoma with FGFR2 fusion/rearrangement | NCT05510427 | Advanced Cholangiocarcinoma with FGFR2 Fusion/Amplification | Not yet recruiting | |
| Pemigatinib | - | tyrosine kinase inhibitor | For metastatic cholangiocarcinoma with FGFR2 fusion/rearrangement | NCT04256980 | Advanced/Metastatic cholangiocarcinoma including FGFR2 rearrangement | Active, not recruiting | |
| Pemigatinib | Versus Gemcitabine and Cisplatin | Tyrosine kinase inhibitor | For metastatic cholangiocarcinoma with FGFR2 fusion/rearrangement | NCT03656536 | Unresectable or Metastatic Cholangiocarcinoma | Recruiting | |
| Pemigatinib | In combination with Gemcitabine and Cisplatin | tyrosine kinase inhibitor | For metastatic cholangiocarcinoma with FGFR2 fusion/rearrangement | NCT04088188 | Unresectable or metastatic cholangiocarcinoma | Recruiting | |
| Pemigatinib | After SBRT | tyrosine kinase inhibitor | For metastatic cholangiocarcinoma with FGFR2 fusion/rearrangement | NCT05565794 | Advanced iCCA with FGFR Fusion/Rearrangement | Not yet recruiting | |
| Futibatinib (Tas-120) | Versus Gemcitabine and Cisplatin | Irreversible FGFR 1-4 inhibitor | For metastatic cholangiocarcinoma with FGFR2 fusion/rearrangement | NCT04093362 | Advanced cholangiocarcinoma with FGFR2 Gene Rearrangements | Active, not recruiting | |
| Futibatinib (Tas-120) | - | irreversible FGFR 1-4 inhibitor | For metastatic cholangiocarcinoma with FGFR2 fusion/rearrangement | NCT05727176 | Advanced cholangiocarcinoma with FGFR2 fusion or rearrangement | Not yet recruiting | |
| Derazantinib | In combination with Atezolizumab | FGFR 1-3 kinase inhibitor | No | NCT05174650 | Advanced iCCA with FGFR Fusion/Rearrangement | Recruiting | |
| E7090 | - | selective FGFR inhibitor | No | NCT04238715 | Advanced or metastatic cholangiocarcinoma with FGFR2 Gene Fusion | Active, not recruiting | |
| 3D185 | - | selective FGFR 1-3 inhibitor | No | NCT05039892 | Locally advanced or metastatic cholangiocarcinoma | Not yet recruiting | |
| Gunagratinib (ICP-192) | - | selective irreversible FGFR inhibitor | No | NCT05678270 | FGFR2-rearranged unresectable or metastatic intrahepatic cholangiocarcinoma | Recruiting | |
| Gunagratinib (ICP-192) | - | selective irreversible FGFR inhibitor | No | NCT04565275 | Advanced solid tumors with FGFR gene alterations | Recruiting | |
| Tinengotinib (TT-00420) | - | multi-kinase inhibitor | No | NCT04919642 | Advanced cholangiocarcinoma with FGFR alterations | Recruiting | |
| HMPL-453 Tartrate | - | selective FGFR 1-3 inhibitor | No | NCT04353375 | Advanced intrahepatic cholangiocarcinoma with FGFR2 Fusion | Not yet recruiting | |
| RLY-4008 | - | selective irreversible FGFR2 inhibitor | No | NCT04526106 | ICC and other advanced solid tumors | Recruiting | |
| Erdafitinib | - | pan-FGFR tyrosine kinase inhibitor | For metastatic urothelial carcinoma | NCT02699606 | Advanced Non-Small-Cell Lung Cancer, Urothelial Cancer, Esophageal Cancer Or Cholangiocarcinoma | Active, not recruiting | |
| KIN-3248 | - | irreversible pan-FGFR inhibitor | No | NCT05242822 | Advanced tumors harboring FGFR2 and/or FGFR3 gene alterations | Recruiting | |
| CPL304110 | - | selective FGFR 1-3 inhibitor | No | NCT04149691 | Advanced Solid Malignancies, including cholangiocarcinoma | Recruiting | |
| KRAS (8%-20%) | Sotorasib | in combination with Anti-PD-1/L1 or Midazolam | KRAS G12C inhibitor | For advanced non-small cell lung cancer (NSCLC) | NCT03600883 | Solid Tumors with KRASG12C Mutations | Active, not recruiting |
| Adagrasib | in combination with Cetuximab, Afatinib, or Cetuximab | KRAS G12C inhibitor | For advanced non-small cell lung cancer (NSCLC) | NCT03785249 | Advanced metastatic cancer | Recruiting | |
| Trametinib | In combination with Hydroxychloroquine (HCQ) | MEK inhibitor | For NSCLC, anaplastic thyroid cancer, and metastatic solid tumors with BRAFv600E mutations | NCT04566133 | Refractory bile tract carcinoma with KRAS mutation | Recruiting | |
| PI3K/AKT/mTOR | Everolimus | - | mTOR inhibitor | For non-functional neuroendocrine tumors (NET) of GI or lung originFor advanced renal cell carcinoma | NCT00973713 | Advanced cholangiocarcinoma | Completed |
| BRAFV600E (2%-7%) | Vemurafenib | in combination with Cobimetinib | BRAF inhibitor | For metastatic solid tumors with BRAFV600E mutation | NCT02091141 | Biliary Tract Cancers | Active, not recruiting |
| ABM-1310 | In combination with Cobimetinib | BRAF inhibitor | No | NCT04190628 | Advanced solid tumors, including Cholangiocarcinoma | Recruiting | |
| ABM-1311 | - | BRAF inhibitor | No | NCT05501912 | BRAF V600-Mutant Advanced Solid Tumors | Recruiting | |
| HER2 (2%-6%) | Zanidatamab | - | HER2-targeted antibody | No | NCT04466891 | HER2-amplified Biliary Tract Cancers | Active, not recruiting |
| Zanidatamab | In combination with established chemotherapy regimens | HER2-targeted antibody | No | NCT03929666 | HER2-expressing GI Cancers, including Biliary Tract Cancer | Recruiting | |
| Trastuzumab deruxtecan | - | HER2-targeted antibody-drug conjugate | For HER2-positive breast cancer and HER2-positive gastric adenocarcinomas | NCT04482309 | Open-Label Study including Biliary Tract Cancer | Active, not recruiting | |
| MRG002 | - | HER2-targeted antibody-drug conjugate | No | NCT04837508 | Advanced or Metastatic Biliary Tract Cancer | Recruiting | |
| IMM2902 | - | HER2/SIRPa bispecific mAb | No | NCT05805956 | Advanced solid tumors expressing HER2 | Recruiting | |
| NTRK (1%-4%) | Larotrectinib | - | pan-NTRK inhibitor | For solid tumors with NTRK gene fusion | NCT02576431 | Solid tumors, including Biliary Tract Cancer | Recruiting |
| Entrectinib | - | NTRK inhibitor | For solid tumors with NTRK gene fusion | NCT02568267 | Basket trial including Cholangiocarcinoma | Active, not recruiting | |
| EGFR (0%-8%) | Erlotinib | In combination with Pemetrexed | EGFR inhibitor | For non-small cell lung cancer (NSCLC) | NCT03110484 | Biliary Tract Cancers | Recruiting |
| BRCA1/2 (1%-3%) | Olaparib | In combination with Pembrolizumab | PARP inhibitor | For breast, ovarian, and pancreatic cancers | NCT04306367 | Advanced cholangiocarcinoma | Active, not recruiting |
| Olaparib | In combination with Ceralasertib (AZD6738) | PARP inhibitor | For breast, ovarian, and pancreatic cancers | NCT04298021 | Bile Duct Cancers | Recruiting | |
| Rucaparib | In combination with Nivolumab | PARP inhibitor | No | NCT03639935 | Biliary Tract Cancers | Active, not recruiting |
Currently actionable GAs
IDH1/2 mutations
In iCCA, IDH1/2 mutations occur in up to ~10%-15% of patients[29]. Isocitrate dehydrogenase (IDH) 1 and 2 are integral enzymes for cellular respiration. IDH1, localized to the cytoplasm, reduces NADPH to NADP+ during the conversion of isocitrate to -ketoglutarate in the citric acid cycle, while IDH2 performs the same function within the mitochondria [Figure 2]. Mutations in IDH1 are most often a single nucleotide variation (SNV) at R132, while in IDH2, the SNVs occur more frequently at residues R140 or R170[27,30]. Mutations in IDH1/2 alter their enzymatic activity and promote the production of the oncometabolite
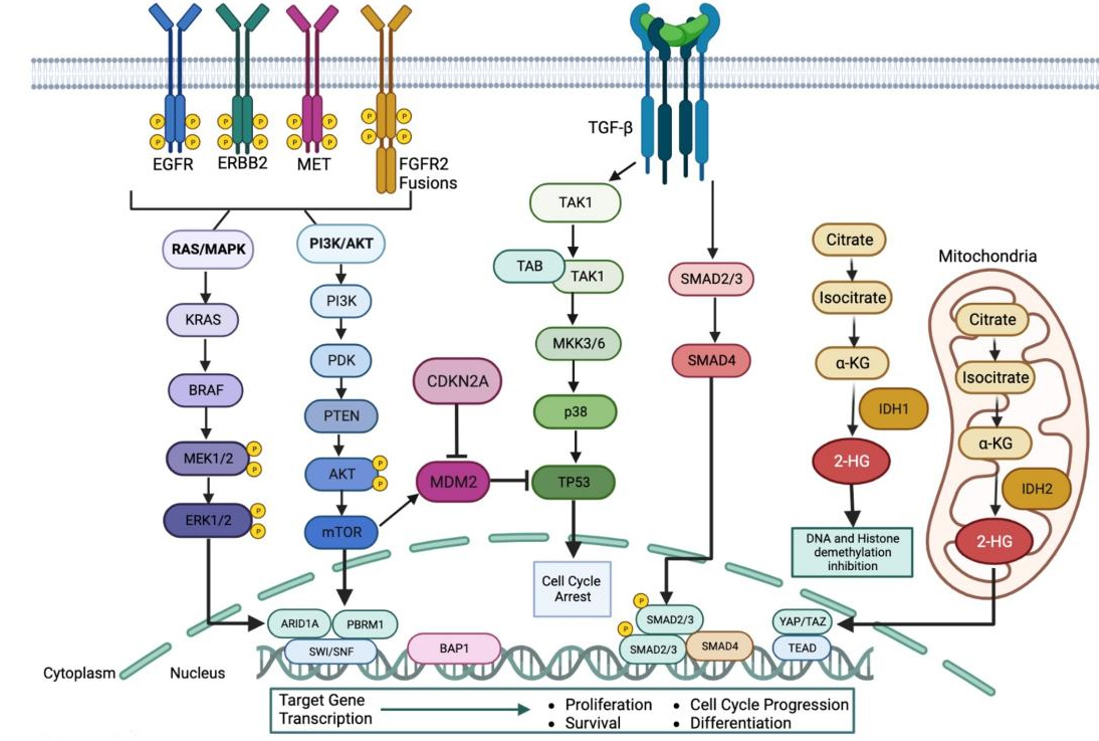
Figure 2. Key signaling pathways in intrahepatic cholangiocarcinoma. A simplified view of pathways activated by genetic alterations identified in iCCA. Mutations in receptor tyrosine kinases like EGFR, ERBB, MET, and FGFR2 fusions dysregulate signaling of the RAS/MAPK and PI3K/AKT pathways. Uncontrolled TGF-β signaling is defined by loss-of-function mutations in SMAD4 and is linked to dysregulated signaling through the TAK1 pathway. Mutations in IDH1/2 lead to increased levels of 2-HG and increased YAP/TAZ-directed transcription. Created with https://www.biorender.com/.
In August of 2021, the FDA approved the use of ivosidenib, an IDH1 inhibitor, for the treatment of patients with IDH1-mutated iCCAs based on the results of the phase III clinical trial, ClarIDHy (NCT02989857), where a significant improvement in progression-free survival was observed compared to placebo (median of 2·7 months vs. 1·4 months), despite a non-significant overall survival[40]. Considering the cytostatic mechanism of action of ivosidenib, the modest clinical benefit is consistent with those reported with other targeted non-cytotoxic drugs. To further explain modest clinical responses, the first mechanisms of acquired resistance have emerged, with two patients enrolled in a phase I trial of ivosidenib as monotherapy (NCT02073994) developing secondary IDH1 mutations, R172K and D279N, at disease progression[41]. These acquired mutations are thought to disrupt the binding of ivosidenib to the IDH1 mutant, rendering the drug ineffective and enabling disease progression[41]. In addition, a third patient enrolled in the clinical trial exhibited persistence of a pre-treatment IDH1 R132C mutation and acquired a novel IDH2 R172V mutation[42]. Future studies further investigating these, and other mechanisms of resistance are needed.
In addition to the downregulation of chromatin modifier genes, an increase in the 2-HG oncometabolite seen in IDH1/2 mutated cancers inhibits proteins associated with DNA damage response, such as lysine-specific demethylase 4A/B (KDM4A/B) and ataxia-telangiectasia mutated (ATM), as well as the DNA repair enzyme alkB homolog (ALKBH)[43,44]. Decreased function in these proteins causes an increase in DNA double-strand breaks and increased DNA damage. This effect, similar to the homologous recombination deficiency (HRD) that is exhibited in BRCA1/2 mutated cancers (referred to as the “BRCAness” phenotype)[45], confers sensitivity to inhibition of DNA repair mechanisms, such as poly(ADP-ribose) polymerase (PARP) inhibition. As IDH1/2 mutations are among the most frequent mutations in iCCA, the PARP inhibitor, Olaparib, is currently being tested in phase II studies (NCT03212274; NCT03878095) to evaluate its efficacy in patients with IDH1/2 mutant solid tumors including CCA[46].
FGFR2 alterations
Fibroblast growth factor receptor 2 (FGFR2) is a member of the family of receptor tyrosine kinases (RTKs) which, upon ligand binding, dimerizes and signals activation of downstream pathways including RAS/MAPK, PI3K/AKT, and JAK/STAT. These pathways play a role in cell proliferation, differentiation, and apoptosis, and when dysregulated, contribute to increased proliferation, tumorigenesis, and metastasis[47-49]. GAs in the FGFR2 gene encompass mutations, gene fusions and in-frame deletions[25,28,40,50-54]. Nearly 80% of all FGFR2 GAs are fusions involving exons 1-18 of the FGFR2 with the kinase domain intact, fused in-frame to diverse gene partners[28,53-56]. This fusion partner leaves FGFR2 without an important proline-rich sequence that allows GRB2 to bind and deactivate downstream signaling[57]. The most prevalent fusion partners are bicaudal C homolog 1 (BICC1), Periphilin 1 (PPHLN1), adenosylhomocysteinase like 1 (AHCYL1), Transforming Acidic Coiled-Coil Containing Protein 3 (TACC3), and meningioma expressed antigen 5 (MGEA5), although over 60 fusion partners have been catalogued[28,50,53-55,58]. Regardless of the partner identity, fusions result in constitutive kinase activity [Figure 2][50,53,54]. Transforming capabilities of FGFR2 fusions have been demonstrated and can be suppressed both in vitro and in vivo with FGFR1-3 inhibitors[50,53], providing the rationale for the first biomarker-driven clinical trials. It has been recently described that while FGFR2 fusions tend to co-occur with alterations in BAP1 and CDKN2A/B, they are usually mutually exclusive with mutations in KRAS[27,39]. Although rare, patients harboring FGFR alterations may present with concomitant alterations which could contribute to resistance mechanisms[56] or, when targetable itself, such as the presence of IDH mutations, make treatment choices difficult. In the latter circumstances, discussion of each clinical case at designated molecular tumor boards should be actively pursued. In this regard, increasing efforts are ongoing to implement local, regional or digital multidisciplinary liver tumor boards to share, discuss and harness the challenges brought by precision oncology[59]. Additional GAs in FGFR2 occur in the form of point mutations or short variants[56], with the most common including gain-of-function mutations (S372, Y375, C382, N549 and F276) that stabilize the active dimerized conformation of FGFR2[27,39]. Extracellular in-frame deletions in the FGFR2 gene resulting in structural and conformational changes that can activate FGFR2 have also been recently documented[51]. These alterations possess transforming capabilities and seem to predict sensitivity to FGFR inhibitors[51].
Based on the results of recent phase 2 clinical trials, three FGFR inhibitors (e.g., infigratinib, pemigatinib, and futibatinib) have received accelerated FDA approval as a treatment option for patients with iCCA harboring a FGFR2 fusion or other alterations[40,60,61]. In particular, infigratinib (BGJ398) and pemigatinib - two reversible ATP-competitive FGFR1-4 inhibitors - showed an objective response rate of 23%[61]
Despite an initial response to these two FGFR inhibitors, the clinical benefit remains modest and short-lived since resistance inevitably emerges, as elegantly described in 4 patients enrolled in a trial for treatment with infigratinib (NCT02150967) where the evaluation of tumor biopsies and plasma at the time of disease progression revealed the onset of secondary mutations in the FGFR2 gene[55,62]. Notably, 3 of the 4 patients developed a mutation in the gatekeeper residue V564, leading to structural change in the FGFR2 binding pocket, ultimately preventing infigratinib from binding. Additionally, these patients acquired mutations in residues V549, E565, K641, and K659, all of which led to kinase activation and increased PI3K/AKT/mTOR signaling, and a mutation in L617 that weakens the stability of an Asp-Phe-Gly conformation that favors inhibitor binding[55,62]. Similarly, patients evaluated after treatment with pemigatinib also acquired secondary mutations in additional residues (N549, E565, K659, L617, and K641), indicating that these mutations confer resistance to multiple FGFR inhibitors[28,63]. In this context, in a phase I clinical trial (NCT02052778), Futibatinib (TAS-120), an irreversible ATP-competitive FGFR1-4 inhibitor, demonstrated efficacy in 4 patients with FGFR2 fusion-positive iCCA who developed resistance to infigratinib or Debio 1347[64]. The application of molecular profiling in circulating tumor DNA (ctDNA) combined with in vitro studies using patient-derived iCCA cells further confirmed that TAS-120 was active against multiple FGFR2 mutations conferring resistance to BGJ398 or Debio 1347, while mutations in the gatekeeper V565 residue continued to show resistance to FGFR inhibition[64]. These results suggest that serial molecular testing could inform sequential treatment with FGFR inhibitors and prolong responses to these agents. However, unlike other GAs, there are limitations to using ctDNA to detect FGFR2 fusions. Berchuck et al. report that FGR2 fusions were detected in serial circulating free DNA (cfDNA) at lower frequencies than in tissues biopsies (1.4% vs. 4.3%), revealing that concordance between them was only 18% in their patient cohort (n = 67 patients with FGFR2 fusions)[65]. Additionally, the sensitivity in their detection assays was higher for the FGFR2-BICC1 fusion versus other fusion partners (58% vs. 2%, respectively), a trend that was not improved by repeated serial sampling[65]. They suggest this is not due to a lack of cfDNA in blood samples, but rather because of the promiscuity of the FGFR2 gene[65]. The abundance and variety of fusion partners in iCCA patients mean that more probes are required to detect those fusions, but limitations on the panel size for cfDNA assays mean increasing the number of probes targeted towards fusion partners is not always feasible[65]. Additionally, Goyal et al. emphasize that while molecular profiling of ctDNA is a useful tool in monitoring a patient’s response to therapy, it is a technique best used in combination with genetic sequencing of tumor biopsies at the multiple stages of treatment and disease progression[64]. Finally, validation with larger clinical cohorts is needed to elucidate whether patterns in resistance mutations can inform treatment strategies[64].
The successful completion of the multinational, open-label, phase 2 study (NCT02052778) confirmed the clinical benefit elicited by futibatinib with a 42% objective response rate[60]. Notably, the use of futibatinib resulted in durable responses and survival that surpassed those indicated by historical data with chemotherapy in patients with refractory iCCA[60]. This agent is being investigated in a phase III clinical trial (NCT04093362) versus the standard of care treatment gemcitabine plus cisplatin. At the same time, several efforts are currently underway to shed light on additional resistance mechanisms to FGFR inhibitors and improve therapeutic responses. In this regard, Wu et al. recently demonstrated that feedback activation of EGFR signaling limits the effectiveness of FGFR inhibitor therapy and drives adaptive resistance in patient-derived models of iCCA carrying FGFR2 fusions, suggesting that the combination of FGFR and EGFR inhibitors could improve responses in this molecularly-defined subgroup of patients[66]. Additionally,
Alterations in KRAS and the MAPK pathway
Kirsten rat sarcoma virus (KRAS) belongs to a group of small GTPases called the Ras family[68]. The protein encoded by KRAS is involved in the regulation of cell growth, differentiation and survival through the RAS/MAPK signaling cascade, and mutations lead to uncontrolled proliferation of cells and tumor growth[69,70] [Figure 2]. Mutations in the KRAS gene are among the most frequent alterations in iCCA
Despite KRAS long being considered "undruggable", recent landmark studies have led to the FDA approval of sotorasib (AMG 510), the first small molecule inhibitor targeting specifically KRASG12C mutation, for advanced non-small cell lung cancer (NSCLC) based on the results of the CodeBreaK 100 trial (NCT03600883)[91]. This study included one biliary cancer patient who achieved disease control[92]. In addition, in December 2022, the FDA granted accelerated approval to adagrasib (MRTX849)[93], another RAS GTPase inhibitor, based on the results of the KRYSTAL-1 study, a multicenter, single-arm, open-label clinical trial (NCT03785249) which included patients with locally advanced or metastatic NSCLC with KRASG12C mutations[94]. An expansion cohort of this study is currently ongoing in patients with advanced tumors, including CCA [Table 1]. Furthermore, inhibitors targeting mutations other than G12C are in the early stages of development. MRTX1133, the KRASG12D inhibitor[95,96], is set to enter a Phase 1 clinical evaluation in 2023 for KRASG12D-driven cancers, including pancreatic, colorectal, lung, and others (NCT05737706). Despite these promising results, acquired resistance is a common pitfall of targeted therapies and KRAS inhibitors are no exception. Acquired resistance to KRAS inhibitors has been recently demonstrated by the identification of multiple treatment-emergent alterations in 27 out of 43 patients with distinct solid cancers (the majority being NSCLC) treated with sotorasib, including secondary alterations in KRAS, NRAS, BRAF, EGFR, FGFR2, MET, MYC and other genes[97-99]. Because of the intense crosstalk between signaling pathways downstream of KRAS and the activation of feedback loops due to treatment-emergent mutations, combining inhibitors targeting multiple redundant signaling pathways seems most promising. For example, genetic or pharmacological targeting of ERK signaling has been shown to enhance the antiproliferative effect of KRASG12C inhibition models with acquired RAS or BRAF mutations[99]. In conclusion, targeting the KRAS pathway holds great promise as a therapeutic approach, but further research is needed to optimize treatment strategies, properly design combination strategies to overcome potential resistance and improve outcomes for iCCA patients.
BRAF
B-raf proto-oncogene (BRAF) is a serine/threonine protein kinase that acts as a downstream effector of KRAS via the ERK/MAPK pathway. Mutations in BRAF result in its constant dimerization, leading to uncontrolled cell proliferation, differentiation, and survival[100-102]. BRAFV600E is the most common variant implicated in many cancers including iCCA where it has been detected in 2%-7% of patients[27,71,74,78,103-106]. Interestingly, in the FoundationCORE database, BRAFV600E and BRAFnon-V600E alterations occurred at similar frequencies but featured a strikingly different mutational spectrum with BRAFV600E mutations showing a predominance of CDKN2A/B deletions over TP53 alterations[27]. In contrast, patients with BRAFnon-V600E alterations had a lower frequency of KRAS, FGFR2 and TERT promoter alterations and an enrichment in ARID1A Gas[27].
While monotherapies against the BRAFV600E mutation have had discouraging results[107], the phase II Rare Oncology Agnostic study (ROAR trial; NCT02034110), including 43 patients with advanced BRAFV600E mutated BTC (iCCA patients, n = 39), showed promising results for the combination of the trametinib MEK 1/2 inhibitor and the BRAF inhibitor, dabrafenib[108]. Based on these results, a combination of dabrafenib and trametinib was approved as a tumor-agnostic therapy for all solid metastatic cancers with BRAFV600E mutations[109,110]. In addition, the BRAF inhibitor ABM-1310 is currently in phase I study (NCT04190628) evaluating its efficacy alone or in combination with cobimetinib for solid tumors with BRAFV600E mutation, including iCCA.
Targeting the ERBB family
The ERBB family encompasses four receptor tyrosine kinases: EGFR (ERBB1), HER2/NEU (ERBB2), ERBB3, and ERBB4[111]. These receptors play a vital role in regulating cellular proliferation, differentiation, and migration by activating MAPK and PI3K/AKT signaling pathways, among others [Figure 2]. Alterations in the ERBB family have been reported in iCCA patients, with EGFR and ERBB2 being the most frequent[73]. EGFR overexpression and HER2 amplification have been reported in iCCA with a frequency of 0%-8% and 1%-6%, respectively, and represent appealing therapeutic targets[19,23,27,58,71,89,112-119].
Various EGFR and HER2 inhibitors have been investigated in early phase trials, including lapatinib, erlotinib, pertuzumab, and trastuzumab[119-122]. However, in various clinical trials, EGFR inhibitors have shown limited benefit. For advanced BTC patients, cetuximab and panitumumab did not show survival benefits when combined with chemotherapy. Erlotinib, an oral EGFR inhibitor, was combined with oxaliplatin and gemcitabine in the phase III trial for advanced, untreated BTC patients, but it did not provide a survival benefit[123]. Currently, Erlotinib is being evaluated in combination with pemetrexed in phase II single-arm prospective study (NCT03110484) in metastatic biliary tract cancer patients.
A few studies are currently ongoing to evaluate HER2-directed therapies in mutant biliary tract cancers. Zanidatamab, a HER2-targeted bispecific antibody, received an FDA breakthrough therapy designation based on an ongoing phase 2b clinical trial (NCT04466891) testing this agent as treatment of patients with locally advanced and/or metastatic HER2-expressing tumors, including biliary tract cancer[124]. In addition, Zanidatamab is being investigated in combination with chemotherapy as first-line treatment in a phase II clinical trial (NCT03929666) for HER2-expressing gastrointestinal cancers, with overall promising preliminary results including a confirmed objective response of 79% and a disease control rate of 92%[125]. The Phase 3 randomized trial HERIZON-GEA-01 (NCT05152147) is ongoing and its successful completion will support the approval for zanidatamab in combination with chemotherapy as an effective treatment option for this molecularly-defined subgroup. In addition, promising results were recently reported in a phase I study testing trastuzumab deruxtecan (T-DXd), a novel HER2-targeted antibody-drug conjugate linked to a topoisomerase I inhibitor, in various solid tumors, including biliary tract cancers[122]. Reported preliminary results for the HERB trial (NCCH1805; JMA-IIA00423) show that HER2 positive iCCA patients (n = 30) treated with T-DXd had longer median PFS vs. the control group (6.1 months vs. 5.1 months) and longer median OS (10.8 months vs. 7.1 months)[126,127]. These results will need to be further evaluated in a larger cohort of patients. Currently, T-DXd is also being tested in an open-label study (NCT04482309) in patients with selected HER2-expressing tumors. Another phase II (NCT04837508) study is currently testing MRG002, a HER2-targeted antibody-drug conjugated to the microtubule-disrupting cytotoxic agent monomethyl auristatin E (MMAE), for advanced or metastatic biliary tract cancer.
NTRK fusions
The neurotrophic tyrosine receptor kinase (NTRK) gene family consists of NTRK1, NTRK2, and NTRK3, which encode tropomyosin receptor kinases (TRK) A, B, and C, respectively. NTRK gene fusions are rare, with a reported frequency of 1%-3.5% in iCCA[21,121]. Oncogenic fusions of the NTRK gene result in a chimeric protein with the N-terminus of the fusion partner joined to the C-terminus of the TRK protein with the catalytic tyrosine kinase domain. These oncogenic fusions lead to constitutive activation of MAPK cascade and PI3K pathway, promoting cell proliferation and survival[128].
Despite being rare, two potent NTRK inhibitors, entrectinib and larotrectinib, have recently emerged as novel therapeutic options in NTRK fusion-positive malignancies, including CCA. In this regard, larotrectinib, which has previously shown promising results in patients expressing TRK fusions, including 2 CCA patients[129], is currently being evaluated in the NAVIGATE phase 2 basket trial (NCT02576431)[130]. Chromosomal rearrangement of ROS1 represents an additional rare oncogenic driver mutation in iCCA (0%-9%)[114,131] and a phase 2 basket study (NCT02568267) is currently evaluating entrectinib in patients with solid tumors including CCA with fusions of the NTRK1/ 2, ROS1 or ALK gene. Notably, both inhibitors - entrectinib and larotrectinib - have been approved for patients with NTRK fusion-positive cancer, independent of their histology[129,132].
DNA mismatch repair and microsatellite instability
DNA mismatch repair (dMMR) is an important system for maintaining genomic integrity through the recognition and repair of DNA replication errors[133]. Mutations in dMMR lead to the accumulation of frameshift mutations in microsatellite-coding genes, leading to microsatellite instability (MSI)[134,135]. Overall, the frequency of MSI is low in iCCA, with MSI-high being reported in about 1%-2% of patients[27,136]. Recent studies have shown that MSI-high iCCA patients exhibit concomitant alterations in genes such as TP53, ARID1A, RNF43, PBRM1, and CDKN2A. In addition, enrichment of alterations related to activation of the WNT signaling pathway and mutations in MLH1 and MSH6 were reported in this subgroup of patients in the FoundationCORE database[27].
Based on the findings from the phase II KEYNOTE-158 trial which demonstrated clinical benefit in MSI-H and dMMR tumors treated with immune checkpoint inhibitors (ICIs)[137], MSI-high status is currently regarded as the best predictive biomarker of ICI responses across all tumor histologies. Despite MSI-H and dMMR being rare events in iCCA, patients in the KEYNOTE-158 trial responded well to pembrolizumab (median PFS 4.2 months, median OS 24.3 months), therefore making these two events a potentially useful biomarker for predicting ICI response in iCCA patients[137].
BRCA1/2 mutations and the potential benefit of PARP inhibitors
Breast Cancer gene 1 (BRCA1) and Breast Cancer gene 2 (BRCA2) are tumor suppressor genes that play a vital role in DNA damage response (DDR) mediated by the homologous recombination pathway. Mutations in BRCA1/2 lead to the accumulation of DNA double-stranded breaks, resulting in genomic instability and tumor growth[138-142]. BRCA1 and BRCA2 mutations have been reported in 1% and 2.4% of iCCA patients, respectively[27,143-145]. iCCA patients with these mutations are concurrently mutated for TP53, CDKN2A/B, KRAS, ARID1A, and PBRM1 amongst others[27]. Studies have shown that poly adenosine diphosphate-ribose polymerase (PARP) inhibitors may be effective for CCA patients with mutations in the DDR genes, including BRCA1/2 alterations[146-148]. Several clinical trials are currently in progress to evaluate the efficacy and safety of PARP inhibitors, either as monotherapy or in combination with other therapies in cholangiocarcinoma. For example, an umbrella study (NCT04298021) is currently evaluating the efficacy of the PARP inhibitor, Olaparib, alone or in combination with durvalumab in advanced biliary tract cancers. A single-arm phase II study (NCT04306367) is testing the safety and efficacy of the combination of pembrolizumab and olaparib to improve the response rate of second-line systemic therapy in patients with advanced CCA who cannot tolerate gemcitabine-based therapy. Another PARP inhibitor, rucaparib, is currently being evaluated in phase II study (NCT03639935) in combination with nivolumab in patients with advanced or metastatic biliary tract cancer following platinum therapy. Additional clinical trials involving more iCCA patients are required to provide definitive evidence of the therapeutic potential of PARP inhibitors for treating iCCA patients.
Other “currently undruggable” alterations
TP53
The tumor suppressor gene tumor protein P53 (TP53) is involved in DNA damage repair, cell cycle arrest, apoptosis, and metabolism[149,150]. Mutations in TP53 have been found in most cancers, including NSCLC, breast cancer, pancreatic ductal cancer, and primary liver cancers (both HCC and iCCA)[27,87,149,151,152]. Typically, mutations in TP53 lead to a loss of function, resulting in increased cell proliferation, metabolic pathway activity, and tumorigenesis[39]. In iCCA, TP53 mutations are the most common alterations occurring in ~20%-42% of patients[27,72,153]. Amplifications of the MDM2 gene, one of the main TP53 negative regulators through ubiquitination and proteasomal degradation, have also been described across many cancers, although only in less than ~4% of iCCA[27]. TP53 mutations frequently co-occur with alterations in KRAS, BRCA1/2, and ERBB2, but are mutually exclusive with FGFR2 fusions[27,39].
Alterations in chromatin remodeling genes
AT-rich interactive domain-containing protein 1A (ARID1A) encodes a key component of the SWI/SNF chromatin-remodeling complex. Loss-of-function mutations in ARID1A frequently occur in iCCA (11%-22%)[27,74,89,106,114,154,155], and lead to aberrant chromatin remodeling, genomic instability, and altered DNA repair, ultimately contributing to CCA progression[156]. Other chromatin remodeling genes such as polybromo-1 (PBRM1) and BRCA1-associated protein 1 (BAP1) are also found mutated in 11%-17%[19,27,86,89] and ~15%-25% of iCCA patients[154,157], respectively, with at least one of the three genes being mutated in 47% of iCCA cases[154]. In particular, loss of function in BAP1 occurs through either chromosomal deletions or inactivating mutations, and leads to tumor formation and progression[158,159]. Germline alterations in BAP1 have also been linked to an increased risk of iCCA and other malignancies[157].
SMAD4 and the TGF-b signaling pathway
Mothers against decapentaplegic homolog 4 (SMAD4) is a regulator of the transforming growth factor- (TGF-) signaling pathway, which controls cell proliferation, differentiation, and other cellular functions[160,161]. In its tumor suppressive role, SMAD4 loss leads to increased TGF- signaling and an increase in transcription of downstream targets[160]. SMAD4 inactivation mutations through chromosomal deletion or point mutations are found in ~5%-10% of iCCA patients[27,72,121], and recent studies have shown that patients with lower SMAD4 expression have poor cell differentiation and more metastatic disease[160]. GAs in other components of the pathway, including both receptors and ligands, have been described at a lower frequency (2%-3%)[83,162].
CDKN2A/B
Deletions in cyclin-dependent kinase inhibitor 2A and 2B (CDKN2A/B) have been reported between 2%-30% for iCCA patients[72,121,163,164]. CDKN2A is a tumor suppressor gene that encodes p16INK4A and p14ARF, while CDKN2B encodes p15INK4B[150,163]. Both p16INK4A and p15INK4B are regulators of the cyclin-dependent kinases CDK4 and CDK6 and play an integral role in inhibiting cell cycle progression through G1 phase[163,165]. The protein p14ARF activates and stabilizes TP53 to activate cell cycle arrest and apoptosis[165,166]. When CDKN2A/B genes are lost, CDK4/6 activity is left unregulated and cell proliferation is promoted[163,165].
TERT
The TERT gene encodes for the catalytic subunit of telomerase enzyme. Telomerase plays a vital role in maintaining genomic integrity and telomere length at the end of the chromosomes, which is vital for cellular replication and stability. TERT acts as a gatekeeper for cellular immortalization while maintaining chromosomal integrity. Unlike HCC, promoter mutations in TERT are found in iCCA at low frequencies (6%)[27,35,85].
MOLECULAR CLASSIFICATIONS OF iCCA
Several classifications have been proposed to date for iCCA patients, building molecular profiles based on transcriptomics, proteogenomics, and genetic characteristics[72,162,167,168]. The overarching goal of these classifications is to ultimately identify groups of patients with similar genomic profiles and use this information to determine a course of treatment. However, despite numerous efforts to establish an effective classification system for iCCA patients that includes genomic, molecular, and immune profiles for subtypes, there is still no consensus on which system to use, mostly due to the lack of validation in prospective clinical trials as well as the difficulties in routinely accessing tissue biopsies. In this paragraph, we will discuss how genetic alterations have contributed to defining distinct molecular and immune subgroups of iCCA. Molecular classifications based on transcriptomics and other -omics have been extensively reviewed elsewhere[169].
Genetic alterations-driven subtypes of iCCA
As the role of massive parallel sequencing has expanded over the past few years, several molecular classes with distinct mutational profiles have emerged[35,72,162,167,168,170-173]. Using genomic and epigenomic data from 489 CCA patients, Jusakul et al. identified four distinct clusters with separate genetic and epigenetic features. Cluster 1 showed enrichment of ARID1A and BRCA1/2 mutations as well as DNA hypermethylation. Both cluster 1 and cluster 2 showed enrichment of TP53 and ERBB2 mutations, but cluster 2 was also enriched with CTNNB1, WNT5B, and AKT1 mutations. In contrast, cluster 3 showed high levels of somatic copy number alterations and upregulation in immune checkpoint genes like PD-1 and PD-L2. Finally, cluster 4 exhibited enrichment in BAP1, IDH1/2, and FGFR alterations. In addition, these clusters were correlated with clinical outcomes, with clusters 3 and 4 having better overall survival compared to clusters 1 and 2[171]. Similarly, Bagante et al. classified patients into two genetic groups after reviewing previously published clustering based on gene mutations[35,170,171]. Their KRAS/TP53 group included patients with mutations in KRAS, NRAS, TP53, and ARID1A, while patients with IDH1/2, BAP1, and PBRM1 were included in the second group. Analysis of pathological characteristics showed that the KRAS/TP53 group had more periductal infiltration while the IDH1-2/BAP1/PBRM1 group exhibited larger tumor sizes. Additionally, they were able to associate the IDH1-2/BAP1/PBRM1 group with longer overall survival versus the KRAS/TP53 group[170]. While both studies had cohorts containing multiple subtypes of CCA (iCCA, eCCA and gallbladder cancer), Nepal et al. proposed a stratification of “only” iCCA patients using the most recurrent mutations identified in a dataset of 496 patients analyzed by whole-exome and targeted exome sequencing[172]. Their subgroups (IDH1, KRAS, TP53, and ‘Undetermined’) exhibited distinct co-mutation profiles as well as unique pathway enrichments. For example, patients in the IDH1 group did not co-occur with TP53 or KRAS mutations but were enriched in BAP1 and BCLAF1 gene alterations and showed enrichment in metabolic pathways. In contrast, patients in the TP53 group had co-occurrences with KRAS mutations, enrichment in alterations in PTEN, RB1 and LATS2, and upregulated MAPK, WNT, and p53 signaling. Consistent with previous studies, Nepal et al. associated their gene mutation groups with patient overall survival, showing that the KRAS and TP53 groups had worse overall survival versus the IDH1 and ‘Undetermined’ groups within the cohort[172]. More recently, Wang et al. expanded existing efforts to analyze a cohort of 1481 iCCAs derived from several publicly available datasets. They identified three clusters based on the most prevalent mutations, with cluster 1 including patients with mutations in KRAS, TP53, and SMAD4, while cluster 2 included patients with IDH1/2 and BAP1 mutations and FGFR2 fusions. Their third cluster, "Wild-Type", included all other patients with enrichment in ARID1A, PBRM1, CDKN2A, PIK3CA, and EPHA2 gene mutations, but exhibiting wild-type genes for those in the other 2 clusters as well as patients with no detectable genetic mutations[173]. These clusters were associated with distinct gene expression patterns, histological features, and clinical prognosis and outcomes. For example, patients in cluster 2 exhibited small-duct type iCCAs with lower tumor burden, better prognosis, and higher expression of growth factors and receptors. In contrast, patients in cluster 1, with KRAS, TP53, and SMAD4 mutations, had higher tumor burden, worse prognosis, and increased expression of genes such as COX-2, MUC1, and IL-6, which are associated with disease progression[173]. Overall, great consistency exists across these studies, with patients with KRAS and TP53 mutations clustering separately from patients with IDH1/2 mutations and FGFR2 fusions. Additionally, a worse prognosis was reported for the subgroups harboring KRAS or TP53 alterations compared to the subgroups including IDH1/2, BAP1, and FGFR2 fusions, indicating that clustering along these lines could provide beneficial information in the clinical setting.
Genotype-immunophenotype correlations in iCCA
With the recent success of immunotherapeutic strategies across several solid cancers, including iCCA, the identification of immune-related subgroups of patients with prognostic/predictive value has gained significant traction[72,174,175]. However, only a few of these studies have paid particular attention to elucidating how the tumor genotype of iCCA affects the immunophenotype and vice versa. Evidence from other solid tumors clearly indicates that tumor-intrinsic alterations, such as oncogenic pathways (i.e., PI3K, MYC) and mutations in driver genes (i.e., gain-of-function mutations in the KRAS), contribute to the immunosuppressive tumor microenvironment that supports cancer growth through various mechanisms and potentially influence responses to ICIs[176-178]. The recent pan-cancer immunogenomic analysis of the intratumoral immune landscape of over 20 solid cancers has further reinforced the concept that tumor-intrinsic genomic alterations determine the tumor immunophenotype and dictate distinct escape mechanisms which are therefore predetermined based on the intra-tumor genetic composition[179]. As an example, the Pan Cancer Immunome ATLAS study concluded that BRAF-mutated tumors may utilize distinct evasion mechanisms compared to KRAS-mutated tumors. Specifically, BRAF-mutated tumors were found to be enriched in immunosuppressive cells, whereas RAS tumors showed downregulation of MHC molecules and immunomodulatory molecules. Nonetheless, whether these associations and underlying mechanisms of immunoevasion differ from one solid tumor to another according to the tissue-specific co-occurring genomic alterations and/or other oncogenic pathways dysregulation remains still unresolved. This is particularly relevant for iCCA, an understudied malignancy whose immune milieu remains poorly understood. Of note, no CCA was included in the Pan Cancer Immunome ATLAS, underlying the dire need to conduct immunogenomic analysis specifically in this disease. A step forward in elucidating the genotype-immunophenotype relationships in iCCA is represented by the recent STIM classification proposed by Martin-Serrano et al., who, through the multi-omics analysis of about 900 iCCA samples, identified five distinct classes, each characterized by a distinct tumor genotype associated with a unique stromal and immune composition[72]. Notably, this immunogenomic analysis suggests that mutations in driver genes such as KRAS and TP53 may play a key role in dictating the immune milieu of iCCA with KRAS-mutant and TP53-mutant tumors being enriched in the inflamed classes of iCCAs, and alterations in FGFR2, IDH1/2 and chromatin remodeling genes being enriched in the non-inflamed classes. Interestingly co-occurrence of TP53 and KRAS mutations seemed to dictate a distinct immune milieu compared to KRAS mutant iCCAs (in absence of co-occurring TP53 mutations) with the former being associated with accumulation of Tregs in poorly inflamed tumors and the latter being enriched in myeloid cells, dysfunctional T cells, myofibroblastic cancer-associated fibroblasts (CAFs) and TGF- signaling in both murine and human iCCAs, all features associated with resistance to immunotherapy. Consistently, Martin-Serrano et al demonstrated that selective KRAS inhibitors can sensitize KRAS-mutant murine iCCAs to ICIs[72]. Clinical trials testing the combination of the newly FDA-approved KRASG12C inhibitor with ICIs with or without ICIs are currently ongoing in lung (NCT04720976, NCT04613596, NCT05472623) and colorectal cancer (NCT04793958). Similar clinical studies are anxiously awaited in iCCA. Consistent results supporting the unique genotype-immunophenotype relationship in iCCA have been recently reported by Lin et al., who applied whole exome sequencing, T-cell receptor sequencing and multiplexed immunofluorescence on 207 tumor regions from 45 patients with iCCA[180]. In this study, highly immune infiltrated subgroups correspond with alterations in KRAS and TP53, while immune "cold" groups correspond with alterations in FGFR2 and BAP1[72,153,180]. Interestingly, while Lin et al. found that patients with IDH1/2 mutations displayed more T-cell infiltration[180], a recent study by Wu et al. has reported that IDH1 mutations could shape a “cold” immune microenvironment in iCCA[181]. Interestingly, in this study, pharmacologic inhibition of IDH1 mutant induced profound immune changes, including CD8+ T-cell recruitment and promotion of TET2-dependent induction of IFNγ response genes in tumor cells, and sensitized murine iCCAs to ICI targeting CTLA4[181]. Overall, these exciting emerging data suggest that a better understanding of the genotype-immunophenotype relationships may provide the rationale for combination immunotherapies to be tested in future clinical trials in molecular-defined subgroups of iCCA patients.
CLINICAL ASSESSMENT OF ACTIONABLE TARGETS
As discussed in the paragraphs above, genetic profiling enables the identification of actionable genetic alterations for targeted therapies. Therefore, both the European Society for Medical Oncology (ESMO) and United States National Comprehensive Cancer Network (NCCN) guidelines recommend molecular testing in all iCCA patients with advanced disease suitable for systemic treatments[182,183]. However, the different nature of GAs, the type of tissue available and the well-known technical challenges of tissue biopsies (i.e., poor quality and/or insufficient material) represent major pitfalls to the routine implementation of molecular testing.
Different testing methods exist due to the different nature of targetable GAs. Conventional methods, which include immunohistochemistry (IHC), fluorescence in situ hybridization and PCR-based DNA or RNA sequence amplification, can be readily and easily applied to detect the presence of ERBB2 amplifications, FGFR2 fusions and BRAFV600 mutations[103,184]. MSI status can also be inferred via IHC to detect the expression of MMR proteins which consist of hMLH1, hPMS2, hMSH2 and hMSH6, or by fragment PCR[185], whereas FGFR2 fusions can be detected by break-apart FISH[186]. However, despite being valuable and relatively inexpensive tools, these methods are limited in scope, lack sensitivity and specificity, do not allow the interrogation of multiple genes, and require prior knowledge of the targeted GAs, ultimately hindering a comprehensive understanding of the iCCA genetic landscape in the clinical setting.
NGS-based technologies allow parallel sequencing of several genes and, thus, should represent the preferred testing modality. NGS approaches effectively detect genetic changes, rearrangements, and copy number alterations in multiple genes in a single analysis. Both WES and RNA-seq allow physicians to perform a comprehensive unbiased molecular screening of individual patients for informed treatment decisions, with RNA-seq being the preferred method for the detection of gene fusions[187,188]. However, major barriers to the routine implementation of these approaches exist, including financial (i.e., high costs and subsequent reimbursement by healthcare payers), turnaround time, and technical challenges (i.e., standardized tissue collection, protocols, etc.). In this regard, recent advances in the field have allowed large gene panels to be assayed with high accuracy, sensitivity, and specificity and at a reduced cost. NGS-based targeted panels such as MSK-IMPACT[187] and FoundationOne CDX offer a high coverage of tumor-specific genes, requiring less input material, easier data analysis and shorter turnaround time. Amplicon-based and anchored multiplex PCR approaches further expand the capabilities of these tests by allowing the identification of gene fusions from known (Oncomine™ Dx Target Test) as well as unknown fusion partners (Archer FusionPlex Solid Tumor Panel). However, iCCA tissue biopsies are often difficult to obtain, or even when a biopsy is feasible, it may yield insufficient material. In this regard, liquid biopsies represent a valid complementary strategy for molecular testing. Liquid biopsy methodologies are noninvasive and encompass NGS, droplet digital PCR, and digital PCR. Liquid biopsies can be used to detect biomarkers circulating tumor cells, circulating tumor DNA/RNA, exosomes, and microRNAs[189]. The clinical applications of liquid biopsies have been recently demonstrated in seminal studies of iCCA where cfDNA sampling successfully informed the sequence of FGFR inhibitors at the time of progression[55]. Nonetheless, the low sensitivity of cfDNA for the detection of FGFR2 fusions, with only 18% concordance between cfDNA and tissue biopsies, requires further optimization before being routinely implemented in clinical practice[65].
FUTURE DIRECTIONS
In summary, significant progress has been made in identifying recurrent and targetable genetic alterations in iCCA, with some being targeted by FDA-approved selective inhibitors while others are being tested in clinical trials. Nonetheless, the prognosis of this disease remains dismal, highlighting the dire need for additional research efforts aimed at identifying new therapeutic targets, understanding how genetic alterations orchestrate the tumor immune microenvironment, and ultimately translating these findings into improved patient outcomes.
The recent application of single cell-based technologies has shed light on the intra-tumor and inter-tumor heterogeneity of iCCA[190,191], although, thus far, only few iCCA samples have been analyzed due to the high costs. It is expected that, in the near future, a more widespread application of these technologies in large human cohorts will allow researchers to identify additional molecular drivers and understand the impact of specific genetic alterations on tumor composition and growth. Within each genetic molecular subgroup of patients (i.e., KRAS-, IDH1-mutant, etc.), it will also be crucial to understand how the specific co-mutational network impacts tumor progression and therapeutic responses to enhance the efficacy of precision medicine.
In a malignancy where 40%-50% of patients harbor at least a targetable alteration, the application of multi-omics (i.e., genomics, proteomics, transcriptomics, etc.) to tissue biopsies before the start of treatment could shed light on mechanisms of resistance to current chemotherapy and targeted therapies. These studies are anxiously awaited since they may identify predictive biomarkers for (1) better patient stratification and (2) informed decisions on sequential treatments when multiple therapeutic options will be available [upon completion of the ongoing phase 3 trials shown in Table 1]. To achieve this goal, education of all medical professionals will play a major role in ensuring universal access to molecular testing for all iCCA patients at diagnosis and collection of serial tumor and blood samples.
An alternative approach to further improve responses to targeted therapies, including FGFR inhibitors, requires the rational design of effective combination strategies. Based on the recent results of the TOPAZ-1 (anti-PD-L1 blockade plus chemotherapy) and KEYNOTE-966 trials (anti-PD1 blockade plus chemotherapy), ICI combinations hold great promise for this malignancy but remain significantly underexplored. To achieve these clinical goals, the development of novel preclinical models that better represent the genetic heterogeneity as well as the tumor-immune interactions of iCCA is urgently needed. Significant efforts should also be devoted to establishing and characterizing precision mouse models, while cross-species analyses should be conducted to assert their clinical relevance. Furthermore, more advanced co-culture models of 3D organoids with other microenvironment components
In conclusion, while a lot more remains to be done, the recent progress may indicate that we are on the right path to improve the outcome for iCCA patients.
DECLARATIONS
Authors’ contributionsConceived the structure of this manuscript, researched the literature and wrote the manuscript: Young SE, Sritharan R, Sia D
Availability of data and materialsNot applicable.
Financial support and sponsorshipDS is supported by the Tisch Cancer Institute (Icahn School of Medicine at Mount Sinai), the F. Klion Award, i3 Genesis Award and the TCI Dev. Funds Award 2023.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
REFERENCES
1. Banales JM, Marin JJG, Lamarca A, et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol 2020;17:557-88.
2. Moeini A, Haber PK, Sia D. Cell of origin in biliary tract cancers and clinical implications. JHEP Rep 2021;3:100226.
3. Vithayathil M, Khan SA. Current epidemiology of cholangiocarcinoma in Western countries. J Hepatol 2022;77:1690-8.
4. Izquierdo-Sanchez L, Lamarca A, La Casta A, et al. Cholangiocarcinoma landscape in Europe: diagnostic, prognostic and therapeutic insights from the ENSCCA Registry. J Hepatol 2022;76:1109-21.
5. Casadio M, Biancaniello F, Overi D, et al. Molecular landscape and therapeutic strategies in cholangiocarcinoma: an integrated translational approach towards precision medicine. Int J Mol Sci 2021;22:5613.
6. Normanno N, Martinelli E, Melisi D, et al. Role of molecular genetics in the clinical management of cholangiocarcinoma. ESMO Open 2022;7:100505.
7. Chung T, Park YN. Up-to-date pathologic classification and molecular characteristics of intrahepatic cholangiocarcinoma. Front Med 2022;9:857140.
8. Meng ZW, Pan W, Hong HJ, Chen JZ, Chen YL. Macroscopic types of intrahepatic cholangiocarcinoma and the eighth edition of AJCC/UICC TNM staging system. Oncotarget 2017;8:101165-74.
9. Akita M, Sawada R, Komatsu M, et al. An immunostaining panel of C-reactive protein, N-cadherin, and S100 calcium binding protein P is useful for intrahepatic cholangiocarcinoma subtyping. Hum Pathol 2021;109:45-52.
10. Lamarca A, Ross P, Wasan HS, et al. Advanced intrahepatic cholangiocarcinoma: post Hoc analysis of the ABC-01, -02, and -03 clinical trials. J Natl Cancer Inst 2020;112:200-10.
11. Okusaka T, Nakachi K, Fukutomi A, et al. Gemcitabine alone or in combination with cisplatin in patients with biliary tract cancer: a comparative multicentre study in Japan. Br J Cancer 2010;103:469-74.
12. Tsukiyama I, Ejiri M, Yamamoto Y, et al. A Cost-effectiveness analysis of gemcitabine plus cisplatin versus gemcitabine alone for treatment of advanced biliary tract cancer in Japan. J Gastrointest Cancer 2017;48:326-32.
13. Valle JW, Furuse J, Jitlal M, et al. Cisplatin and gemcitabine for advanced biliary tract cancer: a meta-analysis of two randomised trials. Ann Oncol 2014;25:391-8.
14. Valle JW, Wasan H, Johnson P, et al. Gemcitabine alone or in combination with cisplatin in patients with advanced or metastatic cholangiocarcinomas or other biliary tract tumours: a multicentre randomised phase II study - the UK ABC-01 study. Br J Cancer 2009;101:621-7.
15. Rizzo A, Brandi G. First-line chemotherapy in advanced biliary tract cancer ten years after the ABC-02 trial: "and yet it moves!". Cancer Treat Res Commun 2021;27:100335.
16. Oh D, He AR, Qin S, et al. A phase 3 randomized, double-blind, placebo-controlled study of durvalumab in combination with gemcitabine plus cisplatin (GemCis) in patients (pts) with advanced biliary tract cancer (BTC): TOPAZ-1. J Clin Oncol 2022;40:378-378.
17. Kelley RK, Ueno M, Yoo C, et al; KEYNOTE-966 Investigators. Pembrolizumab in combination with gemcitabine and cisplatin compared with gemcitabine and cisplatin alone for patients with advanced biliary tract cancer (KEYNOTE-966): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2023;401:1853-65.
18. Javle M, Bekaii-Saab T, Jain A, et al. Biliary cancer: utility of next-generation sequencing for clinical management. Cancer 2016;122:3838-47.
19. Lowery MA, Ptashkin R, Jordan E, et al. Comprehensive molecular profiling of intrahepatic and extrahepatic cholangiocarcinomas: potential targets for intervention. Clin Cancer Res 2018;24:4154-61.
21. Ross JS, Wang K, Gay L, et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist 2014;19:235-42.
22. Hezel AF, Deshpande V, Zhu AX. Genetics of biliary tract cancers and emerging targeted therapies. J Clin Oncol 2010;28:3531-40.
23. Sia D, Hoshida Y, Villanueva A, et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 2013;144:829-40.
24. Sia D, Tovar V, Moeini A, Llovet JM. Intrahepatic cholangiocarcinoma: pathogenesis and rationale for molecular therapies. Oncogene 2013;32:4861-70.
25. Liu PCC, Koblish H, Wu L, et al. INCB054828 (pemigatinib), a potent and selective inhibitor of fibroblast growth factor receptors 1, 2, and 3, displays activity against genetically defined tumor models. PLoS One 2020;15:e0231877.
26. Storandt MH, Jin Z, Mahipal A. Pemigatinib in cholangiocarcinoma with a FGFR2 rearrangement or fusion. Expert Rev Anticancer Ther 2022;22:1265-74.
27. Kendre G, Murugesan K, Brummer T, Segatto O, Saborowski A, Vogel A. Charting co-mutation patterns associated with actionable drivers in intrahepatic cholangiocarcinoma. J Hepatol 2023;78:614-26.
28. Silverman IM, Hollebecque A, Friboulet L, et al. Clinicogenomic analysis of fgfr2-rearranged cholangiocarcinoma identifies correlates of response and mechanisms of resistance to pemigatinib. Cancer Discov 2021;11:326-39.
29. Borger DR, Tanabe KK, Fan KC, et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 2012;17:72-9.
30. Rimini M, Fabregat-Franco C, Burgio V, et al. Molecular profile and its clinical impact of IDH1 mutated versus IDH1 wild type intrahepatic cholangiocarcinoma. Sci Rep 2022;12:18775.
31. Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009;462:739-44.
32. Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010;17:225-34.
33. Xu W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011;19:17-30.
34. Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009;360:765-73.
35. Farshidfar F, Zheng S, Gingras MC, et al. Cancer Genome Atlas Network. Integrative genomic analysis of cholangiocarcinoma identifies distinct idh-mutant molecular profiles. Cell Rep 2017;18:2780-94.
36. Wang P, Dong Q, Zhang C, et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene 2013;32:3091-100.
37. Zhao S, Lin Y, Xu W, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 2009;324:261-5.
38. Saha SK, Parachoniak CA, Ghanta KS, et al. Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature 2014;513:110-4.
39. Dong L, Lu D, Chen R, et al. Proteogenomic characterization identifies clinically relevant subgroups of intrahepatic cholangiocarcinoma. Cancer Cell 2022;40:70-87.e15.
40. Abou-Alfa GK, Sahai V, Hollebecque A, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol 2020;21:671-84.
41. Cleary JM, Rouaisnel B, Daina A, et al. Secondary IDH1 resistance mutations and oncogenic IDH2 mutations cause acquired resistance to ivosidenib in cholangiocarcinoma. NPJ Precis Oncol 2022;6:61.
42. Harding JJ, Lowery MA, Shih AH, et al. Isoform switching as a mechanism of acquired resistance to mutant isocitrate dehydrogenase inhibition. Cancer Discov 2018;8:1540-7.
43. Sulkowski PL, Corso CD, Robinson ND, et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med 2017;9:eaal2463.
44. Molenaar RJ, Radivoyevitch T, Nagata Y, et al. IDH1/2 mutations sensitize acute myeloid leukemia to PARP inhibition and this is reversed by IDH1/2-mutant inhibitors. Clin Cancer Res 2018;24:1705-15.
45. Hachem S, Kassis Y, Hachem MC, Zouein J, Gharios J, Kourie HR. BRCAness in biliary tract cancer: a new prognostic and predictive biomarker? Biomark Med 2023;17:51-7.
46. Fanucci K, Pilat MJ, Shyr D, et al. Multicenter phase II trial of the parp inhibitor olaparib in recurrent IDH1- and IDH2-mutant glioma. Cancer Res Commun 2023;3:192-201.
47. Ornitz DM, Itoh N. The fibroblast growth factor signaling pathway. Wiley Interdiscip Rev Dev Biol 2015;4:215-66.
48. Rizvi S, Yamada D, Hirsova P, et al. A hippo and fibroblast growth factor receptor autocrine pathway in cholangiocarcinoma. J Biol Chem 2016;291:8031-47.
49. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 2010;10:116-29.
50. Arai Y, Totoki Y, Hosoda F, et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014;59:1427-34.
51. Cleary JM, Raghavan S, Wu Q, et al.
52. Makawita S, K Abou-Alfa G, Roychowdhury S, et al. Infigratinib in patients with advanced cholangiocarcinoma with
53. Sia D, Losic B, Moeini A, et al. Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat Commun 2015;6:6087.
54. Wu YM, Su F, Kalyana-Sundaram S, et al. Identification of targetable
55. Goyal L, Saha SK, Liu LY, et al. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov 2017;7:252-63.
56. Kendre G, Marhenke S, Lorz G, et al. The co-mutational spectrum determines the therapeutic response in murine FGFR2 fusion-driven cholangiocarcinoma. Hepatology 2021;74:1357-70.
57. Lin CC, Melo FA, Ghosh R, et al. Inhibition of basal FGF receptor signaling by dimeric Grb2. Cell 2012;149:1514-24.
58. Carotenuto M, Sacco A, Forgione L, Normanno N. Genomic alterations in cholangiocarcinoma: clinical significance and relevance to therapy. Explor Target Antitumor Ther 2022;3:200-23.
59. Tamborero D, Dienstmann R, Rachid MH, et al. Cancer Core Europe consortium. The molecular tumor board portal supports clinical decisions and automated reporting for precision oncology. Nat Cancer 2022;3:251-61.
60. Goyal L, Meric-Bernstam F, Hollebecque A, et al. FOENIX-CCA2 Study Investigators. Futibatinib for FGFR2-rearranged intrahepatic cholangiocarcinoma. N Engl J Med 2023;388:228-39.
61. Javle M, Roychowdhury S, Kelley RK, et al. Infigratinib (BGJ398) in previously treated patients with advanced or metastatic cholangiocarcinoma with FGFR2 fusions or rearrangements: mature results from a multicentre, open-label, single-arm, phase 2 study. Lancet Gastroenterol Hepatol 2021;6:803-15.
62. Krook MA, Lenyo A, Wilberding M, et al. Efficacy of FGFR inhibitors and combination therapies for acquired resistance in FGFR2-fusion cholangiocarcinoma. Mol Cancer Ther 2020;19:847-57.
63. Krook MA, Bonneville R, Chen HZ, et al. Tumor heterogeneity and acquired drug resistance in FGFR2-fusion-positive cholangiocarcinoma through rapid research autopsy. Cold Spring Harb Mol Case Stud 2019;5:a004002.
64. Goyal L, Shi L, Liu LY, et al. TAS-120 Overcomes resistance to ATP-competitive FGFR inhibitors in patients with FGFR2 fusion-positive intrahepatic cholangiocarcinoma. Cancer Discov 2019;9:1064-79.
65. Berchuck JE, Facchinetti F, DiToro DF, et al. The clinical landscape of cell-free DNA alterations in 1671 patients with advanced biliary tract cancer. Ann Oncol 2022;33:1269-83.
66. Wu Q, Zhen Y, Shi L, et al. EGFR inhibition potentiates FGFR inhibitor therapy and overcomes resistance in FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov 2022;12:1378-95.
67. Subbiah V, Sahai V, Maglic D, et al. RLY-4008, the first highly selective FGFR2 inhibitor with activity across FGFR2 alterations and resistance mutations. Cancer Discov 2023;23:OF1-OF20.
68. Chang EH, Gonda MA, Ellis RW, Scolnick EM, Lowy DR. Human genome contains four genes homologous to transforming genes of Harvey and Kirsten murine sarcoma viruses. Proc Natl Acad Sci USA 1982;79:4848-52.
69. Buscail L, Bournet B, Cordelier P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat Rev Gastroenterol Hepatol 2020;17:153-68.
70. Jancík S, Drábek J, Radzioch D, Hajdúch M. Clinical relevance of KRAS in human cancers. J Biomed Biotechnol 2010;2010:150960.
71. Andersen JB, Spee B, Blechacz BR, et al. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology 2012;142:1021-1031.e15.
72. Martin-Serrano MA, Kepecs B, Torres-Martin M, et al. Novel microenvironment-based classification of intrahepatic cholangiocarcinoma with therapeutic implications. Gut 2023;72:736-48.
73. Momoi H, Itoh T, Nozaki Y, et al. Microsatellite instability and alternative genetic pathway in intrahepatic cholangiocarcinoma. J Hepatol 2001;35:235-44.
74. Simbolo M, Fassan M, Ruzzenente A, et al. Multigene mutational profiling of cholangiocarcinomas identifies actionable molecular subgroups. Oncotarget 2014;5:2839-52.
75. Tannapfel A, Benicke M, Katalinic A, et al. Frequency of p16(INK4A) alterations and K-ras mutations in intrahepatic cholangiocarcinoma of the liver. Gut 2000;47:721-7.
76. Xu RF, Sun JP, Zhang SR, et al. KRAS and PIK3CA but not BRAF genes are frequently mutated in Chinese cholangiocarcinoma patients. Biomed Pharmacother 2011;65:22-6.
77. Zheng Y, Qin Y, Gong W, et al. Specific genomic alterations and prognostic analysis of perihilar cholangiocarcinoma and distal cholangiocarcinoma. J Gastrointest Oncol 2021;12:2631-42.
78. Zhu AX, Borger DR, Kim Y, et al. Genomic profiling of intrahepatic cholangiocarcinoma: refining prognosis and identifying therapeutic targets. Ann Surg Oncol 2014;21:3827-34.
79. Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol 2008;9:517-31.
80. Bamford S, Dawson E, Forbes S, et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer 2004;91:355-8.
81. Stolze B, Reinhart S, Bulllinger L, Fröhling S, Scholl C. Comparative analysis of KRAS codon 12, 13, 18, 61, and 117 mutations using human MCF10A isogenic cell lines. Sci Rep 2015;5:8535.
82. Zhou SL, Xin HY, Sun RQ, et al. Association of KRAS variant subtypes with survival and recurrence in patients with surgically treated intrahepatic cholangiocarcinoma. JAMA Surg 2022;157:59-65.
83. Zou S, Li J, Zhou H, et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat Commun 2014;5:5696.
84. Yokoyama M, Ohnishi H, Ohtsuka K, et al. KRAS mutation as a potential prognostic biomarker of biliary tract cancers. Jpn Clin Med 2016;7:33-9.
85. Tian W, Hu W, Shi X, et al. Comprehensive genomic profile of cholangiocarcinomas in China. Oncol Lett 2020;19:3101-10.
86. Zhang Y, Ma Z, Li C, et al. The genomic landscape of cholangiocarcinoma reveals the disruption of post-transcriptional modifiers. Nat Commun 2022;13:3061.
87. Guo C, Liu Z, Yu Y, et al. TP53 /KRAS Co-mutations create divergent prognosis signatures in intrahepatic cholangiocarcinoma. Front Genet 2022;13:844800.
88. Chen TC, Jan YY, Yeh TS. K-ras mutation is strongly associated with perineural invasion and represents an independent prognostic factor of intrahepatic cholangiocarcinoma after hepatectomy. Ann Surg Oncol 2012;19 Suppl 3:S675-81.
89. Churi CR, Shroff R, Wang Y, et al. Mutation profiling in cholangiocarcinoma: prognostic and therapeutic implications. PLoS One 2014;9:e115383.
90. Robertson S, Hyder O, Dodson R, et al. The frequency of KRAS and BRAF mutations in intrahepatic cholangiocarcinomas and their correlation with clinical outcome. Hum Pathol 2013;44:2768-73.
91. Hong DS, Fakih MG, Strickler JH, et al. KRAS(G12C) inhibition with sotorasib in advanced solid tumors. N Engl J Med 2020;383:1207-17.
92. Nakajima EC, Drezner N, Li X, et al. FDA approval summary: sotorasib for KRAS G12C-mutated metastatic NSCLC. Clin Cancer Res 2022;28:1482-6.
93. Hallin J, Engstrom LD, Hargis L, et al. The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov 2020;10:54-71.
94. Jänne PA, Riely GJ, Gadgeel SM, et al. Adagrasib in non-small-cell lung cancer harboring a KRASG12C mutation. N Engl J Med 2022;387:120-31.
95. Hallin J, Bowcut V, Calinisan A, et al. Anti-tumor efficacy of a potent and selective non-covalent KRASG12D inhibitor. Nat Med 2022;28:2171-82.
96. Wang X, Allen S, Blake JF, et al. Identification of MRTX1133, a noncovalent, potent, and selective KRASG12D inhibitor. J Med Chem 2022;65:3123-33.
97. Awad MM, Liu S, Rybkin II, et al. Acquired resistance to KRASG12C inhibition in cancer. N Engl J Med 2021;384:2382-93.
98. Koga T, Suda K, Fujino T, et al. KRAS secondary mutations that confer acquired resistance to KRAS G12C inhibitors, sotorasib and adagrasib, and overcoming strategies: insights from
99. Zhao Y, Murciano-Goroff YR, Xue JY, et al. Diverse alterations associated with resistance to KRASG12C inhibition. Nature 2021;599:679-83.
100. Dai X, Zhang X, Yin Q, et al. Acetylation-dependent regulation of BRAF oncogenic function. Cell Rep 2022;38:110250.
101. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949-54.
102. Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010;363:809-19.
103. Goeppert B, Frauenschuh L, Renner M, et al. BRAF V600E-specific immunohistochemistry reveals low mutation rates in biliary tract cancer and restriction to intrahepatic cholangiocarcinoma. Mod Pathol 2014;27:1028-34.
104. Li W, Cui Y, Yin F, et al. BRAF mutation in Chinese biliary tract cancer patients. J Clin Oncol 2020;38:e16678.
105. Tannapfel A, Sommerer F, Benicke M, et al. Mutations of the BRAF gene in cholangiocarcinoma but not in hepatocellular carcinoma. Gut 2003;52:706-12.
106. Tomczak A, Springfeld C, Dill MT, et al. Precision oncology for intrahepatic cholangiocarcinoma in clinical practice. Br J Cancer 2022;127:1701-8.
107. Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373:726-36.
108. Subbiah V, Lassen U, Élez E, et al. Dabrafenib plus trametinib in patients with BRAFV600E-mutated biliary tract cancer (ROAR): a phase 2, open-label, single-arm, multicentre basket trial. Lancet Oncol 2020;21:1234-43.
109. Wen PY, Stein A, van den Bent M, et al. Dabrafenib plus trametinib in patients with BRAFV600E-mutant low-grade and high-grade glioma (ROAR): a multicentre, open-label, single-arm, phase 2, basket trial. Lancet Oncol 2022;23:53-64.
110. Salama AKS, Li S, Macrae ER, et al. Dabrafenib and trametinib in patients with tumors with BRAFV600E mutations: results of the NCI-MATCH trial subprotocol H. J Clin Oncol 2020;38:3895-904.
111. Zhang H, Berezov A, Wang Q, et al. ErbB receptors: from oncogenes to targeted cancer therapies. J Clin Invest 2007;117:2051-8.
112. Galdy S, Lamarca A, McNamara MG, et al. HER2/HER3 pathway in biliary tract malignancies; systematic review and meta-analysis: a potential therapeutic target? Cancer Metastasis Rev 2017;36:141-57.
113. Li M, Zhang Z, Li X, et al. Whole-exome and targeted gene sequencing of gallbladder carcinoma identifies recurrent mutations in the ErbB pathway. Nat Genet 2014;46:872-6.
114. Moeini A, Sia D, Bardeesy N, Mazzaferro V, Llovet JM. Molecular pathogenesis and targeted therapies for intrahepatic cholangiocarcinoma. Clin Cancer Res 2016;22:291-300.
115. Nakazawa K, Dobashi Y, Suzuki S, Fujii H, Takeda Y, Ooi A. Amplification and overexpression of c-erbB-2, epidermal growth factor receptor, and c-met in biliary tract cancers. J Pathol 2005;206:356-65.
116. Sirica AE. Role of ErbB family receptor tyrosine kinases in intrahepatic cholangiocarcinoma. World J Gastroenterol 2008;14:7033-58.
117. Terada T, Nakanuma Y, Sirica AE. Immunohistochemical demonstration of MET overexpression in human intrahepatic cholangiocarcinoma and in hepatolithiasis. Hum Pathol 1998;29:175-80.
118. Ukita Y, Kato M, Terada T. Gene amplification and mRNA and protein overexpression of c-erbB-2 (HER-2/neu) in human intrahepatic cholangiocarcinoma as detected by fluorescence in situ hybridization,
119. Yoshikawa D, Ojima H, Iwasaki M, et al. Clinicopathological and prognostic significance of EGFR, VEGF, and HER2 expression in cholangiocarcinoma. Br J Cancer 2008;98:418-25.
120. Junttila TT, Akita RW, Parsons K, et al. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell 2009;15:429-40.
121. Lamarca A, Barriuso J, McNamara MG, Valle JW. Molecular targeted therapies: Ready for "prime time" in biliary tract cancer. J Hepatol 2020;73:170-85.
122. Tsurutani J, Iwata H, Krop I, et al. Targeting HER2 with trastuzumab deruxtecan: a dose-expansion, phase I study in multiple advanced solid tumors. Cancer Discov 2020;10:688-701.
123. Lee J, Park SH, Chang HM, et al. Gemcitabine and oxaliplatin with or without erlotinib in advanced biliary-tract cancer: a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 2012;13:181-8.
124. Pant S, Ducreux M, Harding JJ, et al. A phase IIb, open-label, single-arm study of zanidatamab (ZW25) monotherapy in subjects with advanced or metastatic HER2-amplified biliary tract cancers. J Clin Oncol 2021;39:TPS352.
125. Elimova E, Ajani JA, Burris Iii HA, et al. Zanidatamab + chemotherapy as first-line treatment for HER2-expressing metastatic gastroesophageal adenocarcinoma (mGEA). J Clin Oncol 2023;41:347.
126. Ohba A, Morizane C, Ueno M, et al. Multicenter phase II trial of trastuzumab deruxtecan for HER2-positive unresectable or recurrent biliary tract cancer: HERB trial. Future Oncol 2022;18:2351-60.
127. Ohba A, Morizane C, Kawamoto Y, et al. Circulating tumor DNA (ctDNA) analyses in patients with HER2-positive biliary tract cancer (BTC) treated with trastuzumab deruxtecan (T-DXd): exploratory results from the HERB trial. J Clin Oncol 2023;41:4097.
128. Amatu A, Sartore-Bianchi A, Bencardino K, Pizzutilo EG, Tosi F, Siena S. Tropomyosin receptor kinase (TRK) biology and the role of NTRK gene fusions in cancer. Ann Oncol 2019;30:viii5-viii15.
129. Drilon A, Laetsch TW, Kummar S, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med 2018;378:731-9.
130. Hong DS, DuBois SG, Kummar S, et al. Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol 2020;21:531-40.
131. Gu TL, Deng X, Huang F, et al. Survey of tyrosine kinase signaling reveals ROS kinase fusions in human cholangiocarcinoma. PLoS One 2011;6:e15640.
132. Doebele RC, Drilon A, Paz-Ares L, et al. trial investigators. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1-2 trials. Lancet Oncol 2020;21:271-82.
133. Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993;363:558-61.
134. Blake C, Tsao JL, Wu A, Shibata D. Stepwise deletions of polyA sequences in mismatch repair-deficient colorectal cancers. Am J Pathol 2001;158:1867-70.
135. Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon. Science 1993;260:816-9.
136. Naganuma A, Sakuda T, Murakami T, et al. Microsatellite instability-high intrahepatic cholangiocarcinoma with portal vein tumor thrombosis successfully treated with pembrolizumab. Intern Med 2020;59:2261-7.
137. Marabelle A, Le DT, Ascierto PA, et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: results from the phase II KEYNOTE-158 study. J Clin Oncol 2020;38:1-10.
138. Bochar DA, Wang L, Beniya H, et al. BRCA1 is associated with a human SWI/SNF-related complex: linking chromatin remodeling to breast cancer. Cell 2000;102:257-65.
139. Hartman AR, Ford JM. BRCA1 induces DNA damage recognition factors and enhances nucleotide excision repair. Nat Genet 2002;32:180-4.
140. Hughes-Davies L, Huntsman D, Ruas M, et al. EMSY links the BRCA2 pathway to sporadic breast and ovarian cancer. Cell 2003;115:523-35.
141. Paull TT, Cortez D, Bowers B, Elledge SJ, Gellert M. Direct DNA binding by Brca1. Proc Natl Acad Sci USA 2001;98:6086-91.
142. Xu B, Kim St, Kastan MB. Involvement of Brca1 in S-phase and G(2)-phase checkpoints after ionizing irradiation. Mol Cell Biol 2001;21:3445-50.
143. Golan T, Raitses-Gurevich M, Kelley RK, et al. Overall survival and clinical characteristics of BRCA-Associated cholangiocarcinoma: a multicenter retrospective study. Oncologist 2017;22:804-10.
144. Montal R, Sia D, Montironi C, et al. Molecular classification and therapeutic targets in extrahepatic cholangiocarcinoma. J Hepatol 2020;73:315-27.
145. Spizzo G, Puccini A, Xiu J, et al. Molecular profile of BRCA-mutated biliary tract cancers. ESMO Open 2020;5:e000682.
146. Cheng Y, Zhang J, Qin SK, Hua HQ. Treatment with olaparib monotherapy for BRCA2-mutated refractory intrahepatic cholangiocarcinoma: a case report. Onco Targets Ther 2018;11:5957-62.
147. Li W, Ma Z, Fu X, et al. Olaparib effectively treats local recurrence of extrahepatic cholangiocarcinoma in a patient harboring a BRCA2-inactivating mutation: a case report. Ann Transl Med 2021;9:1487.
148. Xiong F, Gong J, Wang Q. Olaparib and pembrolizumab treatment for BRCA1-mutated and PD-L1-positive intrahepatic cholangiocarcinoma recurrence and metastasis: a case report. Onco Targets Ther 2020;13:6385-91.
149. Aubrey BJ, Strasser A, Kelly GL. Tumor-suppressor functions of the TP53 pathway. Cold Spring Harb Perspect Med 2016;6:a026062.
150. Sicklick JK, Fanta PT, Shimabukuro K, Kurzrock R. Genomics of gallbladder cancer: the case for biomarker-driven clinical trial design. Cancer Metastasis Rev 2016;35:263-75.
151. Chirravuri-Venkata R, Dam V, Nimmakayala RK, et al. MUC16 and TP53 family co-regulate tumor-stromal heterogeneity in pancreatic adenocarcinoma. Front Oncol 2023;13:1073820.
152. Hao F, Gu L, Zhong D. TP53 mutation mapping in advanced non-small cell lung cancer: a real-world retrospective cohort study. Curr Oncol 2022;29:7411-9.
153. Mody K, Jain P, El-Refai SM, et al. Clinical, genomic, and transcriptomic data profiling of biliary tract cancer reveals subtype-specific immune signatures. JCO Precis Oncol 2022;6:e2100510.
154. Jiao Y, Pawlik TM, Anders RA, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat Genet 2013;45:1470-3.
155. Sasaki M, Nitta T, Sato Y, Nakanuma Y. Loss of ARID1A expression presents a novel pathway of carcinogenesis in biliary carcinomas. Am J Clin Pathol 2016;145:815-25.
156. Zhao S, Xu Y, Wu W, et al. ARID1A variations in cholangiocarcinoma: clinical significances and molecular mechanisms. Front Oncol 2021;11:693295.
157. Rizzo A, Carloni R, Ricci AD, et al. Molecular profile and prognostic value of BAP1 mutations in intrahepatic cholangiocarcinoma: a genomic database analysis. J Pers Med 2022;12:1247.
158. Chen XX, Yin Y, Cheng JW, et al. BAP1 acts as a tumor suppressor in intrahepatic cholangiocarcinoma by modulating the ERK1/2 and JNK/c-Jun pathways. Cell Death Dis 2018;9:1036.
159. Han A, Purwin TJ, Aplin AE. Roles of the BAP1 tumor suppressor in cell metabolism. Cancer Res 2021;81:2807-14.
160. Yan XQ, Zhang W, Zhang BX, Liang HF, Zhang WG, Chen XP. Inactivation of Smad4 is a prognostic factor in intrahepatic cholangiocarcinoma. Chin Med J 2013;126:3039-43.
161. Zhao M, Mishra L, Deng CX. The role of TGF-β/SMAD4 signaling in cancer. Int J Biol Sci 2018;14:111-23.
162. Nakamura H, Arai Y, Totoki Y, et al. Genomic spectra of biliary tract cancer. Nat Genet 2015;47:1003-10.
163. Arsenijevic T, Coulonval K, Raspé E, Demols A, Roger PP, Van Laethem JL. CDK4/6 inhibitors in pancreatobiliary cancers: opportunities and challenges. Cancers 2023;15:968.
164. Song H, Huang Y, Jiang X. Mutation spectrum associated with metastasis of advanced cholangiocarcinoma. J Int Med Res 2022;50:3000605221102080.
165. Seo J, Seong D, Lee SR, Oh DB, Song J. Post-translational regulation of ARF: perspective in cancer. Biomolecules 2020;10:1143.
166. Tannapfel A, Sommerer F, Benicke M, et al. Genetic and epigenetic alterations of the INK4a-ARF pathway in cholangiocarcinoma. J Pathol 2002;197:624-31.
167. Cho SY, Hwang H, Kim YH, et al. Refining classification of cholangiocarcinoma subtypes via proteogenomic integration reveals new therapeutic prospects. Gastroenterology 2023;164:1293-309.
168. Silvestri M, Nghia Vu T, Nichetti F, et al. Comprehensive transcriptomic analysis to identify biological and clinical differences in cholangiocarcinoma. Cancer Med 2023;12:10156-68.
169. Bramel ER, Sia D. Novel insights into molecular and immune subtypes of biliary tract cancers. Adv Cancer Res 2022;156:167-99.
170. Bagante F, Ruzzenente A, Conci S, et al. Patterns of gene mutations in bile duct cancers: is it time to overcome the anatomical classification? HPB 2019;21:1648-55.
171. Jusakul A, Cutcutache I, Yong CH, et al. Whole-genome and epigenomic landscapes of etiologically distinct subtypes of cholangiocarcinoma. Cancer Discov 2017;7:1116-35.
172. Nepal C, O'Rourke CJ, Oliveira DVNP, et al. Genomic perturbations reveal distinct regulatory networks in intrahepatic cholangiocarcinoma. Hepatology 2018;68:949-63.
173. Wang XY, Zhu WW, Wang Z, et al. Driver mutations of intrahepatic cholangiocarcinoma shape clinically relevant genomic clusters with distinct molecular features and therapeutic vulnerabilities. Theranostics 2022;12:260-76.
174. Job S, Rapoud D, Dos Santos A, et al. Identification of four immune subtypes characterized by distinct composition and functions of tumor microenvironment in intrahepatic cholangiocarcinoma. Hepatology 2020;72:965-81.
175. Lin J, Dai Y, Sang C, et al. Multimodule characterization of immune subgroups in intrahepatic cholangiocarcinoma reveals distinct therapeutic vulnerabilities. J Immunother Cancer 2022;10:e004892.
176. Kortlever RM, Sodir NM, Wilson CH, et al. Myc cooperates with ras by programming inflammation and immune suppression. Cell 2017;171:1301-1315.e14.
177. Liao W, Overman MJ, Boutin AT, et al. KRAS-IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer Cell 2019;35:559-572.e7.
178. Wellenstein MD, de Visser KE. Cancer-cell-intrinsic mechanisms shaping the tumor immune landscape. Immunity 2018;48:399-416.
179. Charoentong P, Finotello F, Angelova M, et al. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep 2017;18:248-62.
180. Lin Y, Peng L, Dong L, et al. Geospatial immune heterogeneity reflects the diverse tumor-immune interactions in intrahepatic cholangiocarcinoma. Cancer Discov 2022;12:2350-71.
181. Wu MJ, Shi L, Dubrot J, et al. Mutant IDH inhibits IFNγ-TET2 signaling to promote immunoevasion and tumor maintenance in cholangiocarcinoma. Cancer Discov 2022;12:812-35.
182. Benson AB, D'Angelica MI, Abbott DE, et al. Hepatobiliary cancers, version 2.2021, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw 2021;19:541-65.
183. Vogel A, Bridgewater J, Edeline J, et al. ESMO Guidelines Committee. Biliary tract cancer: ESMO clinical practice guideline for diagnosis, treatment and follow-up. Ann Oncol 2023;34:127-40.
184. Bartley AN, Washington MK, Ventura CB, et al. HER2 testing and clinical decision making in gastroesophageal adenocarcinoma: guideline from the college of american pathologists, american society for clinical pathology, and american society of clinical oncology. Am J Clin Pathol 2016;146:647-69.
185. Cheah PL, Li J, Looi LM, et al. Screening for microsatellite instability in colorectal carcinoma: Practical utility of immunohistochemistry and PCR with fragment analysis in a diagnostic histopathology setting. Malays J Pathol 2019;41:91-100.
186. Saborowski A, Lehmann U, Vogel A. FGFR inhibitors in cholangiocarcinoma: what's now and what's next? Ther Adv Med Oncol 2020;12:1758835920953293.
187. Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn 2015;17:251-64.
188. Tsongalis GJ, Peterson JD, de Abreu FB, et al. Routine use of the ion torrent AmpliSeq™ cancer hotspot panel for identification of clinically actionable somatic mutations. Clin Chem Lab Med 2014;52:707-14.
189. Ettrich TJ, Schwerdel D, Dolnik A, et al. Genotyping of circulating tumor DNA in cholangiocarcinoma reveals diagnostic and prognostic information. Sci Rep 2019;9:13261.
190. Ma L, Wang L, Khatib SA, et al. Single-cell atlas of tumor cell evolution in response to therapy in hepatocellular carcinoma and intrahepatic cholangiocarcinoma. J Hepatol 2021;75:1397-408.
191. Ma L, Hernandez MO, Zhao Y, et al. Tumor cell biodiversity drives microenvironmental reprogramming in liver cancer. Cancer Cell 2019;36:418-430.e6.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Young SE, Sritharan R, Sia D. Genomic alterations in intrahepatic cholangiocarcinoma. Hepatoma Res 2023;9:34. http://dx.doi.org/10.20517/2394-5079.2023.37
AMA Style
Young SE, Sritharan R, Sia D. Genomic alterations in intrahepatic cholangiocarcinoma. Hepatoma Research. 2023; 9: 34. http://dx.doi.org/10.20517/2394-5079.2023.37
Chicago/Turabian Style
Young, Sara E., Ramja Sritharan, Daniela Sia. 2023. "Genomic alterations in intrahepatic cholangiocarcinoma" Hepatoma Research. 9: 34. http://dx.doi.org/10.20517/2394-5079.2023.37
ACS Style
Young, SE.; Sritharan R.; Sia D. Genomic alterations in intrahepatic cholangiocarcinoma. Hepatoma. Res. 2023, 9, 34. http://dx.doi.org/10.20517/2394-5079.2023.37
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 4 clicks
Cite This Article 4 clicks

 Like This Article 3
likes
Like This Article 3
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.