
Genomic alterations and targeted therapies in extrahepatic cholangiocarcinoma
 0
0
Abstract
The global morbimortality of biliary tract cancer (BTC) is steadily increasing and accounts for ~10% of all primary liver cancer. Distinct anatomical locations of BTC have singularities in their etiopathogenesis, which are translated into differences in their molecular fingerprints and the associated therapeutic approaches. Extrahepatic cholangiocarcinoma (eCCA), arising in the large and distal bile ducts, presents recurrent activating mutations of KRAS and loss-of-function alterations in TP53, SMAD4, and CDKN2A/B. Despite being highly prevalent, no targeted therapies are yet available for these oncogenic drivers. ERBB2 mutations and amplifications, on the other hand, are the most recurrent actionable alterations for eCCA, with several clinical trials aiming to provide benefits in biomarker-enriched populations. In addition, integrative multi-omics analysis of eCCA has allowed the identification of novel molecular classes of this disease that could be therapeutically exploited. Beyond that, the highly immunosuppressive tumor microenvironment of eCCA has prevented until now the success of immune checkpoint inhibitors, recently approved in combination with cytotoxic chemotherapy. Further characterization of eCCA at the molecular level would potentially foster treating patients based on a precision oncology approach in order to increase the clinical outcomes for this challenging disease.
Keywords
INTRODUCTION
Globally, hepatobiliary cancer is the third most common cause of cancer-related death after lung and esophagogastric cancer and ranks sixth in terms of incident cases (more than 1 million new cases each year)[1]. Biliary tract cancer (BTC) is the second most common liver malignancy after hepatocellular carcinoma (HCC)[2]. The estimated worldwide incidence of BTC, including cholangiocarcinoma (CCA) and gallbladder cancer (GBC), is 184,000 new cases annually[1]. Of note, the global mortality rate for CCA has increased during recent decades[3]. CCAs are divided into different subtypes depending on their anatomical origin: intrahepatic (iCCA) or extrahepatic (eCCA), with the second-order bile ducts acting as the separation point[4,5]. In addition, eCCA has been divided into perihilar (pCCA) and distal (dCCA) at the level of the cystic duct[4,5].These subtypes differ in their etiopathogenesis as well as in their clinical management.
The most common clinical manifestation of eCCA is the presence of jaundice as a result of a bile duct stricture[6]. Pathological diagnosis should be obtained before treatment, preferably by endoscopic retrograde cholangiopancreatography-guided biopsies[7]. The 8th edition AJCC/UICC TNM classification established a different staging system for pCCA and dCCA, but despite providing prognostic information, it cannot allow evaluation of the local resectability of the tumor. In this regard, several alternative classifications have been proposed, such as the one described by Bismuth and Corlette for classifying bile duct involvement in pCCA[7].
A global treatment algorithm for BTC is usually presented[7] although the clinical strategy varies for each anatomical subtype. At early stages of eCCA, the only potentially curative option is surgical resection with lymphadenectomy. Neoadjuvant chemoradiotherapy followed by liver transplantation in locally unresectable pCCA was proposed by the Mayo Clinic[8]; however, the level of evidence supporting this approach is insufficient to establish it as a standard of care. The recurrence rate following surgery is high, with 5-year overall survival in the range of 11%-41% for pCCA and 27%-37% for dCCA[6]. The role of adjuvant chemotherapy or chemoradiation therapy in patients with resected eCCA is poorly defined[7], but adjuvant chemotherapy with capecitabine should be considered[9]. For patients who are not candidates for curative treatment, systemic therapy is indicated to relieve symptoms and prolong life. Cytotoxic chemotherapy with cisplatin and gemcitabine is still the cornerstone of treatment[10]. However, the addition of durvalumab (anti-PDL1) to this treatment strategy has recently improved clinical outcomes and may become the new standard for first-line treatment[11].
Molecular profiling of BTC identified FGFR2 fusions and IDH1/2 mutations as candidate targets[12]. Recently published trials confirm the benefit of inhibiting these signaling pathways in patients with tumors harboring these genetic alterations[13,14]. Unfortunately, these actionable genetic alterations are almost exclusively seen in iCCA. Regarding eCCA, no molecular targeted therapies have been approved for its treatment. Therefore, studies are necessary to elucidate molecular pathways driving eCCA progression that would facilitate a precision oncology approach for this disease. The present review aims to provide a comprehensive overview of the current molecular knowledge of eCCA, describing its singularities in terms of molecular pathogenesis, genetic aberrations, molecular classifications, and features of the tumor microenvironment.
RISK FACTORS AND MOLECULAR PATHOGENESIS
BTC rarely occurs before the fourth decade of life and men are at slightly higher risk than women[15]. Long established risk factors for eCCA, such as hepatobiliary flukes, primary sclerosing cholangitis and hepatolithiasis, are associated with chronic biliary inflammation and increased cellular turnover[16]. On the other hand, cirrhosis due to hepatitis B and C virus chronic infection is a major risk factor for iCCA[17]. Nevertheless, in the vast majority of BTC cases, the disease is sporadic and known risk factors are not present[18]. The different geographical distribution of risk factors explains the observed heterogeneous incidence of BTC worldwide.
Liver flukes (Opistorchis viverrine and Clonorchis sinensis), endemic in Asia, can infect humans by the consumption of raw or undercooked fish and the subsequent deposition of eggs in the biliary tract[19]. The parasite persists over the years and progressively accumulates in the biliary system causing mechanical damage and an inflammatory response leading to eCCA[20]. This etiology explains the highest incidence of BTC in Northeast Thailand (> 80 per 100,000 population)[4]. In most Western countries, BTC is a low prevalent cancer (incidence < 6 cases per 100,000 people)[6]. In this scenario, primary sclerosing cholangitis is the most common biliary condition leading to eCCA, produced through peribiliary gland cell proliferation, mucinous metaplasia, and dysplasia to cancer progression within bile ducts[21].
Cholangiocarcinogenesis is a multifactorial process[15]. A variety of cytokines, growth factors, tyrosine kinases, and bile acids can contribute to the alterations in proliferation, apoptosis, senescence, and cell-cycle regulation required for carcinogenesis[22]. Large bile duct eCCA has been proposed to arise from the biliary columnar epithelium and peribiliary glands[6,23], which are also implicated in the origin of precursor lesions (such as intraductal papillary neoplasm)[24]. Tumoral cells gradually adopt invasive phenotypes by changing to a mesenchymal-like phenotype, which increases their migratory and invasion capabilities, and eventually deposit at distant sites[25]. Several signaling pathways are dysregulated in BTC[25], including the inflammation-related IL-6-JAK-STAT3[26], MAPK-ERK[27], PI3K-AKT-mTOR[28], Hedgehog[29], Wnt[30], Notch[31] and others.
STRUCTURAL GENETIC ALTERATIONS
Several studies have identified recurrent genetic alterations in BTC by next-generation sequencing[32]. The median numbers of non-silent somatic mutations in iCCA, eCCA, and GBC are 39, 35, and 64, respectively[33].
In terms of chromosomal aberrations in CCA, broad gains of 5q, 7p, 8q, 13q, 17q, and 20q and losses of 3p, 6q, 9p, and 17p have been reported[34,35]. These aberrations also underlie high-level focal amplification of oncogenes such as ERBB2 or deletion of tumor suppressor genes such as CDKN2A and TP53. The proportion of the most prevalent aberrations in eCCA is provided in Table 1[33,36-38]. These genes converge into four main oncogenic signaling pathways[38]: RTK-RAS-PI3K (altered in 53% of tumors), TP53-RB (47%), histone modification (22%), and transforming growth factor-β (TGFβ, 18%).
Recurrent structural genetic alterations in extrahepatic cholangiocarcinoma.
| Gene | Alteration type | Percentage (range) |
| TP53 | Mut/Del | 26%-45% |
| KRAS | Mut | 10%-43% |
| CDKN2A | Del | 5%-28% |
| SMAD4 | Mut | 10%-16% |
| ARID1A | Mut | 7%-15% |
| CDKN2B | Del | 15% |
| APC | Mut | 4%-11% |
| ELF3 | Mut | 7%-10% |
| EPHA2 | Mut | 10% |
| ERBB2 | Mut/Amp | 4%-9% |
| ARID2 | Mut | 5%-8% |
| PTEN | Mut | 5%-7% |
| ARID1B | Mut | 4%-7% |
| STK11 | Mut | 2%-7% |
| SF3B1 | Mut | 6% |
| YEATS4 | Amp | 6% |
| NF1 | Mut | 5%-6% |
| ATM | Mut | 5%-6% |
| MYC | Amp | 4%-6% |
| ACVR2A | Mut | 3%-6% |
| FGF19 | Amp | 5% |
| CCND1 | Amp | 5% |
| GNAS | Mut | 5% |
| MDM2 | Amp | 5% |
| KMT2D | Mut | 5% |
| RNF43 | Mut | 5% |
| PIK3CA | Mut | 4%-5% |
| FBXW7 | Mut | 4%-5% |
| CCNE1 | Amp | 3%-5% |
| NRAS | Mut | 1%-5% |
Activating mutations of KRAS, particularly on the G12D hotspot[38], and loss-of-function mutations in TP53, SMAD4 and CDKN2A/B are the most prevalent throughout different anatomical locations. In addition, mutations in several chromatin-remodeling genes, including ARID1A, have also been frequently observed in BTC[39]. The repertoire of some less prevalent alterations, however, varies across the different anatomical BTC subtypes, which is in line with the previously mentioned differences in etiopathogenesis.
FGFR2 translocations have been uncovered in approximately 20% of iCCA[12,33,40,41]. Multiple fusion partners to a consistent breakpoint within the FGFR2 gene have been reported. The mechanism by which FGFR2 fusions drive oncogenesis has been associated with the ligand-independent constitutive activation of the fusion protein and the subsequent canonical downstream signaling. Furthermore, mutations in IDH1 and IDH2 are found to be exclusively in around 14% of iCCA[12,33,42]. These IDH1/2 mutations are associated with hypermethylation of CpG shores, followed by global deregulation of transcriptional programs regulating differentiation. Indeed, they have been proposed to promote iCCA by blocking hepatocyte differentiation through the deregulation of HNF4α[43].
ERBB2 mutations and amplifications have been described in BTC, with a higher incidence in GBC and eCCA[44,45]. Activating alterations of this gene leads to downstream oncogenic pathway signaling, including the MAPK. Of note, ERBB2 structural genetic alterations are more prevalent in tumors with papillary histology[38]. On the other hand, novel gene fusions involving PRKACA/B, which encode catalytic subunits of protein kinase A, have been detected only in eCCA, albeit in a low proportion[33].
The same technological advances that have enabled the construction of a comprehensive catalog of cancer genes are becoming increasingly available for diagnostic purposes with reasonable costs and timeframes. Thus, clinicians will need to distinguish genomic data that could effectively be targeted with matched drugs based on available evidence in order to facilitate the implementation of precision medicine[46,47].
MOLECULAR CLASSIFICATIONS
Studies conducted in the setting of The Cancer Genome Atlas (TCGA)[42]and the International Cancer Genome Consortium (ICGC)[33]consortiums analyzed together all types of BTC in order to propose an integrative clustering of these tumors. However, eCCA was underrepresented in these projects (4 and 40 patients in the TCGA[42] and ICGC[33], respectively). The proposed molecular classifications highlighted the critical role of anatomical location in the biological landscape of this disease[33]. Molecular landscapes also differed by etiology (liver flukes)[36], underscoring how BTC subtypes may arise through different extrinsic and intrinsic carcinogenic processes. According to these observations, a molecular classification for each specific subtype of BTC seems a better approach in order to capture the biological peculiarities of each disease.
Molecular profiling of iCCA as a single entity has allowed the discovery of two distinct transcriptome-based classes[48-50]: an Inflammation class with predominant activation of STAT3 and overexpression of cytokines and a Proliferation class with activation of classic oncogenic pathways (including RAS and MET) and specific copy number alterations that correlate with worse outcome[48]. Furthermore, an IDH mutant-enriched subtype of iCCA has been uncovered, with distinct molecular features including low expression of chromatin modifiers, elevated expression of mitochondrial genes, and increased mitochondrial DNA copy number[42].
A comprehensive genomic analysis of 189 eCCA from Western countries proposed the existence of four transcriptome-based well-defined molecular classes [Figure 1][38]: the Metabolic class, determined by disruption of bile acid and fatty acid metabolism, which may favor tumor progression and the acquisition of a HNF4A-driven hepatocyte-like phenotype; the Proliferation class, with activation of the cell cycle, mTOR and ERBB2 as key features; the Mesenchymal class, defined by EMT, TGFβ signaling activation and a desmoplastic reaction observed on pathological analysis, resulting into poor clinical outcomes; and finally, the Immune class, with a higher lymphocytic infiltration and increased immune checkpoint expression.
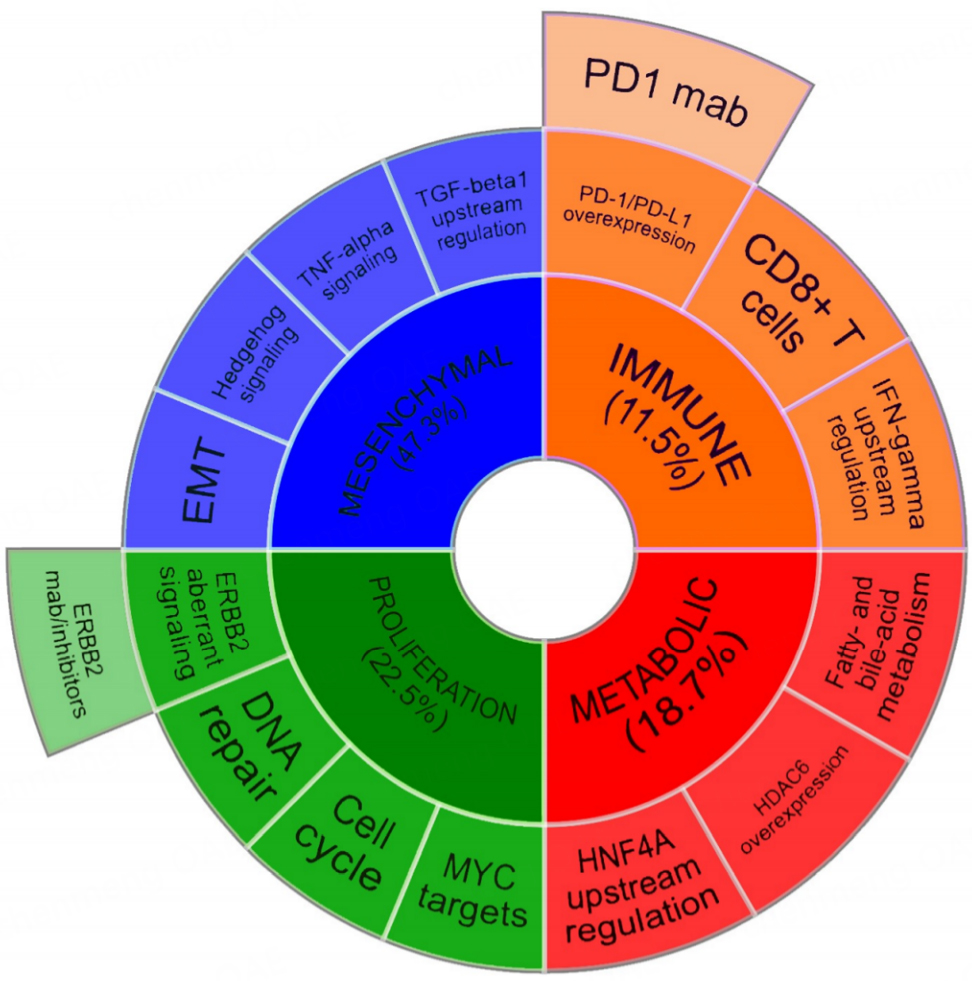
Figure 1. Molecular classification of extrahepatic cholangiocarcinoma.Tumor and microenvironment molecular features of transcriptome-based Metabolic, Proliferation, Mesenchymal and Immune classes discovered in eCCA[38]. Candidate targeted therapies with a potential benefit in Proliferation and Immune classes are proposed. Figure adapted from Montal et al. J. Hepatol 2020[38].
Tumor and microenvironment molecular features of transcriptome-based Metabolic, Proliferation, Mesenchymal and Immune classes discovered in eCCA[38]. Candidate targeted therapies with a potential benefit in Proliferation and Immune classes are proposed. Figure adapted from Montal et al. J. Hepatol 2020[38].
This molecular classification of eCCA aligns well with the known molecular landscape of gastrointestinal tract tumors[51], with some overlaps that suggest recurring oncogenic pathways among different tissues of origin. Indeed, the four molecular classes were subsequently validated in an external cohort[33] using a gene-expression eCCA classifier[38], indicating cross-validity of the model independently of geographic region.
TUMOR MICROENVIRONMENT
BTC is characterized by a dense and reactive desmoplastic stroma containing mainly cancer-associated fibroblasts (CAFs), endothelial cells, and a complex group of inflammatory cells, including macrophages, neutrophils, natural killer cells, and T cells[24].
Although the precise origin of CAFs is still not clear, they are probably derived from tissue-resident portal fibroblasts or hepatic stellate cells[52]. CAFs promote tumor progression via reciprocal communication with cancer cells and stromal cells. They secrete molecules such as PDGF and Rho GTPases that promote cancer progression by enhancing proliferation, survival, and angiogenesis[52-55]. Indeed, a stromal signature with up-regulated genes related to cell cycle, extracellular matrix, and TGFβ pathways was found to be associated with poor CCA prognosis[56]. Likewise, the previously mentioned and predominant Mesenchymal class of eCCA[38] presented overexpression of periostin, which is produced by activated CAFs and associated with the promotion of cell invasion[52].
Tumor-associated macrophages (TAMs), originating from circulating monocytes, are the most representative infiltrating immune cells of the BTC stromal compartment. They produce several molecules with well-known tumorigenic effects, such as matrix metalloproteinases, interleukins, VEGFA, TNF, TGFβ and Wnt ligands, which establishes a crucial crosstalk with the rest of the tumor microenvironment to create an immunosuppressive milieu[6].
The emerging therapeutic role of immune checkpoint inhibitors (ICIs) in cancer has increased the interest in analyzing the T cell immune infiltration of tumors. Immunohistochemical analyses have demonstrated a predominant presence of CD8 + T cells within the tumor and CD4 + T cells in invasive margin[57]. A small subset of eCCA, included in the Immune class[38], displayed marked lymphocytic infiltration. However, this T cell tumor infiltration was dysfunctional, which may explain another mechanism of tumor immune evasion. Indeed, interferon IFN-γ was a key regulator of this class, which has been proposed to predict clinical responses to ICIs[58]. PDL1, a commonly used biomarker for predicting response to ICIs, was expressed in 57% of tumors from the Immune class in comparison to 28% in the rest of the molecular classes of eCCA. Overall, there are still a lot of questions to be answered about the cellular and molecular mechanisms by which the anti-tumor immune response is triggered and maintained[59].
ACTIONABLE MOLECULAR ALTERATIONS AND TARGETED THERAPIES
Molecular profiling of BTC has characterized the genomic landscape of this disease and has proposed candidate targets for drug development [Figure 2][32]. Targeted therapies were initially explored in BTC for all comers with disappointing results. Phase II trials assessing everolimus (mTOR inhibitor)[60], selumetinib[61] (MEK inhibitor) and erlotinib[62] (EGFR inhibitor) showed objective responses in just 5-10% of patients, indicating the need for developing clinical trials with a biomarker-enrichment design. Subsequent clinical trials have improved patient selection based on molecular features as well as anatomical location. Actionable genomic alterations are less common in eCCA than in iCCA, with only ~25% of tumors having an associated potential targeted therapy[38]. In Table 2, there is a list of ongoing phase II-III clinical trials assessing targeted therapies for patients with eCCA.

Figure 2. Molecular targeted therapies with ongoing clinical trials for extrahepatic cholangiocarcinoma.Targeted therapies identified in Table 2 are represented according to their signaling pathways.
Ongoing trials of targeted therapies for extrahepatic cholangiocarcinoma.
| Drug | Target | Treatment setting | Biomarker | Combination therapies | NCT reference | |
| Immune checkpoint inhibitors | Camrelizumab | PD1 | Locally advanced | Radiotherapy | NCT03898895 | |
| Durvalumab | PDL1 | Neoadjuvant, Adjuvant, Advanced | Cisplatin/Gemcitabine, Tremelimumab/Capecitabine, Tremelimumab/Radiotherapy, Olaparib, Ceralasertib, TACE/Bevacizumab/Tremelimumab | NCT03482102, NCT05239169, NCT04308174, NCT05222971, NCT04298008, NCT04298021, NCT03937830 | ||
| Envafolimab | PDL1 | Advanced | Gemcitabine/Oxaliplatin | NCT03478488 | ||
| Nivolumab | PD1 | Advanced | DKN-01, Rucaparib | NCT04057365, NCT03639935 | ||
| Pembrolizumab | PD1 | Advanced | PDL1 CPS > 1%, MSI-H | SMT-NK injection, Olaparib, Lenvatinib | NCT05429697, NCT04306367, NCT03895970, NCT04550624 | |
| Tislelizumab | PD1 | Adjuvant, Locally advacned, Advanced | Capecitabine/Lenvatinib, Lenvatinib/Gemcitabine/Oxaliplatin, Lenvatinib/Cisplatin/Gemcitabine, Sitravatinib, Radiotherapy, Levnatinib/Oxaliplatin/Capecitabine | NCT05532059, NCT04727996, NCT05254847, NCT05156788, NCT04866836, NCT05291052 | ||
| Toripalimab | PD1 | Advanced | Gemcitabine/S1, Lenvatinib, TACE, HAIC, Gemcitabine/Oxaliplatin, Axitinib | NCT03796429, NCT04211168, NCT05448183, NCT04217954, NCT04191343, NCT04010071 | ||
| Tremelimumab | CTLA4 | Adjuvant, Advanced | Durvalumab/Capecitabine, Durvalumab/Radiotherapy, Durvalumab/Bevacizumab/TACE | NCT03482102, NCT05239169, NCT03937830 | ||
| TQB2450 | PDL1 | Advanced | Antolinib | NCT04809142 | ||
| Sintilimab | PD1 | Advanced | Bevacizumab/Gemcitabine/Oxaliplatin | NCT04984980 | ||
| XmAb20717 | PD1/CTLA4 | Advanced | NCT05297903 | |||
| VEGFR/FGFR signaling | Anlotinib | VEGFR/FGFR | Advanced | TQB2450 | NCT04809142 | |
| Apatinib | VEGFR | Advanced | NCT03427242 | |||
| Axitinib | VEGFR | Advanced | Toripalimab | NCT04010071 | ||
| Bevacizumab | VEGFR | Advanced | Sintilimab/Gemcitabine/Oxaliplatin, Durvalumab//Tremelimumab/TACE | NCT04984980, NCT03937830 | ||
| Infigratinib | FGFR | Advanced | FGFR2 fusion | NCT03773302 | ||
| Lenvatinib | VEGFR/FGFR | Adjuvant, Locally advanced, Advanced | Capecitabine/Tislelizumab, Tislelizumab/Gemcitabine/Oxaliplatin, Paclitaxel, Toripalimab, Tislelizumab/Cisplatin/Gemcitabine, Pembrolizumab, Tislelizumab/Oxaliplatin/Capecitabine | NCT05170438, NCT04211168, NCT05532059, NCT05254847, NCT05509478, NCT05156788, NCT03895970, NCT05291052, NCT04550624 | ||
| Pemigatinib | FGFR | Advanced | FGFR2 fusion | NCT03656536 | ||
| Sitravatinib | VEGFR | Tislelizumab | NCT04727996 | |||
| Tasurgratinib | FGFR | Advanced | FGFR2 fusion | NCT04238715 | ||
| TT-00420 | VEGFR/FGFR | Advanced | FGFR2 alterations | NCT04919642 | ||
| MAPK/ERK signaling | Erlotinib | EGFR | Advanced | Pemetrexed | NCT03110484 | |
| MRG002 | HER2 | Advanced | HER2 overexpression | NCT04837508 | ||
| MRG003 | EGFR | Advanced | EGFR positive | NCT04838964 | ||
| RC48-ADC | HER2 | Advanced | HER2 overexpression | NCT04329429 | ||
| Trametinib | MEK1/2 | Advanced | KRAS mutation | Hydroxychloroquine | NCT04566133 | |
| Trastuzumab | HER2 | Advanced | HER2 overexpression | Oxaliplatin/5FU | NCT04722133 | |
| Zanidatamab | HER2 | Advanced | HER2 overexpression | Cisplatin/Gemcitabine | NCT03929666 | |
| Miscellaneous | Abemaciclib | CDK4/CDK6 | Advanced | NCT04003896 | ||
| Ceralasertib | ATR | Advanced | Durvalumab, Olaparib | NCT04298008, NCT04298021 | ||
| DKN-01 | DKK1 | Advanced | Nivolumab | NCT04057365 | ||
| HA121-28 | RET | Advanced | NCT04784520 | |||
| Olaparib | PARP | Advanced | DDR mutation | Pembrolizumab, Durvalumab, Ceralasertib | NCT04306367, NCT05222971, NCT04298021 | |
| Rucaparib | PARP | Advanced | Nivolumab | NCT03639935 | ||
| TST001 | CLDN18.2 | Advanced | CLDN18.2 overexpression | NCT05190575 |
The discovery of recurrent FGFR2 fusions in patients with CCA stimulated the pharmaceutical appearance of FGFR inhibitors. Infigratinib, the first of its class, showed a promising 15% objective response rate (ORR) in patients with FGFR alterations[63]. As seen with other oncogene-addicted tumors, acquired resistance to this drug limited the durability of response and has been related to the emergence of polyclonal secondary mutations in the FGFR2 kinase domain[64]. Other selective FGFR inhibitors have followed these studies, highlighting the case of pemigatinib, approved by regulatory agencies on the basis of 35% ORR and median OS of 21 months in patients with FGFR2 fusions whose disease had progressed while receiving prior therapy[13]. Phase III clinical trials (NCT03656536, NCT03773302) are comparing FGFR-inhibition versus cisplatin and gemcitabine in the first-line setting. Even though eCCA is not an exclusion criterion for these trials, the presence of FGFR2 fusions in this anatomical location is marginal in comparison to iCCA.
The finding that IDH1/2 mutations occur frequently in CCA led to the development of inhibitors specific to the individual mutant alleles (e.g., to IDH1R132 and IDH2R172). Ivosidenib, an IDH1 inhibitor, has improved progression-free survival (PFS) from 1.4 months with placebo to 2.7 months in patients previously treated with unresectable or metastatic CCA with an IDH1 mutation[64]. However, as seen with FGFR2 alterations, IDH mutations are rarely seen in eCCA, and thus, the clinical development of drugs against IDH is focused on iCCA.
Another promising target with ongoing clinical trials in patients with eCCA is ERBB2. In GBC and eCCA, ERBB2 overexpression, amplification, or mutation can occur in up to 15% of cases. Small CCA cohorts treated with trastuzumab plus pertuzumab[65] or with neratinib have detected partial responses in some patients[66]. Novel therapeutic approaches against ERBB2 are in clinical development, such as antibody-drug conjugates (MRG002 and RC48-ADC) or bispecific antibodies directed against two non-overlapping domains of ERBB2 (zanidatamab).
A basket trial observed up to 36% ORR with dabrafenib (BRAF inhibitor) in combination with trametinib (MEK inhibitor) for CCA harboring BRAF-V600E mutations[67]. However, the low prevalence of this mutation in eCCA (< 2%) hampers the development of large clinical trials. Much more common is the presence of mutations in KRAS (up to 43% depending on the cohort), a traditionally undruggable target that would be the focus of clinical research during the following years due to the recent appearance of KRASG12D inhibitors[68]. Another target of the MAP/ERK signaling pathway is EGFR, with mutations in a small subset of eCCA patients.
The presence of somatic or germline mutations in DNA damage repair genes such as ATM o BRCA1/2 in eCCA has facilitated the development of trials with PARP inhibitors (olaparib and rucaparib), specifically targeting these genetic aberrations. Beyond structural genetic alterations, CLDN18.2 overexpression has been used as a biomarker for selecting the eventual clinical efficacy of monoclonal antibodies against this protein (TST001). Other targeted therapies, mostly tyrosine kinase inhibitors against VEGFR/FGFR signaling pathways[69], are in clinical development for eCCA without a biomarker-enrichment design and typically in combination with chemotherapy or ICIs.
The clinical experience with ICIs in monotherapy for BTC is limited in comparison to other solid tumors[19]. A phase II trial evaluating pembrolizumab
CONCLUSION
Cancer is a multifactorial disease harboring a cocktail of altered oncogenes and tumor suppressors that work in concert with specific molecular pathways leading to the carcinogenic process. In view of this, oncological translational research has benefited from worldwide efforts aimed at delineating the genetic and molecular fingerprints of thousands of human samples spanning all major cancer types. However, molecular characterization of BTC has been traditionally challenged by the scarcity of available samples[42] and by the heterogeneity among different anatomical subtypes[33,42]. Indeed, the inclusion of surgically resected patients has been traditionally the only source of samples for integrative genomic analysis of BTC[33,36,38,42,48], which inherently questions the reproducibility of the molecular fingerprint depicted in localized tumors in metastatic tumors. In this scenario, liquid biopsy is envisioned as a valuable tool for dynamic molecular testing to guide the selection of precision therapies[72].
As opposed to other solid malignancies, structural genetic alterations detected in eCCA were of limited clinical relevance. However, recent discoveries offer new hope for these tumors. Studies analyzing hundreds of samples from eCCA[33,36-38,42]identified TP53, KRAS, SMAD4 and ARID1A as the most prevalent mutations in eCCA, whereas recurrent deletions include mainly CDKN2A/B. Mutations in IDH1/2 and FGFR2 translocations, of interest for their associated targeted therapies, are predominant in iCCA[12]. ERBB2 mutations and amplifications are the most recurrent actionable alterations for eCCA. Together with other low prevalent genetic alterations (BRCA1/2, EGFR, ERBB2, CDK4, BRAF, NRAS, PIK3CA, and MDM2), around 25% of eCCA display at least one putative actionable driver[38]. Beyond that, four novel transcriptome-based eCCA classes have been identified and linked with proposed treatment strategies[38], notably, ERBB2 inhibitors for the Proliferation class based on the aberrant activation of this target and its downstream signaling pathways, and anti-PD-1/PD-L1 inhibitors for the Immune class according to the high CD8 + lymphocytic infiltration of these tumors. Altogether, the uncovered genomic traits of eCCA provide the rationale for analyzing novel treatment strategies for biomarker-enriched populations.
Besides the tumor intrinsic biological features, it is worth highlighting the dense desmoplastic stroma observed in eCCA that, as observed in pancreatic cancer, is responsible for creating a mechanical barrier that prevents appropriate vascularization and thus limits exposure to systemic treatments[73]. At the same time, it confers a unique immunosuppressive microenvironment that has challenged the emergence of immunotherapies[6]. To date, single-agent ICIs have been unsuccessful in eCCA and other ‘non-immunogenic’ tumors, partly owing to the restrains in immune cell infiltration[74]. However, a small percentage (4%) of hypermutated eCCA show promise due to MSI-H/dMMR[38,70]. To achieve clinical success, future approaches should consider combing therapies that target multiple aspects of the tumor microenvironment. In fact, the combination of cytotoxic chemotherapy and ICIs has recently improved the clinical outcome of advanced eCCA patients[11]. In addition, the identification of accurate predictive biomarkers of response to ICIs remains an unmet medical need, as it could facilitate the effective use of these drugs in eCCA.
A large number of clinical trials assessing targeted therapies are in development for BTC, a substantial part of them still without a companion biomarker for the optimal selection of patients that may benefit from the intervention. Furthermore, anatomical location of BTC is not considered as an inclusion criterion for the vast majority of studies, despite its fundamental implications according to the described different biological landscapes of eCCA, iCCA and GBC. The most frequent strategy of ongoing clinical trials is the combination of ICIs together with inhibitors against VEGFR/FGFR signaling pathways, according to the synergistic effect observed in other solid tumors such as HCC[75]. New drug approvals for eCCA are expected to come as a result of the successful achievement of different steps, including molecular profiling of tumors, identification of oncogenic driver alterations, discovery of novel targeted therapies, and demonstration of clinical benefit for a biomarker-enriched population.
Overall, targeted therapies have had a profound effect on cancer medicine, although there are still scientific obstacles to the broad implementation of precision oncology. Of note, only approximately 20% of oncogenic or tumor suppressor proteins can be targeted by currently available medicines[76]. Recent advances, however, have allowed the clinical development of inhibitors against mutant KRAS, the paradigm of unactionable target[77], and altered in almost half of eCCA. On the other hand, tumor heterogeneity adds a new level of complexity that is likely to have an impact on the efficacy of targeted therapies, something that might be better understood thanks to the emergence of single-cell sequencing technologies[78]. In addition, genomic alterations are only one of several biologic drivers of cancer. As a consequence, DNA sequencing would need to be complemented by other high-throughput technologies such as DNA methylation, RNA sequencing or phosphoprotein profiling in order to expand the current therapeutic armamentarium of eCCA.
To conclude, in contrast to the historic "one-size-fits-all" chemotherapy strategy used for the treatment of eCCA, the comprehensive molecular profiling of this disease conducted over the last decade is approaching a precision oncology discipline[79], where patient characteristics are combined with their tumor genomic landscape to enable matching with molecularly targeted agents in order to maximize treatment efficacy and minimize toxicity.
DECLARATIONS
Authors’ contributionsAll authors made substantial contributions to each stage of the preparation of this manuscript for publication: Oronich A, Pallisé O, Salud A, Montal R
Availability of data and materialsNot applicable.
Financial support and sponsorshipRM acknowledges the support from ISCIII (PI21/01619 research project and Juan Rodés contract); SEOM (research project; TTD (research project); and Fundación MERCK Salud (research project).
Conflicts of interestRM has received consulting and lecture fees from Servier, Roche and BMS and travel and education funding from MSD, Eli Lilly, Bayer, Roche, and AstraZeneca.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
REFERENCES
1. Fitzmaurice C, Akinyemiju TF, Al Lami FH, et al. Global burden of disease cancer collaboration. global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 29 cancer groups, 1990 to 2016: a systematic analysis for the global burden of disease study. JAMA Oncol 2018;4:1553-68.
2. Everhart JE, Ruhl CE. Burden of digestive diseases in the United States part III: liver, biliary tract, and pancreas. Gastroenterology 2009;136:1134-44.
3. Bertuccio P, Malvezzi M, Carioli G, et al. Global trends in mortality from intrahepatic and extrahepatic cholangiocarcinoma. J Hepatol 2019;71:104-14.
4. Bridgewater J, Galle PR, Khan SA, et al. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J Hepatol 2014;60:1268-89.
5. Blechacz B, Komuta M, Roskams T, Gores GJ. Clinical diagnosis and staging of cholangiocarcinoma. Nat Rev Gastroenterol Hepatol 2011;8:512-22.
6. Banales JM, Cardinale V, Carpino G, et al. Cholangiocarcinoma: current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat Rev Gastroenterol Hepatol 2016;13:261-80.
7. Vogel A, Bridgewater J, Edeline J, et al. ESMO guidelines committee. Biliary tract cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol 2023;34:127-40.
8. Darwish MS, Kim WR, Harnois DM, et al. Efficacy of neoadjuvant chemoradiation, followed by liver transplantation, for perihilar cholangiocarcinoma at 12 US centers. Gastroenterology 2012;143:88-98.
9. Primrose JN, Fox RP, Palmer DH, et al. BILCAP study group. Capecitabine compared with observation in resected biliary tract cancer (BILCAP): a randomised, controlled, multicentre, phase 3 study. Lancet Oncol 2019;20:663-73.
10. Valle J, Wasan H, Palmer DH, et al. ABC-02 trial investigators. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med 2010;362:1273-81.
11. Oh D, Ruth He A, Qin S, et al. Durvalumab plus gemcitabine and cisplatin in advanced biliary tract cancer. NEJM Evidence 2022;1:EVIDoa2200015.
12. Sia D, Losic B, Moeini A, et al. Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat Commun 2015;6:6087.
13. Abou-Alfa GK, Sahai V, Hollebecque A, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol 2020;21:671-84.
14. Abou-Alfa GK, Macarulla T, Javle MM, et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): a multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol 2020;21:796-807.
16. Clements O, Eliahoo J, Kim JU, Taylor-Robinson SD, Khan SA. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma: a systematic review and meta-analysis. J Hepatol 2020;72:95-103.
17. Palmer WC, Patel T. Are common factors involved in the pathogenesis of primary liver cancers? A meta-analysis of risk factors for intrahepatic cholangiocarcinoma. J Hepatol 2012;57:69-76.
20. Haswell-Elkins MR, Satarug S, Tsuda M, et al. Liver fluke infection and cholangiocarcinoma: model of endogenous nitric oxide and extragastric nitrosation in human carcinogenesis. Mutat Res 1994;305:241-52.
21. Carpino G, Cardinale V, Renzi A, et al. Activation of biliary tree stem cells within peribiliary glands in primary sclerosing cholangitis. J Hepatol 2015;63:1220-8.
22. Rizvi S, Gores GJ. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology 2013;145:1215-29.
23. Moeini A, Sia D, Bardeesy N, Mazzaferro V, Llovet JM. Molecular pathogenesis and targeted therapies for intrahepatic cholangiocarcinoma. Clin Cancer Res 2016;22:291-300.
24. Banales JM, Marin JJG, Lamarca A, et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol 2020;17:557-88.
26. Servais FA, Kirchmeyer M, Hamdorf M, et al. Modulation of the IL-6-Signaling pathway in liver cells by miRNAs targeting gp130, JAK1, and/or STAT3. Mol Ther Nucleic Acids 2019;16:419-33.
27. Clapéron A, Mergey M, Nguyen Ho-Bouldoires TH, et al. EGF/EGFR axis contributes to the progression of cholangiocarcinoma through the induction of an epithelial-mesenchymal transition. J Hepatol 2014;61:325-32.
28. Pant K, Richard S, Peixoto E, Gradilone SA. Role of glucose metabolism reprogramming in the pathogenesis of cholangiocarcinoma. Front Med 2020;7:113.
29. Tang L, Tan YX, Jiang BG, et al. The prognostic significance and therapeutic potential of hedgehog signaling in intrahepatic cholangiocellular carcinoma. Clin Cancer Res 2013;19:2014-24.
30. Boulter L, Guest RV, Kendall TJ, et al. WNT signaling drives cholangiocarcinoma growth and can be pharmacologically inhibited. J Clin Invest 2015;125:1269-85.
31. Guest RV, Boulter L, Dwyer BJ, et al. Notch3 drives development and progression of cholangiocarcinoma. Proc Natl Acad Sci U S A 2016;113:12250-5.
32. Valle JW, Lamarca A, Goyal L, Barriuso J, Zhu AX. New horizons for precision medicine in biliary tract cancers. Cancer Discov 2017;7:943-62.
33. Nakamura H, Arai Y, Totoki Y, et al. Genomic spectra of biliary tract cancer. Nat Genet 2015;47:1003-10.
34. Homayounfar K, Gunawan B, Cameron S, et al. Pattern of chromosomal aberrations in primary liver cancers identified by comparative genomic hybridization. Hum Pathol 2009;40:834-42.
35. Deenonpoe R, Sa-Ngiamwibool P, Watcharadetwittaya S, et al. Fluorescence
36. Jusakul A, Cutcutache I, Yong CH, et al. Whole-Genome and epigenomic landscapes of etiologically distinct subtypes of cholangiocarcinoma. Cancer Discov 2017;7:1116-35.
37. Lee H, Wang K, Johnson A, et al. Comprehensive genomic profiling of extrahepatic cholangiocarcinoma reveals a long tail of therapeutic targets. J Clin Pathol 2016;69:403-8.
38. Montal R, Sia D, Montironi C, et al. Molecular classification and therapeutic targets in extrahepatic cholangiocarcinoma. J Hepatol 2020;73:315-27.
39. Jiao Y, Pawlik TM, Anders RA, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat Genet 2013;45:1470-3.
40. Arai Y, Totoki Y, Hosoda F, et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014;59:1427-34.
41. Wu YM, Su F, Kalyana-Sundaram S, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov 2013;3:636-47.
42. Farshidfar F, Zheng S, Gingras MC, et al. Cancer Genome Atlas Network. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-Mutant molecular profiles. Cell Rep 2017;18:2780-94.
43. Saha SK, Parachoniak CA, Ghanta KS, et al. Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature 2014;513:110-4.
44. Galdy S, Lamarca A, McNamara MG, et al. HER2/HER3 pathway in biliary tract malignancies; systematic review and meta-analysis: a potential therapeutic target? Cancer Metastasis Rev 2017;36:141-57.
45. Li M, Zhang Z, Li X, et al. Whole-exome and targeted gene sequencing of gallbladder carcinoma identifies recurrent mutations in the ErbB pathway. Nat Genet 2014;46:872-6.
46. Mateo J, Chakravarty D, Dienstmann R, et al. A framework to rank genomic alterations as targets for cancer precision medicine: the ESMO Scale for Clinical Actionability of molecular Targets (ESCAT). Ann Oncol 2018;29:1895-902.
47. Chakravarty D, Gao J, Phillips SM, et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol 2017:2017.
48. Sia D, Hoshida Y, Villanueva A, et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 2013;144:829-40.
49. Andersen JB, Spee B, Blechacz BR, et al. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology 2012;142:1021-1031.e15.
50. Andersen JB, Thorgeirsson SS. Genomic decoding of intrahepatic cholangiocarcinoma reveals therapeutic opportunities. Gastroenterology 2013;144:687-90.
51. Bijlsma MF, Sadanandam A, Tan P, Vermeulen L. Molecular subtypes in cancers of the gastrointestinal tract. Nat Rev Gastroenterol Hepatol 2017;14:333- 42.
52. Sirica AE. The role of cancer-associated myofibroblasts in intrahepatic cholangiocarcinoma. Nat Rev Gastroenterol Hepatol 2011;9:44-54.
53. Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013;501:346-54.
54. Mertens JC, Fingas CD, Christensen JD, et al. Therapeutic effects of deleting cancer-associated fibroblasts in cholangiocarcinoma. Cancer Res 2013;73:897-907.
55. Cadamuro M, Nardo G, Indraccolo S, et al. Platelet-derived growth factor-D and Rho GTPases regulate recruitment of cancer-associated fibroblasts in cholangiocarcinoma. Hepatology 2013;58:1042-53.
56. Sulpice L, Rayar M, Desille M, et al. Molecular profiling of stroma identifies osteopontin as an independent predictor of poor prognosis in intrahepatic cholangiocarcinoma. Hepatology 2013;58:1992-2000.
57. Kasper HU, Drebber U, Stippel DL, Dienes HP, Gillessen A. Liver tumor infiltrating lymphocytes: comparison of hepatocellular and cholangiolar carcinoma. World J Gastroenterol 2009;15:5053-7.
58. Ayers M, Lunceford J, Nebozhyn M, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 2017;127:2930-40.
59. Kraehenbuehl L, Weng CH, Eghbali S, Wolchok JD, Merghoub T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat Rev Clin Oncol 2022;19:37-50.
60. Buzzoni R, Pusceddu S, Bajetta E, et al. Activity and safety of RAD001 (everolimus) in patients affected by biliary tract cancer progressing after prior chemotherapy: a phase II ITMO study. Ann Oncol 2014;25:1597-603.
61. Bekaii-Saab T, Phelps MA, Li X, et al. Multi-institutional phase II study of selumetinib in patients with metastatic biliary cancers. J Clin Oncol 2011;29:2357-63.
62. Philip PA, Mahoney MR, Allmer C, et al. Phase II study of erlotinib in patients with advanced biliary cancer. J Clin Oncol 2006;24:3069-74.
63. Javle M, Lowery M, Shroff RT, et al. Phase II study of BGJ398 in patients with FGFR-Altered advanced cholangiocarcinoma. J Clin Oncol 2018;36:276-82.
64. Goyal L, Saha SK, Liu LY, et al. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov 2017;7:252-63.
65. Hainsworth JD, Meric-Bernstam F, Swanton C, et al. Targeted therapy for advanced solid tumors on the basis of molecular profiles: results from mypathway, an open-label, phase IIa multiple basket study. J Clin Oncol 2018;36:536-42.
66. Hyman DM, Piha-Paul SA, Won H, et al. HER kinase inhibition in patients with HER2- and HER3-mutant cancers. Nature 2018;554:189-94.
67. Subbiah V, Lassen U, Élez E, et al. Dabrafenib plus trametinib in patients with BRAF(V600E)-mutated biliary tract cancer (ROAR): a phase 2, open-label, single-arm, multicentre basket trial. Lancet Oncol 2020;21:1234-43.
68. Hallin J, Bowcut V, Calinisan A, et al. Anti-tumor efficacy of a potent and selective non-covalent KRAS(G12D) inhibitor. Nat Med 2022;28:2171-82.
69. Mariotti V, Fiorotto R, Cadamuro M, Fabris L, Strazzabosco M. New insights on the role of vascular endothelial growth factor in biliary pathophysiology. JHEP Rep 2021;3:100251.
70. Marabelle A, Le DT, Ascierto PA, et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: results from the phase II KEYNOTE-158 study. J Clin Oncol 2020;38:1-10.
71. Piha-Paul SA, Oh DY, Ueno M, et al. Efficacy and safety of pembrolizumab for the treatment of advanced biliary cancer: results from the KEYNOTE-158 and KEYNOTE-028 studies. Int J Cancer 2020;147:2190-8.
72. Corcoran RB, Chabner BA. Application of cell-free DNA analysis to cancer treatment. N Engl J Med 2018;379:1754-65.
73. Ho WJ, Jaffee EM, Zheng L. The tumour microenvironment in pancreatic cancer - clinical challenges and opportunities. Nat Rev Clin Oncol 2020;17:527-40.
74. Karamitopoulou E. Tumour microenvironment of pancreatic cancer: immune landscape is dictated by molecular and histopathological features. Br J Cancer 2019;121:5-14.
75. Finn RS, Qin S, Ikeda M, et al. IMbrave150 Investigators. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med 2020;382:1894-905.
77. Canon J, Rex K, Saiki AY, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019;575:217-23.
78. Song G, Shi Y, Meng L, et al. Single-cell transcriptomic analysis suggests two molecularly distinct subtypes of intrahepatic cholangiocarcinoma. Nat Commun 2022;13:1642.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Oronich A, Pallisé O, Salud A, Montal R. Genomic alterations and targeted therapies in extrahepatic cholangiocarcinoma. Hepatoma Res 2023;9:26. http://dx.doi.org/10.20517/2394-5079.2023.04
AMA Style
Oronich A, Pallisé O, Salud A, Montal R. Genomic alterations and targeted therapies in extrahepatic cholangiocarcinoma. Hepatoma Research. 2023; 9: 26. http://dx.doi.org/10.20517/2394-5079.2023.04
Chicago/Turabian Style
Oronich, Arnau, Ona Pallisé, Antonia Salud, Robert Montal. 2023. "Genomic alterations and targeted therapies in extrahepatic cholangiocarcinoma" Hepatoma Research. 9: 26. http://dx.doi.org/10.20517/2394-5079.2023.04
ACS Style
Oronich, A.; Pallisé O.; Salud A.; Montal R. Genomic alterations and targeted therapies in extrahepatic cholangiocarcinoma. Hepatoma. Res. 2023, 9, 26. http://dx.doi.org/10.20517/2394-5079.2023.04
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 4 clicks
Cite This Article 4 clicks

 Like This Article 4
likes
Like This Article 4
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.