
Targeting the interactions between lymphocytes and liver cancer stem cells in combination with immunotherapy is a promising therapeutic strategy


Abstract
Hepatocellular carcinoma (HCC) is one of the most common malignant tumors worldwide, with a poor prognosis and high recurrence rate. Liver cancer stem cells (LCSCs), a small subset of HCC cells, have the capacity for self-renewal and the property of treatment resistance, suggesting that LCSCs are key factors in causing poor prognosis for HCC patients. In addition, LCSCs interact with immune cells to participate in the formation of an immunosuppressive microenvironment and escape the immune surveillance in HCC, especially lymphocytes. At present, immunotherapies for HCC are mainly based on reactivating the lymphocyte system, including immune checkpoint inhibitors, multifunctional antibodies, and adoptive cell therapy. Therefore, blocking the interactions between lymphocytes and LCSCs in combination with immunotherapy may be a promising therapeutic strategy. This review summarizes the interaction mechanisms of lymphocytes and LCSCs and the current exploration of combination therapy in HCC.
Keywords
INTRODUCTION
HCC is the most common type of primary liver cancer and a leading cause of cancer-related death worldwide[1]. HCC has a poor prognosis, and its 5-year overall survival (OS) rate is less than 20%, making it the second most lethal tumor after pancreatic cancer[2]. Early-stage HCC patients can largely improve 5-year OS rates through liver transplantation or radical surgery[3]. However, only 15% of HCC patients benefit from it, and most patients are diagnosed at an advanced stage[4]. Acquired drug resistance and tumor recurrence after treatment are the key factors leading to the poor prognosis of advanced HCC patients[5,6].
Cancer stem cells (CSCs) are a small subset of cancer cells with stem cell properties that play a major role in tumor growth, metastasis, recurrence, and resistance to therapy[7]. Liver cancer stem cells (LCSCs) often express biomarkers such as CD44, CD47, CD24, EpCAM, and CD133[8]. Targeting LCSCs therapy based on their specific biomarkers may be an effective strategy to eliminate tumor recurrence at the source. LCSCs are involved in the formation of an immunosuppressive microenvironment, altering or impairing the natural function of immune cells, thereby evading immune surveillance[9-13]. Mobilizing or reactivating the immune system can effectively eliminate cancer. Immunotherapy, especially the strategy for activating the immune activity of lymphocytes, is currently a promising tumor treatment[14]. Immune checkpoint inhibitors (ICIs), multifunctional antibodies and adoptive cell therapy are increasingly used for immunotherapy in the lymphocyte system, but the high drug resistance and relapse rate are still unsolved problems[15]. Combining immunotherapy and LCSCs targeted therapy may better solve the current problem. This review briefly summarizes the research on the mutual regulatory mechanism of lymphocytes and LCSCs and some explorations of combination therapy in HCC.
THE INTERACTIONS BETWEEN LYMPHOCYTES AND LCSCS
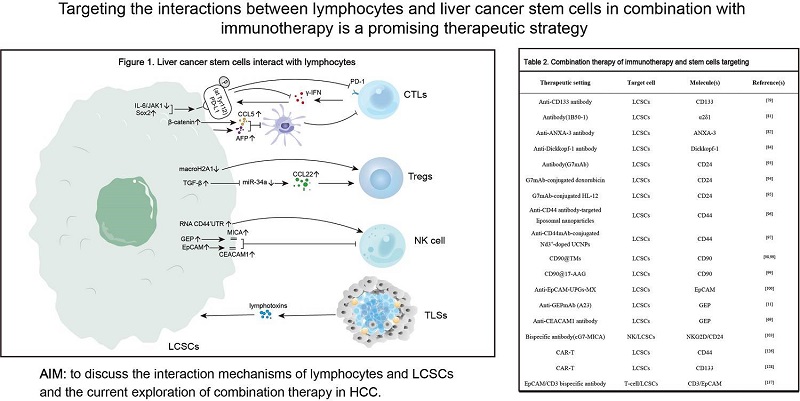
Lymphocytes, the main population targeted by immunotherapy, play an important role in the development and progression of HCC. LCSCs, although only a small fraction of HCC cells, possess the ability of self-renewal and tumor formation and are a key factor leading to poor prognosis[16]. This section describes the progress of research on the interactions of lymphocytes with LCSCs and non-LCSCs [Table 1 and Figure 1].

Figure 1. Liver cancer stem cells interact with lymphocytes.
Interaction between lymphocytes and liver cancer stem cells
| LCSCs | Molecule(s) | Activity of Lymphocytes | References |
| SOX2+ LCSCs | PD-L1↑ | TILs↓ | [35,36] |
| CK19+ LCSCs SALL4+ LCSCs | PD-L1↑ | TILs↓ | [109] |
| CD133+ LCSCs | Galectin-3↑ | CD8+ T cells↓ | [134] |
| CD44+ LCSCs | Histone macroH2A1↓ | Tregs↑ | [10] |
| CD44+ LCSCs | TGF-β-miR-34a-CCL22↑ | Tregs↑ | [60,61] |
| GEP+ LCSCs | MICA↑ | NK↓ | [12] |
| EpCAM+ LCSC | CEACAM1↑ | NK↓ | [11] |
| CD133+ LCSCs SOX2+ LCSCs | HMBOX1↑ | NK↑ | [69] |
| CD44+ LCSC | ceRNA CD44 3' UTR↑ | NK↑ | [70] |
CD8+ cytotoxic T lymphocytes
CD8+ T cells are important lymphocytes in the tumor-adaptive immune response. CD8+ T cells are modified into antigen-specific cytotoxic T lymphocytes (CTLs) via the T-cell receptor (TCR), which recognizes the major histocompatibility complex I (MHCI) on the antigen-presenting cells[17]. On the one hand, CTLs directly cause cell destruction by releasing cytotoxic substances such as granzyme, perforin and interferon
Single-cell RNA-sequencing studies have revealed that the infiltrating T cells are characterized by an enrichment of regulatory T cells (Tregs) and exhausted CD8+ T-cell clones. This landscape exemplifies an immunosuppressive microenvironment. 37% of exhausted CD8+ T-cell clones shared TCR β chain with other CD8+ T-cell clusters, suggesting that exhausted CD8+ T cells were likely derived from other CD8+ T cells in HCC microenvironment[23,24]. Subsequently, Wang et al. identified a T-cell exhaustion mechanism in which thymocyte selection-associated high mobility group box protein (TOX) in T cells promotes its own entry into a state of functional failure by stimulating the PD-1 endocytic cycle[25]. The latest study revealed that HCC patients with enrichment of severely exhausted CD8+ T cells (TEX) had a lower survival rate, while those with a predominance of CD103+ tissue-resident memory cells (TRM) had a higher survival rate. Dynamic changes in TEX and TRM affect the prognosis of HCC patients[26]. Therefore, reactivation of exhausted T-cell activity is important for treatment of HCC. Using antibodies to block immune checkpoints, including PD-1, CTLA-4, Lag-3, TIGIT and Tim-3, can reverse the function of exhausted T cells and restore the antitumor activity of tumor-infiltrating T cells[27,28]. Removing the inhibitory effect of immune microenvironment suppressor cells may also be a strategy to reshape the antitumor activity of T cells. Hypoxia-inducible factor-1α (HIF-1α) induces the expression of triggering receptor-1 expressed on myeloid cells-1 (TREM-1) in tumor-associated macrophages (TAMs), which reverses the activity of dysfunctional CD8+ T cells and reduces resistance to programmed cell death ligand-1 (PD-L1) blockade[29]. Icaritin, an adjuvant, also inhibits myeloid-derived suppressor cells (MDSCs) to improve HCC therapeutic efficacy[30].
CSCs are one of the important factors contributing to immunosuppression. Reduction of MHC-I expression on CSCs suppresses CTL-mediated tumor killing[31]. Meanwhile, LCSCs recruit M2-type macrophages by activating Yes-associated protein to evade immune clearance[32]. Compared to non-LCSCs, LCSCs express more PD-L1 and show stronger immunosuppressive effects. In addition to generating negative regulatory signals to PD-1+ effector T cells, PD-L1 derived from tumor cells also releases signals into themselves that enhance the expression of stemness-associated genes like cytokeratin 19 (CK19) and regulate LCSCs number and function[33,34]. Several studies have revealed that microenvironmental regulatory factors can upregulate PD-L1 expression on LCSCs to support their growth. On the one hand, the transcription factor Sox2 enhances PD-L1 transcriptional activity by binding to the PD-L1 promoter region to promote LCSCs survival[35,36]. On the other hand, the IL-6/JAK1 signaling pathway drives PD-L1 Y112 phosphorylation, which recruits the endoplasmic reticulum-associated N-glycosyltransferase STT3A to catalyze PD-L1 glycosylation and maintain PD-L1 stability[37]. Upregulation of PD-L1 expression impairs IFN-γ secretion from CTLs by a negative feedback regulatory mechanism, enhancing the ability of LCSCs to protect against T-cell killing and immune evasion[38]. Therefore, targeting the above sites, such as Sox2 combined with anti-PD-L1, may contribute to antitumor therapy.
Dendritic cells (DCs) pulsed with total HCC RNA induce effector T lymphocyte activation. Activated effector T lymphocytes enhance the ability to kill tumors and secrete more IFN-γ[39,40]. LCSCs can inhibit cytotoxic T-cell activity by inhibiting the recruitment of DCs and promoting tumor cell growth.
Regulatory T Cells (Tregs)
Tregs are a subpopulation of CD4+ T cells that are characterized by the expression of CD25 and Foxp3[45,46]. Tregs are the main subpopulations that maintain the body’s immune tolerance to tumors. Tregs derived from peripheral blood and tumor-infiltrating lymphocytes (TILs) of patients with HCC are increasing and are more inhibited than those derived from normal subjects[47,48]. An elevated level of Tregs in the tumor microenvironment is associated with poor prognosis[49]. Mechanistically, intratumor Tregs upregulate the expression of glucocorticoid-induced tumor necrosis factor receptors (GITRs) and inhibit tumor-specific T-cell activity[50,51]. In addition, Tregs stimulate their own differentiation and promote HCC immune escape via the upregulation of lnc-EGFR[52].
Tregs interact with other immune cells via cell contact and cytokine secretion to inhibit their activity and thus participate in the formation of the immunosuppressive tumor microenvironment. For example, Tregs mediate the loss of the human leukocyte antigen-DR isotype (HLA-DR) on type 2 conventional dendritic cells (cDC2s), which impairs DC antigen presentation and thus suppresses antitumor immunity in HCC[53]. Indeed, HCC cells can also secrete cytokines or nutrients to maintain and enhance the immunosuppression of Tregs. Mechanistically, HCC-derived growth differentiation factor 15 (GDF15), by recognizing the receptor CD48, promotes the production of induced Tregs and enhances the suppressive function of natural Tregs, which induces tumor immunosuppression[54]. In addition, tumors phagocytose nutrients that are necessary for effector T cells to exert cytotoxicity while flushing out lactate to provide nutrients to Tregs. Tregs use the lactate transporter protein monocarboxylate transporter 1 (MCT1) to convert lactate into energy and maintain a state of tumor immune tolerance[55].
Both CSCs and Tregs are responsible for tumor immune tolerance and tumor recurrence. The preoperative peripheral blood quantity of EpCAM+ circulating tumor cells and Tregs is positively correlated with the risk of postoperative recurrence and metastasis in HCC patients[56]. The interactions between CSCs and Tregs further lead to tumor immunosuppression. Enriched LGR5+ LCSCs are associated with poor prognosis in HCC, whereas Tregs increased LGR5 of CSCs expression in gastric cancer through TGF-β1[57-59]. Tregs and LGR5+ CSCs may also be associated in HCC. For instance, CD44+ LCSCs regulate TGF-β1 expression, and TGF-β1 has been shown to recruit Tregs through the miR-34a-CCL22 axis, which promotes immune escape[60,61]. In addition, downregulation of histone macroH2A1 in HCC cells increases LCSCs and
Natural killer cells
Natural killer (NK) cells, a subpopulation of normal human peripheral blood lymphocytes, can nonspecifically kill tumor cells. This natural killing activity requires neither specific antigen nor MHC restriction. NK cells express a large number of immune recognition receptors, including kill-activated receptors (KARs) and inhibitory receptors, to recognize ligands on normal cells and tumor cells, thus maintaining a balance between the immune response and immune tolerance of NK cells. Correspondingly, abnormal NK cell receptors could promote HCC development[62].
Natural killer group 2D (NKG2D) is a typical KAR on the surface of NK cells, and its expression plays a significant role in the innate immunity of NK cells against the malignant transformation of HCC[63]. Increased expression of the soluble form of MHC class I-related chain A (MICA) in patients with advanced HCC correlates with downregulation of NKG2D expression and impairs NK-cell function[64]. However, recent studies have found that activation of the NKG2D system perhaps contributes to a strong inflammatory response that exacerbates liver tissue damage. Cadoux et al. discovered for the first time that NKG2D ligands MICA/B and ULBP1/2 are linked with poor prognosis and early tumor recurrence in HCC[65]. In addition, β-catenin signaling downregulates the expression of murine NKG2D ligands Rae-1 through binding to TCF4, which attenuates the invasive capacity of HCC[65]. The above findings suggest that targeting NK cells to treat HCC is a double-edged sword. We should pay attention to indicators of liver injury and inflammatory response in the treatment.
Cytokine-induced killer (CIK) cells recognize and kill LCSCs via the NKG2D system. Intravenous infusion of CIK cells can significantly retard tumor growth[66]. However, LCSCs can attenuate the toxicity of NK cells by synthesizing inhibitory proteins, such as granulin-epithelin precursor (GEP). GEP is a hallmark of the LCSCs and functionally increases the stemness of CSCs by regulating the expression of stemness-associated signaling molecules, including β-catenin, Oct4, Nanog and Sox2[67]. In addition, GEP confers the ability of HCC cells to evade NK cytotoxicity by regulating MICA expression[12]. Another study found that EpCAMHigh LCSCs also resisted NK-cell-mediated cytotoxicity by upregulating carcinoembryonic antigen-associated cell adhesion molecule 1 (CEACAM1)[11]. Restoring the sensitivity of LCSCs to NK-cell-mediated cytotoxicity can enhance innate immune suppression in HCC. Blocking the expression of GEP and CEACAM1 restores the natural killing activity of NK cells[11,12,68]. In addition, LCSCs overexpressing HMBOX1 suppress their self-renewal and improve NK-cell-mediated antitumor immune responses[69]. Recently, researchers also found that LCSCs enhance their own sensitivity to NK cells by upregulating the competitive endogenous RNA (ceRNA) CD44 3' UTR[70].
Tertiary lymphoid structures
Tertiary lymphoid structures (TLSs) are classically defined as lymphoid aggregates formed in nonhematopoietic organs. TLSs do not exist under physiological conditions but are formed during chronic and non-resolving inflammatory processes such as infection, transplant rejection, autoimmune diseases and cancer[71]. TLSs are characterized by CD20+ B surrounded by CD3+ T-cell structures, similar to the lymphoid follicles in secondary lymphoid organs. In addition, TLSs contain components such as distinct DC populations, dense stromal networks, and specialized vascular systems provided by peripheral node addressin (PNAd)-positive high endothelial venules (HEVs) that are thought to mediate lymphocyte recruitment[72].
TLSs are involved in antigen-specific antitumor immune responses mainly by promoting the induction of effector T cells, central memory T cells and plasma cells. In almost all solid tumors, TLSs are associated with a reduction in recurrence risk and an improvement in survival rate[73]. However, the role of TLSs in the pathogenesis of HCC is controversial, as they may promote the growth of tumor progenitor cells. Finkin
COMBINATION OF IMMUNOTHERAPY AND TARGETING LCSCS
Multifunctional antibody against LCSCs
Several LCSCs-specific markers have been directly discovered, such as CD24, CD44, CD47, CD90, CD133, EpCAM and ANXA-3. Antibody-mediated targeted therapeutic strategies can effectively target CSC subpopulations and inhibit tumor growth or recurrence [Table 2]. CD44 is a surface feature of CSCs, and its expression level correlates with poor patient prognosis. Anti-CD44 antibody effectively eliminates CSCs from tumors and prevents pancreatic cancer tumor formation and tumor recurrence after radiotherapy[77]. Under low glucose conditions, CD133 antibody induces apoptosis of LCSCs by inhibiting autophagy and enhancing chemotherapeutic efficacy[78]. IgG1-iS18, an antibody targeting the 37 kDa/67 kDa laminin receptor (LRP/LR), impairs the adhesion and invasive ability of HCC cells and is a strategy for the treatment of metastatic HCC[79]. Antibody 1B50-1, which targets calcium channel α2δ1, reduces the self-renewal and tumorigenic capacity of LCSCs by affecting intracellular calcium signaling, thereby inhibiting HCC recurrence[80]. An anti-ANXA-3 antibody blocks the MKK4/JNK pathway in CD133+ LCSCs, attenuating cell self-renewal and inhibiting tumor growth[81]. When combined with sorafenib, the antitumor effect of anti-ANXA-3 antibodies was better. In addition, ANXA-3 can stratify resistance to sorafenib, which is beneficial to help HCC patients develop better treatment plans on their own[82]. Anti-Dickkopf-1 (DKK-1) inhibits angiogenesis and cancer cell proliferation in vitro and suppresses LCSCs growth in vivo[83].
Combination therapy of immunotherapy and stem cells targeting
| Therapeutic setting | Target cell | Molecule(s) | References |
| Anti-CD133 antibody | LCSCs | CD133 | [78] |
| Antibody(1B50-1) | LCSCs | α2δ1 | [80] |
| Anti-ANXA-3 antibody | LCSCs | ANXA-3 | [81] |
| Anti-Dickkopf-1 antibody | LCSCs | Dickkopf-1 | [83] |
| Antibody(G7mAb) | LCSCs | CD24 | [92] |
| G7mAb-conjugated doxorubicin | LCSCs | CD24 | [93] |
| G7mAb-conjugated HL-12 | LCSCs | CD24 | [94] |
| Anti-CD44 antibody-targeted liposomal nanoparticles | LCSCs | CD44 | [95] |
| Anti-CD44mAb-conjugated Nd3+-doped UCNPs | LCSCs | CD44 | [96] |
| CD90@TMs | LCSCs | CD90 | [97,98] |
| CD90@17-AAG | LCSCs | CD90 | [98] |
| Anti-EpCAM-UPGs-MX | LCSCs | EpCAM | [99] |
| Anti-GEPmAb (A23) | LCSCs | GEP | [11] |
| Anti-CEACAM1 antibody | LCSCs | GEP | [68] |
| Bispecific antibody(cG7-MICA) | NK/LCSCs | NKG2D/CD24 | [102] |
| CAR-T | LCSCs | CD44 | [125] |
| CAR-T | LCSCs | CD133 | [127] |
| EpCAM/CD3 bispecific antibody | T-cell/LCSCs | CD3/EpCAM | [136] |
The interaction between CD47 and SIRPα inhibits the phagocytosis of CSCs by macrophages[84,85]. By blocking the binding of CD47 and SIRPα, an anti-CD47 antibody restores the phagocytic activity of macrophages and inhibits HCC tumor growth[86]. Lo et al. found that treatment with chemotherapeutic drugs or sorafenib in combination with an anti-CD47 antibody reduced the occurrence of drug resistance and enhanced the antitumor effect of the drugs[87,88]. Recently, Du et al. invented a bispecific antibody targeting the HCC-associated antigen Glypican-3 (GPC3) and CD47, which could inhibit HCC development by enhancing the innate immune response involving macrophages and neutrophils[89]. In addition, blockade of CD47 can increase microenvironmental CD8+ cytotoxic T-cell infiltration as well as enhance tumor cell sensitivity to radiation therapy[90]. Anti-CD47 antibodies combined with T-cell immune checkpoint inhibitors may be an effective therapeutic strategy to achieve stronger antitumor benefits by exploiting the respective advantages.
Antibody-drug couples use specific monoclonal antibodies to transport cytotoxic agents, which can selectively kill tumor cells[91]. He et al. designed a CD24-targeting monoclonal antibody, G7 mAb, and used its combination drug to target LCSCs[92]. G7 mAb-conjugated doxorubicin (Dox) was shown to effectively inhibit tumor growth with less systemic toxicity in tumor-bearing nude mice[93]. G7 mAb can also form an immunoconjugate targeting CD24+ HCC with NO donor HL-12. This immunoconjugate not only induces apoptosis of LCSCs via antibody-dependent cytotoxicity, but also inhibits tumor growth by upregulating intratumor NO levels, exerting effective antitumor effects in vivo and in vitro[94].
Antibodies can also be conjugated to biomaterials such as nanoparticles or thermosensitive liposomes to achieve local-specific imaging and elimination of tumors. Anti-CD44 antibody-conjugated liposomal nanoparticles can specifically deliver the chemotherapeutic drug Dox or killer genes to target cells and induce their apoptosis. Such nanoparticles can also be combined with bioluminescence imaging to quantify the effect of in vivo killer gene therapy, thereby dynamically monitoring the tumor treatment process[95]. Nd3+-doped core-shell upconversion nanoparticles (Nd-CSUCNPs) are a novel multimodal imaging reporter combining magnetic resonance (MR) and real-time upconversion luminescence (UCL). Anti-CD44 antibody conjugated with Nd-CSUCNP can deliver the imaging tool to HCC and improve the accuracy of preoperative multimodal imaging-guided HCC surgical resection[96]. Thermosensitive magnetoliposomes (TMs) loaded with anti-CD90 antibodies (CD90@TMs), which are transported to LCSCs in a targeted manner, effectively kill CD90+ LCSCs by magnetic hyperthermia therapy[97]. Heat shock protein (HSP) inhibitors encapsulated in thermosensitive magnetic liposomes make LCSCs continuously sensitive to hyperthermia and then induce apoptosis[98]. Han et al. invented a nanoparticle micelle combining mitoxantrone (MX) and anti-EpCAM antibody, which can synergistically treat EpCAM+ HCC cells with drugs and optical targeting. Nanoparticle micelles can be used for MR/UCL dual-mode imaging, which can help to diagnose HCC more accurately[99]. A variety of specific antibodies have been shown to have good antitumor effects in preclinical trials, but their application in the clinical setting needs to be further explored. On this basis, the application of first-line drugs for the clinical treatment of HCC, such as sorafenib combined with LCSCs-specific antibodies, may be a promising therapeutic strategy. The combination therapy now seems to be effective, at least in preclinical trials, greatly inhibiting tumor growth and recurrence.
Lymphocytes targeting LCSCs
NK cells
NK cells protect the organism that produces them from damage while recognizing and killing harmful foreign substances such as viruses and tumors by expressing a series of activated and inhibitory receptors[100]. A preclinical trial found that the NK-cell-activated receptor ligands MICA/MICB, Fas, and DR5 were upregulated in CSCs, suggesting that NK cells preferentially target cancer stem cells[101]. Enhancing the strength and targeting of NK cells to kill CSCs may be an effective therapeutic strategy. Several studies have shown that NK cells can effectively kill LCSCs through antibody-dependent cell-mediated cytotoxicity (ADCC). As previously described, both GEP and CEACAM1 are upregulated in LCSCs[67]. Anti-GEP monoclonal antibody A23 or anti-CEACAM1 antibody can enhance the killing activity of NK cells and promote tumor regression[11,68]. cG7-MICA, a bispecific antibody against CD24 and NKG2D, recruits NK cells to the periphery of CSCs and promotes the release of IFN-γ and TNF-α from NK cells. In addition, cG7-MICA can enhance the inhibitory effect of sorafenib on tumors, suggesting that
The infusion of autologous or allogeneic NK cells after activation, culture and expansion in vitro can break the immune tolerance of the body and enhance the antitumor immune response[103]. The
T cells
Immune checkpoint
ICI therapy is an important approach in current immunotherapy, especially targeting the PD-1/PD-L1 pathways. Calderaro et al. found that PD-L1 was highly expressed in a subset of LCSCs[108]. Nishida et al. also demonstrated that PD-L1 expression in HCC was positively correlated with the presence of tumor stem cell markers, including cytokeratin 19 (CK19) and Sal-like protein 4 (SALL4)[34]. In addition, the accumulation of PD-L1 on CSCs promotes the occurrence of immune evasion[109]. Considering that CSCs induce CTLs apoptosis by binding to PD-1, immune checkpoint inhibitors may indirectly inhibit the growth of CSCs. Atezolizumab selectively targets PD-L1, restoring T cells’ ability to destroy tumor cells. The Food and Drug Administration (FAD) authorized the combination immunotherapy of atezolizumab and bevacizumab for the treatment of hepatocellular cancer in 2020. The combined drug group outperformed the sorafenib group in terms of OS rates, progression-free survival, and objective response rate in the Phase III clinical study (NCT03434379)[110]. Single ICI immunotherapy improved OS rates but did not reach prespecified statistical significance[111,112]. Combining ICIs with other treatments may be more effective and have more durable responses[113]. Antiangiogenic drugs can improve the microenvironment of solid tumors and enhance the sensitivity of ICI therapy to tumors, thereby improving their efficacy. Several clinical trials (NCT03434379, NCT03006926, NCT03794440, NCT03463876, etc.) have shown that anti-PD-1/PD-L1 inhibitors combined with antiangiogenic drugs have good tumor effects and prolong the survival of HCC patients[114-118].
An increased frequency of mutations in genes involved in the phosphatidyl inositol 3-kinase (PI3K)-Akt pathway was also observed in HCC with high PD-L1 expression[34]. In glioma, loss of phosphatase and tensin homolog (PTEN) function and activation of the PI3K pathway lead to upregulation of PD-L1 in tumor cells[119]. Correspondingly, downregulation of PTEN and upregulation of PI3KCA in HCC increased the expression of LCSC markers, such as CK19 and CD133[120]. Therefore, activation of the PI3K-Akt pathway may lead to PD-L1 overexpression and increased tumor self-renewal. Activation of the PI3K-Akt pathway is one of the hallmarks of CSCs, and PI3K inhibitors preferentially reduce the levels of CSCs in tumors[121]. Therefore, the combined targeting of PI3K-Akt pathway inhibitors and anti-PD-1/PD-L1 inhibitors may help suppress HCC growth, especially the LCSCs. This treatment may be a promising strategy for relapse-prone HCC patients, which needs to be confirmed by further preclinical studies.
Chimeric antigen receptor-specific T (CAR-T) cells
CAR-T cells express specific antigen receptors and recognize tumor-associated antigens (TAAs), thereby targeting tumor areas to perform cytotoxic T-cell functions. CAR-T-cell therapies targeting CD19 are efficient against hematological tumors, so the FDA has approved two related CAR-T products[122,123]. However, for solid tumors, CAR-T-cell efficacy still faces many challenges, such as virus-related risks and appropriate targets[124]. CSCs have the ability of self-renewal and differentiation and play an important role in tumor initiation and recurrence. Therefore, an increasing number of studies use markers of CSCs as TAAs recognized by CAR-T cells. After CAR-T cells targeting CD44+ LCSCs were infused into CD44+ HCC xenograft mice, they accumulated in CD44+ tumor regions and effectively inhibited tumor growth[125]. NKG2D-BBz CAR-T cells can effectively eliminate NKG2DL+ HCC cells in vitro and in vivo[126]. A phase I clinical trial involving CD133+ advanced metastatic malignancies (NCT02541370) revealed that CD133-specific CAR-T cells (CART-133) were a feasible and effective therapeutic strategy to stabilize the disease and prolong patient survival[127].
Tumor heterogeneity is a key factor affecting the efficacy of CAR-T therapy. Biopsy specimens from two patients revealed that CART-133 effectively eliminated CD133+ tumor cells, but some CD133- tumor cells that were not eliminated may cause tumor progression and recurrence[127]. This suggests that it is essential to inhibit the rapid growth of antigen-negative cells while improving the efficacy of CAR-T targeting antigen-positive cells. A phase I clinical trial (NCT02414269) revealed that the efficacy of CAR-T cells in the treatment of malignant pleural disease can be enhanced with the help of anti-PD-1 drug[128]. PD-1 blockade works through endogenous tumor-specific T cells, potentially eliminating antigen-negative cells left behind after CAR-T-cell treatment. The combination of the two complementary advantages is a promising antitumor strategy[129].
The limited expansion of CAR-T cells in vivo is also one of the important factors affecting their antitumor efficacy[130,131]. Coexpression of cytokines enhances the ability of CAR-T cells to persist against tumors in vivo. Batra et al. found that coexpression of IL15 and IL21 enhanced the antitumor strength and persistence of GP3-CAR T cells. Two clinical trials (NCT02932956 and NCT02905188) are exploring the therapeutic efficacy of this modified GP3-CAR T biohybrid in HCC patients[131]. Another clinical trial (NCT03198546) showed that CAR-T cells with upregulated IL-7 and CCL19 expression increased patient survival[132,133]. Hence, modification of CSC-specific CAR-T cells by coexpressing cytokines may enhance their efficacies for patients with HCC.
Other approaches
Therapeutic modalities such as small molecule inhibitors, bispecific antibodies, and cancer vaccines can also enhance the immune response of cytotoxic T cells to tumors. LDN193189, a small molecule compound, relieves the inhibition of Galectin-3 on the proliferation activity of CD8+ T cells by downregulating the expression of Galectin-3 in CSCs, thereby inhibiting the immune evasion ability of LCSCs[134]. Bispecific antibodies can simultaneously bind to two different specific antigens, such as TAAs of tumor cells and CD3 of T cells, which facilitates the accumulation of cytotoxic T cells at the tumor site. Anti-GPC3/CD3 bispecific antibodies rely on GPC3 to recruit cytotoxic T cells to the HCC xenograft region and effectively inhibit tumor growth[135]. Zhang et al. developed a bispecific antibody targeting EpCAM and CD3-specific antigens that induced strong cytotoxicity and inhibited the expression of stemness-associated genes in LCSCs. An EpCAM/CD3 bispecific antibody significantly inhibited the growth of HCC xenografts in vivo[136]. Catumaxomab, a trifunctional receptor targeting EpCAM and CD3, enhances antitumor activity by recruiting T cells and Fcγ receptor (FcγR)-positive helper cells to tumor sites. Several clinical studies have shown that catumaxomab can effectively remove tumor cells and prevent ascites accumulation in patients with ovarian cancer and gastric cancer[137,138]. Furthermore, compared with paracentesis alone, catumaxomab can prolong the survival time and maintain a better quality of life in malignant patients[139]. Catumaxomab has a good effect in the prevention and treatment of malignant patients, but its efficacy in solid tumors, including HCC, needs to be further explored in clinical trials.
Pulsed DCs carrying specific antigens can induce the activation of naive T lymphocytes to generate tumor cell antigen-specific CTLs, thereby inhibiting tumor growth in a targeted manner[140]. DCs loaded with CD133+ LCSCs RNA stimulate specific CTL proliferation and IFN-γ secretion in vivo and effectively inhibit tumor growth[141]. Choi et al. demonstrated that using EpCAM peptides as loaders for DC vaccines can effectively promote specific CTL activation and kill EpCAM+ LCSCs[142]. In addition to using proteins and nucleic acids as loaders for DC vaccines, fusion of LCSCs and DCs is also a potential strategy that may effectively activate multiple antigen-specific CILs for the immune response. Pang et al. revealed that CD90+HepG2/DC fusion cells upregulate T lymphocyte-mediated specific antitumor immune responses both in vivo and ex vivo[143]. Attenuated listeria monocytogenes (LM) is also a promising tumor vaccine vector that induces the activation and proliferation of antigen-specific T lymphocytes in vivo by upregulating the MHC-I and MHC-II pathways. Yang et al. prepared a novel cancer vaccine using LM replication-deficient LMΔdalΔdat strain-loaded CD24, which can reduce the number of Tregs in tumor TILs and enhance the activity of specific CD8+ T cells[144]. This vaccine effectively inhibits tumor growth by altering the balance between cells in the immunosuppressive tumor microenvironment and is a promising therapeutic tool. Cancer vaccines targeting CSCs produce antigen-specific CTLs in vivo and potently kill LCSCs. This therapeutic strategy may be effective in killing tumors and inhibiting tumor recurrence, and its efficacy needs to be further explored in the clinic.
PROSPECTIVE AND CONCLUSION
In the process of antitumor immunity, lymphocytes play an important role in eliminating tumors. CSCs are currently recognized as a key factor in tumor heterogeneity and tumor recurrence. During HCC progression, LCSCs participate in the formation of an immunosuppressive microenvironment by damaging or altering lymphocyte phenotypes, thereby evading immune surveillance. Understanding the interaction mechanisms between lymphocytes and LCSCs and reversing the immunosuppressive microenvironment could help develop more effective treatment strategies.
With the development of technologies such as single-cell sequencing and lineage tracing, we have the opportunity to further explore the origin of lymphocytes and CSCs in the tumor immune microenvironment and their interaction mechanisms in tumorigenesis and development. The current strategy of immunotherapy is mainly to reshape the immune function of lymphocytes, including immune checkpoint inhibitor therapy, CAR-T therapy, multifunctional antibodies, etc. Although immunotherapy has a good effect, high drug resistance and recurrence rates are still problems that need to be solved. Combination immunotherapy and other treatments, such as antiangiogenic drugs, can effectively improve the treatment effect and prolong the survival time of patients. Immunotherapy targeting LCSCs is also a promising treatment modality. This strategy not only kills tumors but also inhibits tumor recurrence in a targeted manner. At present, some preclinical experiments have shown that immunotherapy targeting LCSCs has good antitumor effects and low toxicity. For instance, anti-ANXA3 antibodies can block
DECLARATIONS
Authors’ contributionWrote the manuscript: Chen WM, Zhang XP
Assistance : Sun X, Yan YY
Supervised the project: Li L
All the authors have read and approved the final manuscript.
Availability of data and materialsNot applicable.
Financial support and sponsorshipThe present work was supported by the National Natural Science Foundation of China (82273474 and 82103680), the Basic and Applied Basic Research Foundation of Guangdong Province (2022A1515010068), and the Guangdong Science and Technology Department (2020B1212060018 and 2020B1212030004).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
REFERENCES
2. Islami F, Ward EM, Sung H, et al. Annual report to the nation on the status of cancer, part 1: national cancer statistics. J Natl Cancer Inst 2021;113:1648-69.
3. Ju MR, Yopp AC. Surgical resection of early stage hepatocellular carcinoma: balancing tumor biology with the host liver. Chin Clin Oncol 2021;10:5.
4. Roxburgh P, Evans TR. Systemic therapy of hepatocellular carcinoma: are we making progress? Adv Ther 2008;25:1089-104.
6. Tang J, Sui CJ, Wang DF, et al. Targeted sequencing reveals the mutational landscape responsible for sorafenib therapy in advanced hepatocellular carcinoma. Theranostics 2020;10:5384-97.
7. Clarke MF, Dick JE, Dirks PB, et al. Cancer stem cells-perspectives on current status and future directions: AACR wrkshop on cancer stem cells. Cancer Res 2006;66:9339-44.
8. Xiang Y, Yang T, Pang BY, Zhu Y, Liu YN. The progress and prospects of putative biomarkers for liver cancer stem cells in hepatocellular carcinoma. Stem Cells Int 2016;2016:7614971.
9. Ruiu R, Tarone L, Rolih V, et al. Cancer stem cell immunology and immunotherapy: Harnessing the immune system against cancer’s source. Cancer Immunotherapy. Elsevier; 2019. p. 119-88.
10. Lo Re O, Mazza T, Giallongo S, et al. Loss of histone macroH2A1 in hepatocellular carcinoma cells promotes paracrine-mediated chemoresistance and CD4+CD25+FoxP3+ regulatory T cells activation. Theranostics 2020;10:910-24.
11. Park DJ, Sung PS, Kim JH, et al. EpCAM-high liver cancer stem cells resist natural killer cell-mediated cytotoxicity by upregulating CEACAM1. J Immunother Cancer 2020;8:e000301.
12. Cheung PF, Yip CW, Wong NC, et al. Granulin-epithelin precursor renders hepatocellular carcinoma cells resistant to natural killer cytotoxicity. Cancer Immunol Res 2014;2:1209-19.
13. Lee TK, Guan XY, Ma S. Cancer stem cells in hepatocellular carcinoma - from origin to clinical implications. Nat Rev Gastroenterol Hepatol 2022;19:26-44.
14. Hao X, Sun G, Zhang Y, et al. Targeting immune cells in the tumor microenvironment of HCC: new opportunities and challenges. Front Cell Dev Biol 2021;9:775462.
15. Sperandio RC, Pestana RC, Miyamura BV, Kaseb AO. Hepatocellular carcinoma immunotherapy. Annu Rev Med 2022;73:267-78.
17. Farhood B, Najafi M, Mortezaee K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: a review. J Cell Physiol 2019;234:8509-21.
18. Bian J, Lin J, Long J et al. T lymphocytes in hepatocellular carcinoma immune microenvironment: insights into human immunology and immunotherapy. Am J Cancer Res 2020;10:4585-606.
19. Bálint Š, Müller S, Fischer R, et al. Supramolecular attack particles are autonomous killing entities released from cytotoxic T cells. Science 2020;368:897-901.
20. Logtenberg MEW, Scheeren FA, Schumacher TN. The CD47-SIRPα immune checkpoint. Immunity 2020;52:742-52.
21. Chiu DK, Yuen VW, Cheu JW, et al. Hepatocellular carcinoma cells up-regulate PVRL1, stabilizing PVR and inhibiting the cytotoxic T-Cell response via TIGIT to mediate tumor resistance to PD1 inhibitors in mice. Gastroenterology 2020;159:609-23.
22. Jansen CS, Prokhnevska N, Master VA, et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 2019;576:465-70.
23. Zheng C, Zheng L, Yoo JK, et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 2017;169:1342-1356.e16.
24. Yang Y, Liu F, Liu W, et al. Analysis of single-cell RNAseq identifies transitional states of T cells associated with hepatocellular carcinoma. Clin Transl Med 2020;10:e133.
25. Wang X, He Q, Shen H, et al. TOX promotes the exhaustion of antitumor CD8+ T cells by preventing PD1 degradation in hepatocellular carcinoma. J Hepatol 2019;71:731-41.
26. Barsch M, Salié H, Schlaak AE, et al. T-cell exhaustion and residency dynamics inform clinical outcomes in hepatocellular carcinoma. J Hepatol 2022;77:397-409.
27. McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol 2019;37:457-95.
28. Zhou G, Sprengers D, Boor PPC, et al. Antibodies against immune checkpoint molecules restore functions of tumor-infiltrating T Cells in hepatocellular carcinomas. Gastroenterology 2017;153:1107-1119.e10.
29. Wu Q, Zhou W, Yin S, et al. Blocking triggering receptor expressed on myeloid cells-1-positive tumor-associated macrophages induced by hypoxia reverses immunosuppression and anti-programmed cell death ligand 1 resistance in liver cancer. Hepatology 2019;70:198-214.
30. Tao H, Liu M, Wang Y, et al. Icaritin induces anti-tumor immune responses in hepatocellular carcinoma by inhibiting splenic myeloid-derived suppressor cell generation. Front Immunol 2021;12:609295.
31. Morrison BJ, Steel JC, Morris JC. Reduction of MHC-I expression limits T-lymphocyte-mediated killing of Cancer-initiating cells. BMC Cancer 2018;18:469.
32. Guo X, Zhao Y, Yan H, et al. Single tumor-initiating cells evade immune clearance by recruiting type II macrophages. Genes Dev 2017;31:247-59.
33. Gupta HB, Clark CA, Yuan B, et al. Tumor cell-intrinsic PD-L1 promotes tumor-initiating cell generation and functions in melanoma and ovarian cancer. Signal Transduct Target Ther 2016;1:16030.
34. Nishida N, Sakai K, Morita M, et al. Association between genetic and immunological background of hepatocellular carcinoma and expression of programmed cell death-1. Liver Cancer 2020;9:426-39.
35. Kaufhold S, Garbán H, Bonavida B. Yin Yang 1 is associated with cancer stem cell transcription factors (SOX2, OCT4, BMI1) and clinical implication. J Exp Clin Cancer Res 2016;35:84.
36. Zhong F, Cheng X, Sun S, Zhou J. Transcriptional activation of PD-L1 by Sox2 contributes to the proliferation of hepatocellular carcinoma cells. Oncol Rep 2017;37:3061-7.
37. Chan LC, Li CW, Xia W, et al. IL-6/JAK1 pathway drives PD-L1 Y112 phosphorylation to promote cancer immune evasion. J Clin Invest 2019;129:3324-38.
38. Huang CY, Wang Y, Luo GY, et al. Relationship between PD-L1 expression and CD8+ T-cell immune responses in hepatocellular carcinoma. J Immunother 2017;40:323-33.
39. Pan K, Zhao JJ, Wang H, et al. Comparative analysis of cytotoxic T lymphocyte response induced by dendritic cells loaded with hepatocellular carcinoma -derived RNA or cell lysate. Int J Biol Sci 2010;6:639-48.
40. Shibolet O, Alper R, Zlotogarov L, et al. NKT and CD8 lymphocytes mediate suppression of hepatocellular carcinoma growth via tumor antigen-pulsed dendritic cells. Int J Cancer 2003;106:236-43.
41. Yamashita T, Ji J, Budhu A, et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology 2009;136:1012-24.
42. Ruiz de Galarreta M, Bresnahan E, Molina-Sánchez P, et al. β-catenin activation promotes immune escape and resistance to Anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov 2019;9:1124-41.
43. Pardee AD, Shi J, Butterfield LH. Tumor-derived α-fetoprotein impairs the differentiation and T cell stimulatory activity of human dendritic cells. The Journal of Immunology 2014;193:5723-32.
44. Pan QZ, Pan K, Wang QJ, et al. Annexin A3 as a potential target for immunotherapy of liver cancer stem-like cells. Stem Cells 2015;33:354-66.
45. Chen X, Du Y, Huang Z. CD4+CD25+ Treg derived from hepatocellular carcinoma mice inhibits tumor immunity. Immunology Letters 2012;148:83-9.
46. Kryczek I, Liu R, Wang G, et al. FOXP3 Defines regulatory T cells in human tumor and autoimmune disease. Cancer Research 2009;69:3995-4000.
47. Cabrera R, Ararat M, Eksioglu EA, et al. Influence of serum and soluble CD25 (sCD25) on regulatory and effector T-cell function in hepatocellular carcinoma. Scand J Immunol 2010;72:293-301.
48. Ormandy LA, Hillemann T, Wedemeyer H, Manns MP, Greten TF, Korangy F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res 2005;65:2457-64.
49. Santoiemma PP, Powell DJ Jr. Tumor infiltrating lymphocytes in ovarian cancer. Cancer Biol Ther 2015;16:807-20.
50. Pedroza-Gonzalez A, Verhoef C, Ijzermans JN, et al. Activated tumor-infiltrating CD4+ regulatory T cells restrain antitumor immunity in patients with primary or metastatic liver cancer. Hepatology 2013;57:183-94.
51. Fu J, Xu D, Liu Z, et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology 2007;132:2328-39.
52. Jiang R, Tang J, Chen Y, et al. The long noncoding RNA lnc-EGFR stimulates T-regulatory cells differentiation thus promoting hepatocellular carcinoma immune evasion. Nat Commun 2017;8:15129.
53. Suthen S, Lim CJ, Nguyen PHD, et al. Hypoxia-driven immunosuppression by Treg and type-2 conventional dendritic cells in HCC. Hepatology 2022;76:1329-44.
54. Wang Z, He L, Li W, et al. GDF15 induces immunosuppression via CD48 on regulatory T cells in hepatocellular carcinoma. J Immunother Cancer 2021;9:e002787.
55. Watson MJ, Vignali PDA, Mullett SJ, et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature 2021;591:645-51.
56. Zhou Y, Wang B, Wu J, et al. Association of preoperative EpCAM Circulating Tumor Cells and peripheral Treg cell levels with early recurrence of hepatocellular carcinoma following radical hepatic resection. BMC Cancer 2016;16:506.
57. Cao W, Li M, Liu J, et al. LGR5 marks targetable tumor-initiating cells in mouse liver cancer. Nat Commun 2020;11:1961.
58. Ko CJ, Li CJ, Wu MY, Chu PY. Overexpression of LGR-5 as a Predictor of Poor Outcome in Patients with Hepatocellular Carcinoma. Int J Environ Res Public Health 2019;16:1836.
59. Liu XS, Lin XK, Mei Y, et al. Regulatory T cells promote overexpression of Lgr5 on gastric cancer cells via TGF-beta1 and confer poor prognosis in gastric cancer. Front Immunol 2019;10:1741.
60. Mima K, Okabe H, Ishimoto T, et al. CD44s regulates the TGF-β-mediated mesenchymal phenotype and is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Res 2012;72:3414-23.
61. Yang P, Li QJ, Feng Y, et al. TGF-β-miR-34a-CCL22 signaling-induced Treg cell recruitment promotes venous metastases of HBV-positive hepatocellular carcinoma. Cancer Cell 2012;22:291-303.
62. Sun C, Sun H, Zhang C, Tian Z. NK cell receptor imbalance and NK cell dysfunction in HBV infection and hepatocellular carcinoma. Cell Mol Immunol 2015;12:292-302.
63. Liu P, Chen L, Zhang H. Natural killer cells in liver disease and hepatocellular carcinoma and the NK cell-based immunotherapy. J Immunol Res 2018;2018:1206737.
64. Jinushi M, Takehara T, Tatsumi T, et al. Impairment of natural killer cell and dendritic cell functions by the soluble form of MHC class I-related chain A in advanced human hepatocellular carcinomas. J Hepatol 2005;43:1013-20.
65. Cadoux M, Caruso S, Pham S, et al. Expression of NKG2D ligands is downregulated by β-catenin signalling and associates with HCC aggressiveness. J Hepatol 2021;74:1386-97.
66. Rong XX, Wei F, Lin XL, et al. Recognition and killing of cancer stem-like cell population in hepatocellular carcinoma cells by cytokine-induced killer cells via NKG2d-ligands recognition. Oncoimmunology 2016;5:e1086060.
67. Cheung PF, Cheng CK, Wong NC, et al. Granulin-epithelin precursor is an oncofetal protein defining hepatic cancer stem cells. PLoS One 2011;6:e28246.
68. Cheung PF, Yip CW, Ng LW, et al. Restoration of natural killer activity in hepatocellular carcinoma by treatment with antibody against granulin-epithelin precursor. Oncoimmunology 2015;4:e1016706.
69. Zhao H, Jia H, Han Q, Zhang J. Homeobox containing 1 inhibits liver cancer progression by promoting autophagy as well as inhibiting stemness and immune escape. Oncol Rep 2018;40:1657-65.
70. Weng J, Han X, Liu K, et al. CD44 3'-Untranslated region functions as a competing endogenous RNA to enhance NK sensitivity of liver cancer stem cell by regulating ULBP2 expression. Int J Biol Sci 2019;15:1664-75.
71. Sautès-Fridman C, Lawand M, Giraldo NA, et al. Tertiary lymphoid structures in cancers: prognostic value, regulation, and manipulation for therapeutic intervention. Front Immunol 2016;7:407.
73. Fridman WH, Zitvogel L, Sautès-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol 2017;14:717-34.
74. Finkin S, Yuan D, Stein I, et al. Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nat Immunol 2015;16:1235-44.
75. Calderaro J, Petitprez F, Becht E, et al. Intra-tumoral tertiary lymphoid structures are associated with a low risk of early recurrence of hepatocellular carcinoma. J Hepatol 2019;70:58-65.
76. Wu R, Guo W, Qiu X, et al. Comprehensive analysis of spatial architecture in primary liver cancer. Sci Adv 2021;7:eabg3750.
77. Li L, Hao X, Qin J, et al. Antibody against CD44s inhibits pancreatic tumor initiation and postradiation recurrence in mice. Gastroenterology 2014;146:1108-18.
78. Chen H, Luo Z, Sun W, et al. Low glucose promotes CD133mAb-elicited cell death via inhibition of autophagy in hepatocarcinoma cells. Cancer Letters 2013;336:204-12.
79. Chetty C, Khumalo T, Da Costa Dias B, et al. Anti-LRP/LR specific antibody IgG1-iS18 impedes adhesion and invasion of liver cancer cells. PLoS One 2014;9:e96268.
80. Zhao W, Wang L, Han H, et al. 1B50-1, a mAb raised against recurrent tumor cells, targets liver tumor-initiating cells by binding to the calcium channel α2δ1 subunit. Cancer Cell 2013;23:541-56.
81. Tong M, Fung TM, Luk ST, et al. ANXA3/JNK signaling promotes self-renewal and tumor growth, and its blockade provides a therapeutic target for hepatocellular carcinoma. Stem Cell Reports 2015;5:45-59.
82. Tong M, Che N, Zhou L, et al. Efficacy of annexin A3 blockade in sensitizing hepatocellular carcinoma to sorafenib and regorafenib. J Hepatol 2018;69:826-39.
83. Suda T, Yamashita T, Sunagozaka H, et al. Dickkopf-1 promotes angiogenesis and is a biomarker for hepatic stem cell-like hepatocellular carcinoma. Int J Mol Sci 2022;23:2801.
84. Chao MP, Weissman IL, Majeti R. The CD47-SIRPα pathway in cancer immune evasion and potential therapeutic implications. Curr Opin Immunol 2012;24:225-32.
85. Lee TK, Cheung VC, Lu P, et al. Blockade of CD47-mediated cathepsin S/protease-activated receptor 2 signaling provides a therapeutic target for hepatocellular carcinoma. Hepatology 2014;60:179-91.
86. Xiao Z, Chung H, Banan B, et al. Antibody mediated therapy targeting CD47 inhibits tumor progression of hepatocellular carcinoma. Cancer Lett 2015;360:302-9.
87. Lo J, Lau EY, So FT, et al. Anti-CD47 antibody suppresses tumour growth and augments the effect of chemotherapy treatment in hepatocellular carcinoma. Liver Int 2016;36:737-45.
88. Lo J, Lau EY, Ching RH, et al. Nuclear factor kappa B-mediated CD47 up-regulation promotes sorafenib resistance and its blockade synergizes the effect of sorafenib in hepatocellular carcinoma in mice. Hepatology 2015;62:534-45.
89. Du K, Li Y, Liu J, et al. A bispecific antibody targeting GPC3 and CD47 induced enhanced antitumor efficacy against dual antigen-expressing HCC. Mol Ther 2021;29:1572-84.
90. Soto-Pantoja DR, Terabe M, Ghosh A, et al. CD47 in the tumor microenvironment limits cooperation between antitumor T-cell immunity and radiotherapy. Cancer Res 2014;74:6771-83.
91. Murali M, Kumar AR, Nair B, et al. Antibody-drug conjugate as targeted therapeutics against hepatocellular carcinoma: preclinical studies and clinical relevance. Clin Transl Oncol 2022;24:407-31.
92. He H, Tu X, Zhang J, et al. A novel antibody targeting CD24 and hepatocellular carcinoma in vivo by near-infrared fluorescence imaging. Immunobiology 2015;220:1328-36.
93. Ma Z, He H, Sun F, et al. Selective targeted delivery of doxorubicin via conjugating to anti-CD24 antibody results in enhanced antitumor potency for hepatocellular carcinoma both in vitro and in vivo. J Cancer Res Clin Oncol 2017;143:1929-40.
94. Sun F, Wang Y, Luo X, et al. Anti-CD24 Antibody-nitric oxide conjugate selectively and potently suppresses hepatic carcinoma. Cancer Research 2019;79:3395-405.
95. Wang L, Su W, Liu Z, et al. CD44 antibody-targeted liposomal nanoparticles for molecular imaging and therapy of hepatocellular carcinoma. Biomaterials 2012;33:5107-14.
96. Lee J, Gordon AC, Kim H, et al. Targeted multimodal nano-reporters for pre-procedural MRI and intra-operative image-guidance. Biomaterials 2016;109:69-77.
97. Yang R, An LY, Miao QF, et al. Effective elimination of liver cancer stem-like cells by CD90 antibody targeted thermosensitive magnetoliposomes. Oncotarget 2016;7:35894-916.
98. An Y, Yang R, Wang X, et al. Facile Assembly of thermosensitive liposomes for active targeting imaging and synergetic chemo-/magnetic hyperthermia therapy. Front Bioeng Biotechnol 2021;9:691091.
99. Han Y, An Y, Jia G, et al. Theranostic micelles based on upconversion nanoparticles for dual-modality imaging and photodynamic therapy in hepatocellular carcinoma. Nanoscale 2018;10:6511-23.
101. Ames E, Canter RJ, Grossenbacher SK, et al. NK Cells preferentially target tumor cells with a cancer stem cell phenotype. J Immunol 2015;195:4010-9.
102. Han Y, Sun F, Zhang X, et al. CD24 targeting bi-specific antibody that simultaneously stimulates NKG2D enhances the efficacy of cancer immunotherapy. J Cancer Res Clin Oncol 2019;145:1179-90.
103. Yu M, Li Z. Natural killer cells in hepatocellular carcinoma: current status and perspectives for future immunotherapeutic approaches. Front Med 2017;11:509-21.
104. Chang YH, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res 2013;73:1777-86.
105. Kamiya T, Chang YH, Campana D. Expanded and activated natural killer cells for immunotherapy of hepatocellular carcinoma. Cancer Immunol Res 2016;4:574-81.
106. Lu SJ, Feng Q. CAR-NK cells from engineered pluripotent stem cells: Off-the-shelf therapeutics for all patients. Stem Cells Transl Med 2021;10 Suppl 2:S10-7.
107. Wrona E, Borowiec M, Potemski P. CAR-NK cells in the treatment of solid tumors. Int J Mol Sci 2021;22:5899.
108. Calderaro J, Rousseau B, Amaddeo G, et al. Programmed death ligand 1 expression in hepatocellular carcinoma: relationship with clinical and pathological features. Hepatology 2016;64:2038-46.
109. Hsu JM, Xia W, Hsu YH, et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat Commun 2018;9:1908.
110. Finn RS, Qin S, Ikeda M, et al. IMbrave150 Investigators. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med 2020;382:1894-905.
111. Finn RS, Ryoo B, Merle P, et al. for the KEYNOTE-240 Investigators. Results of KEYNOTE-240: phase 3 study of pembrolizumab (Pembro) vs best supportive care (BSC) for second line therapy in advanced hepatocellular carcinoma (HCC). JCO 2019;37:4004-4004.
112. Yau T, Park J, Finn R, et al. CheckMate 459: a randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Ann Oncol 2019;30:v874-5.
113. Nishida N, Kudo M. Immune phenotype and immune checkpoint inhibitors for the treatment of human hepatocellular carcinoma. Cancers (Basel) 2020;12:1274.
114. Finn RS, Qin S, Ikeda M, et al. IMbrave150: updated overall survival (OS) data from a global, randomized, open-label phase III study of atezolizumab (atezo) + bevacizumab (bev) versus sorafenib (sor) in patients (pts) with unresectable hepatocellular carcinoma (HCC). JCO 2021;39:267-267.
115. Zhu AX, Finn RS, Ikeda M, et al. A phase Ib study of lenvatinib (LEN) plus pembrolizumab (PEMBRO) in unresectable hepatocellular carcinoma (uHCC). JCO 2020;38:4519-4519.
116. Ren Z, Xu J, Bai Y, et al. ORIENT-32 study group. Sintilimab plus a bevacizumab biosimilar (IBI305) versus sorafenib in unresectable hepatocellular carcinoma (ORIENT-32): a randomised, open-label, phase 2-3 study. Lancet Oncol 2021;22:977-90.
117. Xu J, Shen J, Gu S, et al. Camrelizumab in combination with apatinib in patients with advanced hepatocellular carcinoma (RESCUE): a nonrandomized, open-label, Phase II trial. Clin Cancer Res 2021;27:1003-11.
118. Zhang Y, Xu J, Shen J, et al. Update on overall survival (OS) of RESCUE: an open-label, phase 2 trial of camrelizumab (C) in combination with apatinib (A) in patients with advanced hepatocellular carcinoma (HCC). JCO 2021;39:4076-4076.
119. Parsa AT, Waldron JS, Panner A, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med 2007;13:84-8.
120. Chen D, Li Z, Cheng Q, et al. Genetic alterations and expression of PTEN and its relationship with cancer stem cell markers to investigate pathogenesis and to evaluate prognosis in hepatocellular carcinoma. J Clin Pathol 2019;72:588-96.
121. Zhou H, Yu C, Kong L, et al. B591, a novel specific pan-PI3K inhibitor, preferentially targets cancer stem cells. Oncogene 2019;38:3371-86.
122. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med 2018;378:439-48.
123. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-Cell therapy in refractory large B-cell lymphoma. N Engl J Med 2017;377:2531-44.
125. Wang H, Ye X, Ju Y, et al. Minicircle DNA-mediated CAR T cells targeting CD44 suppressed hepatocellular carcinoma both in vitro and in vivo. Onco Targets Ther 2020;13:3703-16.
126. Sun B, Yang D, Dai H, et al. Eradication Of Hepatocellular Carcinoma by NKG2D-Based CAR-T cells. Cancer Immunol Res 2019;7:1813-23.
127. Wang Y, Chen M, Wu Z, et al. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. Oncoimmunology 2018;7:e1440169.
128. Adusumilli PS, Zauderer MG, Rivière I, et al. A Phase I trial of regional mesothelin-targeted cAR T-cell therapy in patients with malignant pleural disease, in combination with the anti-PD-1 agent pembrolizumab. Cancer Discov 2021;11:2748-63.
129. Davies JS, Karimipour F, Zhang L, et al. Non-synergy of PD-1 blockade with T-cell therapy in solid tumors. J Immunother Cancer 2022;10:e004906.
130. Zhai B, Shi D, Gao H, et al. A phase I study of anti-GPC3 chimeric antigen receptor modified T cells (GPC3 CAR-T) in Chinese patients with refractory or relapsed GPC3+ hepatocellular carcinoma (r/r GPC3+ HCC). JCO 2017;35:3049-3049.
131. Batra SA, Rathi P, Guo L, et al. Glypican-3-specific CAR T cells coexpressing IL15 and IL21 have superior expansion and antitumor activity against hepatocellular carcinoma. Cancer Immunol Res 2020;8:309-20.
132. Pang N, Shi J, Qin L, et al. IL-7 and CCL19-secreting CAR-T cell therapy for tumors with positive glypican-3 or mesothelin. J Hematol Oncol 2021;14:118.
133. Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol 2018;36:346-51.
134. Liang Z, Wu B, Ji Z, et al. The binding of LDN193189 to CD133 C-terminus suppresses the tumorigenesis and immune escape of liver tumor-initiating cells. Cancer Lett 2021;513:90-100.
135. Yu L, Yang X, Huang N, et al. A novel targeted GPC3/CD3 bispecific antibody for the treatment hepatocellular carcinoma. Cancer Biol Ther 2020;21:597-603.
136. Zhang P, Shi B, Gao H, et al. An EpCAM/CD3 bispecific antibody efficiently eliminates hepatocellular carcinoma cells with limited galectin-1 expression. Cancer Immunol Immunother 2014;63:121-32.
137. Burges A, Wimberger P, Kümper C, et al. Effective relief of malignant ascites in patients with advanced ovarian cancer by a trifunctional anti-EpCAM x anti-CD3 antibody: a phase I/II study. Clin Cancer Res 2007;13:3899-905.
138. Heiss MM, Murawa P, Koralewski P, et al. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: Results of a prospective randomized phase II/III trial. Int J Cancer 2010;127:2209-21.
139. Wimberger P, Gilet H, Gonschior AK, et al. Deterioration in quality of life (QoL) in patients with malignant ascites: results from a phase II/III study comparing paracentesis plus catumaxomab with paracentesis alone. Ann Oncol 2012;23:1979-85.
140. Wierecky J, Mueller M, Brossart P. Dendritic cell-based cancer immunotherapy targeting MUC-1. Cancer Immunol Immunother 2006;55:63-7.
141. Sun JC, Pan K, Chen MS, et al. Dendritic cells-mediated CTLs targeting hepatocellular carcinoma stem cells. Cancer Biol Ther 2010;10:368-75.
142. Choi YJ, Park SJ, Park YS, Park HS, Yang KM, Heo K. EpCAM peptide-primed dendritic cell vaccination confers significant anti-tumor immunity in hepatocellular carcinoma cells. PLoS One 2018;13:e0190638.
143. Pang YB, He J, Cui BY, et al. A potential antitumor effect of dendritic cells fused with cancer stem cells in hepatocellular carcinoma. Stem Cells Int 2019;2019:5680327.
144. Yang Y, Hou J, Lin Z, et al. Attenuated Listeria monocytogenes as a cancer vaccine vector for the delivery of CD24, a biomarker for hepatic cancer stem cells. Cell Mol Immunol 2014;11:184-96.
145. Héninger E, Krueger TE, Lang JM. Augmenting antitumor immune responses with epigenetic modifying agents. Front Immunol 2015;6:29.
146. Bai X, Zhou Y, Yokota Y, et al. Adaptive antitumor immune response stimulated by bio-nanoparticle based vaccine and checkpoint blockade. J Exp Clin Cancer Res 2022;41:132.
147. Woller N, Gürlevik E, Fleischmann-Mundt b, et al. viral infection of tumors overcomes resistance to PD-1-immunotherapy by broadening neoantigenome-directed T-cell responses. Mol Ther 2015;23:1630-40.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Chen WM, Zhang XP, Yan YY, Sun X, Li L. Targeting the interactions between lymphocytes and liver cancer stem cells in combination with immunotherapy is a promising therapeutic strategy. Hepatoma Res 2023;9:2. http://dx.doi.org/10.20517/2394-5079.2022.52
AMA Style
Chen WM, Zhang XP, Yan YY, Sun X, Li L. Targeting the interactions between lymphocytes and liver cancer stem cells in combination with immunotherapy is a promising therapeutic strategy. Hepatoma Research. 2023; 9: 2. http://dx.doi.org/10.20517/2394-5079.2022.52
Chicago/Turabian Style
Chen, Wo-Ming, Xiao-Ping Zhang, Yuan-Yuan Yan, Xiao Sun, Lei Li. 2023. "Targeting the interactions between lymphocytes and liver cancer stem cells in combination with immunotherapy is a promising therapeutic strategy" Hepatoma Research. 9: 2. http://dx.doi.org/10.20517/2394-5079.2022.52
ACS Style
Chen, W.M.; Zhang X.P.; Yan Y.Y.; Sun X.; Li L. Targeting the interactions between lymphocytes and liver cancer stem cells in combination with immunotherapy is a promising therapeutic strategy. Hepatoma. Res. 2023, 9, 2. http://dx.doi.org/10.20517/2394-5079.2022.52
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 10 clicks
Cite This Article 10 clicks

 Like This Article 25
likes
Like This Article 25
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.