
Molecular links between non-alcoholic fatty liver disease and hepatocellular carcinoma


Abstract
Non-alcoholic fatty liver disease (NAFLD) and its advanced complication, non-alcoholic steatohepatitis (NASH), have become leading causes of hepatocellular carcinoma (HCC) worldwide. In this review, we discuss the role of metabolic, gut microbial, immune and endocrine mediators which promote the progression of NAFLD to HCC. In particular, this progression involves multiple hits resulting from lipotoxicity, oxidative stress, inhibition of hepatic autophagy and inflammation. Furthermore, dysbiosis in the gut associated with obesity also promotes HCC via induction of proinflammatory cytokines and Toll like receptor signalling as well as altered bile metabolism. Additionally, compromised T-cell function and impaired hepatic hormonal action promote the development of NASH-associated HCC. Lastly, we discuss the current challenges involved in the diagnosis and treatment of NAFLD/NASH-associated HCC.
Keywords
Introduction
Hepatocellular carcinoma (HCC) is the primary form of liver cancer and is a leading cause of cancer-related mortality worldwide[1]. It is predominantly known to occur in patients suffering from underlying chronic liver disease and cirrhosis. Hepatitis B and C virus (HBV and HCV, respectively) infections, excessive consumption of alcohol and non-alcoholic fatty liver disease (NAFLD) historically have been recognised as the major causes of HCC; however, the incidence of virus-associated HCC is expected to decrease in the near future due to the development of effective and inexpensive vaccines for HBV and potent anti-HCV drugs[1,2]. In contrast, the prevalence of non-viral hepatitis continues to rise and has become the major cause for liver transplantation in Europe and the USA[3]. The increased prevalence of metabolic disorders, particularly diabetes, NAFLD and obesity, have led to changes in the epidemiology and aetiology of HCC[4]. Obesity is considered a risk factor for hepatic complications such as NAFLD and HCC[5-9]. Although 17%-33% of the general population is estimated to be affected by NAFLD, it reaches 75% in obese individuals and is even higher in patients with type II diabetes mellitus (T2DM)[10,11]. Moreover, T2DM itself is associated with an increased risk of liver damage[12], including HCC[13-15]. Chronic damage to liver metabolism caused by alcohol and poor nutrition leads to alcoholic liver disease that can co-exist with NAFLD/non-alcoholic steatohepatistis (NASH), and thereby increases both the progression of NAFLD and the risk for NAFLD/NASH-associated HCC[2,3].
This review focuses on NAFLD-associated HCC, and describes its epidemiology and the clinical, cellular, metabolic, microbiome and endocrine factors that promote the development of HCC from NAFLD. We also examine the molecular pathways that lead to progression from NAFLD to HCC as well as the challenges and future directions for its treatment and prevention.
NAFLD increases the risk of liver cancer
NAFLD encompasses a spectrum of liver pathologies which involve an accumulation of triglycerides in the hepatocytes, hepatocyte apoptosis, liver inflammation and fibrosis termed as NASH, and, in extreme cases, it can progress to cirrhosis and HCC[16]. NAFLD is the most common cause of HCC across the globe[16-28]. Although the progression of NAFLD to HCC involves NASH and cirrhosis, the direct development of HCC from benign steatosis or non-cirrhotic NASH has also been reported[29,30]. The increased prevalence of the underlying liver disease in the general population has led to an increase of 9% in the annual rates of incidence of NAFLD-associated HCC[31]. Interestingly, HCC can progress from NASH as well as cirrhosis. In a study cohort based on 756 patients, Piscaglia et al.[32] reported that 46.2% of the NAFLD associated HCC cases occurred without cirrhosis. Similar results were reported by a Japanese study, in which 49% of NAFLD associated HCC cases arose without cirrhosis[33], and a German study where 41.7% of the cases arose without cirrhosis[34]. Furthermore, in animal models, diet-induced NAFLD leads to spontaneous HCC[35].
Cellular mechanisms involved in NAFLD pathogenesis
NAFLD is a complex disease with multiple modifiers such as diet, lifestyle and gut microbiota which act in a susceptible genetic/epigenetic environment and modulate response to calorific excess[36,37]. The role of insulin resistance is central to this pathophysiological process and causes an increase in hepatic fat accumulation by increased deposition of free fatty acids (FFAs)[38]. This leads to oxidative stress, protein misfolding, autophagy inhibition and mitochondrial damage within hepatocytes, termed as “lipotoxicity”[38]. Chronic lipotoxicity challenges hepatocytes with both oxidative and endoplasmic reticulum (ER) stress. Oxidative stress mediated by reactive oxygen/nitrogen species (ROS/RNS) play a major role in NAFLD/NASH pathogenesis and complications. The high production of ROS causes mitochondrial damage, lipid peroxidation and low-density lipoprotein oxidation culminating into inflammation, activation of hepatic stellate cells (HSCs) leading to fibrogenesis, necrosis, cirrhosis and HCC[39].
ER stress is cell activated to regulate protein synthesis and restore homeostatic equilibrium in response to accumulation of unfolded or misfolded proteins. However, deregulated or insufficient responses to ER stress in liver may lead to lipid accumulation, insulin resistance, inflammation and apoptosis, all of which play important roles in the pathogenesis of NAFLD[40]. These events lead to inflammation and fibrosis as macrophage infiltration, hepatic progenitor cell activation and fibrogenesis ensue[41,42]. There are multiple factors that contribute to the pathogenesis of NAFLD and its progression. These include: dysregulated lipid metabolism, oxidative stress, ER stress, mitochondrial dysfunction, altered immune function, and gut-microbiota imbalance acting together in a genetic/epigenetic environment [Figure 1].
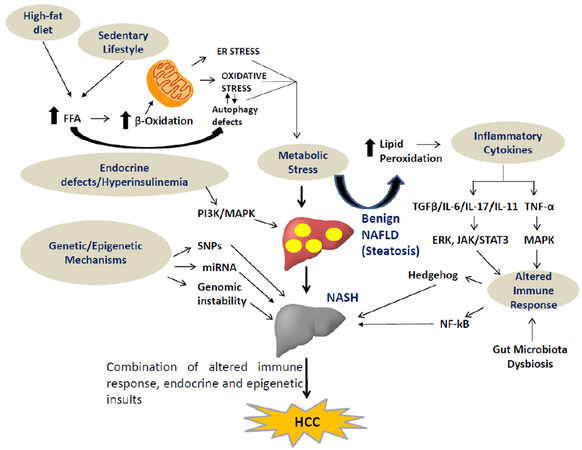
Figure 1. Multiple hits lead to onset and progression of NAFLD/NASH to HCC. Diverse signalling pathways involved in metabolic stress such as FFAs ER-stress, cytokine production (IL-6, IL-17, IL-11 and TGF-β), altered immune response, pro-fibrogenic mediators (hedgehog and NF-κB), gut dysbiosis and endocrine defects drive the development of NAFLD/NASH-associated HCC. NAFLD: non-alcoholic fatty liver disease; NASH: non-alcoholic steatohepatitis; HCC: hepatocellular carcinoma; FFAs: free fatty acids; ER: endoplasmic reticulum; IL-6: interleukin-6; IL-17: interleukin-17; IL-11: interleukin-11; TGF-β: transforming growth factor β; SNPs: single nucleotide polymorphisms; miRNA: micro RNA; PI3K: phosphatidylinositol 3-kinases; MAPK: mitogen-activated protein kinase; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B-cells; TNF-α: tumour necrosis factor-alpha; ERK: extracellular receptor kinase; JAK: Janus kinase; STAT: signal transducer and activator of transcription
Molecular mechanisms leading to progression from NAFLD to HCC
Studies on the development of HCC suggest that carcinogenesis in hepatocytes is a consequence of genetic/epigenetic alterations as well as complex changes in energy metabolism, cell growth and proliferation and immune signalling pathways. These changes in the cells lead to inflammation, hepatocyte injury, fibrosis and progression to HCC [Figure 1].
Genetic/epigenetic mechanisms
Several single nucleotide polymorphisms (SNPs) have been associated with the occurrence of NAFLD and its progression to advanced fibrosis[36]. The patatin-like phospholipase domain-containing protein 3 (PNPLA3) gene polymorphism is associated with the progression of NASH-associated HCC[43]. PNPLA3 impairs triglyceride mobilisation from lipid droplets. Patients carrying the PNPLA3 polymorphism are reported to have a three-fold increased risk of developing HCC[44,45]. Transmembrane 6 superfamily member 2 gene (TM6SF2) mutations are also prevalent in NASH patients[46]. They are believed to be linked to liver injury in the pathogenesis of NASH-associated HCC[47]. Recently, membrane bound O-acetyl transferase domain containing 7 (MBOAT7) rs641738 variant associated with NAFLD progression has also been linked to HCC susceptibility[48][Figure 1].
In addition to the SNPs, genetic instability is also believed to stimulate the progression of NASH to HCC. Mutations in oncogenic genes, such as the human telomerase reverse transcriptase (hTERT) gene which catalyses the addition of nucleotides to the ends of eukaryotic chromosomes, tumour protein p53, cyclin dependent kinase inhibitor 2 A, albumin, catenin beta-1 and axis inhibition protein 1 (involved in Wnt/β-catenin signalling), are prevalent in exome-sequencing analysis of HCC[49]. Aberrant DNA methylation is also an important mechanism in NASH progression[50], and can lead to silencing of genes involved in DNA repair, lipid metabolism, glucose metabolism and progression of fibrosis[50]. In particular, the epigenetic changes in the gene encoding chromodomain helicase DNA-binding protein 1 are reported to be linked to NASH-associated HCC[51].
The expression of several microRNAs (miRNAs) also is reported to be dysregulated in many types of cancer, including NASH-associated HCC[52]. The miRNAs are small noncoding RNAs that down-regulate gene expression by interfering with transcription and/or translation. These miRNAs are involved in cell signalling pathways associated with oncogenesis, such as transforming growth factor (TGF)-β, Wnt/β-catenin, mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinases (PI3K)/AKT/mTOR pathways, which can be activated in HCC[53]. In particular, miRNAs known to target the inhibitors of PI3K/AKT pathway are found in HCC. In this connection, steatosis, hepatomegaly and HCC have been observed in phosphatase and tensin-deficient mice[54]. Several miRNAs are differentially expressed in a high fat diet mouse model during the NAFLD-NASH-HCC transitions[55]. Hepatic miR-340-5p, miR-484, miR-574-3p and miR-720 were expressed in NAFLD, NASH and HCC, and miR-125a-5p and miR-182 showed early and significant dysregulation during hepatic tissue damage[55].
Metabolic pathways
The association of obesity, high-fat diet and diabetes to NAFLD/NASH and its progression to HCC suggests the existence of a molecular link between energy metabolism and cell cycle control in the hepatocytes, which may be a key mechanism driving the progression of NASH to HCC. Several animal studies have been conducted to investigate NASH-associated HCC. These studies showed that the progression of NASH-associated HCC may be due to abnormal lipid metabolism, oxidative stress, ER stress and mitochondrial dysfunction acting independently or in tandem[56,57][Figure 1].
It is worth noting that mitochondrial activities such as β-oxidation, electron transfer, ATP production and ROS generation regulate the fat metabolism and energy homeostasis in hepatocytes[58]. During the early hepatosteatoic phase of NAFLD when there is fatty acid accumulation in hepatic cells, mitochondria prevent oxidative stress and help facilitate the partition of lipotoxic FFAs into stable triglycerides that can be stored in fat droplet, and thereby prevent oxidative stress[59]. However, chronic high-fat or high-fructose diet leads to lipid over-accumulation in the hepatocytes due to cellular metabolic reprogramming and accumulation of toxic metabolites[60]. These changes lead to imbalances in hepatic metabolism that result in excessive production of FFAs, which can cause lipotoxicity[61].
The excessive accumulation of these fatty acids increases β-oxidation and ROS production, which can limit mitochondrial function[62]. When these mitochondrial abnormalities are accompanied by diminished intracellular antioxidant protection in NASH, pathways of fatty acid metabolism are altered[63], which, in turn, can cause metabolic stress[63]. Overproduction of ROS frequently occurs in cancer, and is believed to play an important role in the development of HCC[64]. Intriguingly, oxidative stress and abnormal methylation of tumour suppressor genes are found the livers of NAFLD patients[65]. ER stress also contributes to hepatocyte injury and carcinogenesis in NASH[66]. Thus, the cross-talk among oxidative stress, ER stress and cell death pathways likely plays a role in the development of NASH and its progression to HCC[67]. Similar to oxidative and ER stress, autophagy dysregulation may be involved in the progression of NASH to HCC[68]. In this regard, impaired autophagy leads to defective lipid metabolism[69], proteotoxicity[70], mitochondrial dysfunction[71] and inflammation[72], all of which can contribute to HCC induction.
Several xenobiotic metabolising genes of the aldo-keto reductase family show parallel induction in NASH and HCC, suggesting a genetic link between NASH and its progression to HCC[73-76]. Thus, disturbance in hepatic cell metabolism can lead to increased cell death, DNA damage, immune cell activation and compensatory proliferation[57]. These changes in hepatic cells activate HSCs and induce fibrosis. If tumour surveillance and DNA damage repair are impaired in NASH, pre-malignant cells can develop, and, after critical genetic/epigenetic changes, they become clones that progress to HCC. Therefore, the cumulative effects of oxidative stress and proliferative response during inflammation and fibrosis are thought to drive the progression of HCC[57].
Gut microbiota
“Gut microbiota” refers to populations of bacteria hosted by the adult human intestine, which maintain a symbiotic relationship with the host and have a key role in the host immune system. They perform various functions in the body such as digestion of inaccessible nutrients, synthesis of vitamins and resistance to pathogens[77]. They are known to ferment carbohydrates such as cellulose and xylans into short-chain fatty acids (SCFAs). The liver is exposed to gut-derived products by portal circulation, which provides a defence against bacterial toxins. SCFAs improve hepatic autophagy and gut barrier function, and reduce the permeability of bacterial toxins. These gut products can reduce pro-inflammatory pathways and insulin resistance, which are associated with the progression of chronic liver disease[78,79]. The gut microbiota composition is dynamic and may be influenced by diet, hygiene and the use of antibiotics[80]. The modification of the normal microbiota termed as “dysbiosis” is believed to be associated with the progression of NAFLD and other chronic metabolic diseases[81-85].
Dysbiosis in gut flora has been associated with HCC incidence in humans and animal models[86]. Mice kept in germ-free conditions or given antibiotics tend to develop fewer and smaller HCCs[87,88]. At the molecular level, dysbiosis of the gut microbiota leads to an increase in secretion of inflammatory cytokines, such as tumour necrosis factor alpha and interleukin-8 (IL-8) along with the activation of toll like receptor (TLR)-4 and TLR-9, resulting in production of IL-1β by Kupffer cells, which are star-shaped (stellate) phagocytic cells located in the liver. IL-1β promotes lipid accumulation and apoptosis in hepatocytes, causing steatosis and inflammation, as well as activation of HSCs to produce fibrogenic mediators, and accelerate HCC establishment[89-92]. Furthermore, dysbiosis promotes the development of NAFLD-associated HCC by modifying bile acid metabolism. Specifically, alterations in the composition of the gut microbiota can result in higher levels of deoxycholic acid and the activation of its receptor farnesoid X receptor, which provokes a senescence-associated secretory phenotype in HSCs, resulting in the secretion of various inflammatory and tumour-promoting factors in the liver, thus promoting the development of HCCs[87,93][Figure 1]. To summarise, the intestinal microbiota may promote the development of NAFLD-associated cirrhosis and HCC by increasing inflammatory cytokine secretion, activating TLR-4 and TLR-9 and modifying bile acid metabolism.
Immunological pathways
Metabolic stress not only leads to increased ROS generation but also triggers the inflammatory responses, which are a pre-requisite for the progression of NASH-associated HCC. Insulin resistance and oxidative stress are known to stimulate nuclear factor kappa-light-chain-enhancer of activated B-cells pathway, which promotes hepatocyte survival[94]. ROS and the products of lipid peroxidation stimulate the release of inflammatory cytokines including tumour necrosis factor-alpha (TNF-α) and IL-6 from hepatic cells[95]. TNF-α is reported to promote hepatocellular carcinogenesis by activating hepatic progenitor cells[67]. IL-6 activates the signal transducer and activator of transcription 3, an oncogenic transcription factor that induces cell proliferation, inhibits apoptotic pathways and may be involved in the development of NASH-associated HCC[96][Figure 1]. Similarly, other cytokines such as IL-17A and IL-11 have also been implicated in NASH and NASH-associated HCC[97-99].
Cellular injury activates the hedgehog signalling pathway, which is involved in repair and regeneration in the liver and replaces damaged hepatocyte. The Hedgehog signalling pathway is implicated in fibrogenic activation and hepatocellular ballooning[100], features associated with the advancement of NASH. Impairment of the Hedgehog pathway also leads to dysregulation of cell repair mechanisms and promotes the malignant transformation involved in the progression of HCC[101]. The TGF-β signalling also mediates the progression of fibrogenesis through regulation of cell death and lipid metabolism in NASH[102,103][Figure 1].
The role of CD8+ and CD4+ T lymphocytes in hepatocyte damage and carcinogenesis has been studied in various animal models[104]. In mouse models and human samples, dysregulation of lipid metabolism in NAFLD causes a selective loss of intrahepatic CD4+ but not CD8+ T lymphocytes, leading to increased inflammation and accelerated development of HCCs[105]. Platelet cargo, platelet adhesion and platelet activation appear to be pivotal for NASH and subsequent hepatocarcinogenesis. In particular, platelet GPIbα is a mediator of hepatic immune cell trafficking and antiplatelet therapy (e.g., aspirin/clopidogrel and ticagrelor) has been demonstrated to prevent NASH and subsequent HCC development[106]. One recently published study used a new zebra fish model and reported that a high fat diet promotes non-resolving inflammation in the liver and enhances cancer progression[107]. The authors found that metformin inhibits high fat diet-induced HCC progression, by reducing inflammation and restoring tumour surveillance[107].
Endocrine pathways
Several hormones play important roles in the pathogenesis of NAFLD and its consequent progression to HCC. One of the most crucial in these is insulin resistance and hyperinsulinemia, which is an associated feature of NAFLD[108]. Insulin resistance and hyperinsulinemia are known to increase the expression of insulin and insulin-like growth factor-1 (IGF-1). Insulin and IGF-1 trigger signalling cascades by binding to their receptors, namely the insulin receptor and the IGF-1 receptor, to activate the PI3K and MAPK pathways, which are crucial in the pathogenesis of HCC since they induce cell proliferation and inhibit apoptosis[109]. In particular, the PI3K pathway mediates the progression of HCC by cyclin D1-dependent control of the cell cycle, mTOR dependent cellular growth and mouse double minute 2 homolog Mdm2/p53-dependent apoptosis[110]. Interestingly, there can be cross-talk between other signalling pathways and PI3K-mediated signalling; e.g., hippo signalling suppresses IRS2/Akt-mediated HCC development in rodent models[111]. Activation of the MAPK pathway by insulin resistance induces the expression of proto-oncogenes, c-fos, and c-jun, and promotes hepatic fibrosis and carcinogenesis by activating the Wnt/β-catenin signalling cascade[112][Figure 1].
NAFLD is also associated with increased circulating levels of leptin[113]. Leptin initiates intracellular signalling of pro-inflammatory cytokines such as TNF-α and IL-6 and activates JAK2/STAT, MAPK and PI3K signalling pathways by binding to its receptor in HCC cells[114]. Furthermore, leptins are known to upregulate hTERT expression, leading to the immortalisation of HCC cells[115]. NAFLD is also associated with decreased sensitivity to thyroid hormones[116] and both NAFLD and HCC are associated with hypothyroidism in humans[117,118]. Furthermore, thyroid hormone treatment decreases hepatosteatosis and the progression of NAFLD in both rodents and humans[119]. Several new studies provide evidence for a potential role of androgen and the androgen receptor pathway in the development of NASH-related HCC and in the treatment of HCC[120]. Gender disparity exists in the incidence of NAFLD associated HCC[121]. Intriguingly, although males are more likely to develop both NAFLD and HCC than females, after the age of 60 this trend is reversed[122-124]. This has been attributed to the loss of the protective effects of oestrogen in females[125]. Besides hormonal stimuli, deregulation of hepatic circadian clock genes also significantly contributes towards the progression of NAFLD to HCC[126].
Future directions and conclusions
Studies from clinical and basic research have provided a better understanding of the aetiology of NASH-associated HCC. Data from various studies reveal that the co-ordinated actions of genetic instability, impaired lipid metabolism, increased oxidative stress altered lipid metabolism, hepatocyte apoptosis, inflammation, fibrosis and altered hormone signalling contribute to the development of HCC. These pathways likely act simultaneously and in combination to activate genetic and epigenetic mechanisms that cause progression of NAFLD and promote the development of NAFLD/NASH-associated HCC. At the clinical level, currently, it is not possible to determine which patients with NASH are most prone to develop HCC. Further studies are required to identify the patients who are at a risk of developing HCC. The identification of specific biomarkers is essential for predicting the transition from NASH to HCC. Currently, there are no pharmacological therapies for the prevention or treatment of NASH and NASH-associated HCC, thus understanding the mechanisms for the pathogenesis of these conditions may lead to the development of novel therapies. Anti-fibrotic, anti-diabetic, anti-inflammatory, antibiotics/probiotics and lipid-lowering drugs either alone or in combination could hold promise for the treatment for NAFLD/NASH-associated HCC.
Declarations
AcknowledgmentsWe wish to acknowledge Dr. Paul M. Yen, Duke-NUS Medical School, Singapore for his advice and suggestion while writing the review.
Authors’ contributionsStudy concept and design: Raza S, Sinha RA
Literature search: Raza S, Rajak S, Anjum B, Sinha RA
Drafting of the manuscript: Raza S, Sinha RA
Critical revision of the manuscript for important intellectual content: Raza S, Sinha RA
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by the ICMR (59/05/2019/ONLINE/BMS/TRM) and Wellcome Trust/DBT India Alliance Fellowship (IA/I/16/2/502691) awarded to Sinha RA.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
1. Akinyemiju T, Abera S, Ahmed M, Alam N, Alemayohu MA, et al; Global Burden of Disease Liver Cancer Collaboration. The burden of primary liver cancer and underlying etiologies from 1990 to 2015 at the global, regional, and national level: results from the global burden of disease study 2015. JAMA Oncol 2017;3:1683-91.
2. Blonski W, Kotlyar DS, Forde KA. Non-viral causes of hepatocellular carcinoma. World J Gastroenterol 2010;16:3603-15.
3. Bertot LC, Adams LA. Trends in hepatocellular carcinoma due to non-alcoholic fatty liver disease. Expert Rev Gastroenterol Hepatol 2019;13:179-87.
4. Swinburn BA, Sacks G, Hall KD, McPherson K, Finegood DT, et al. The global obesity pandemic: shaped by global drivers and local environments. Lancet 2011;378:804-14.
5. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 2018;15:11-20.
6. Bhaskaran K, Douglas I, Forbes H, dos-Santos-Silva I, Leon DA, et al. Body-mass index and risk of 22 specific cancers: a population-based cohort study of 5.24 million UK adults. Lancet 2014;384:755-65.
7. Milic S, Lulic D, Stimac D. Non-alcoholic fatty liver disease and obesity: biochemical, metabolic and clinical presentations. World J Gastroenterol 2014;20:9330-7.
9. Petrucciani N, Gugenheim J. Molecular pathways between obesity, non-alcoholic steatohepatitis (NASH) and hepatocellular carcinoma (HCC). Hepatobiliary Surg Nutr 2019;8:395-7.
10. Xia MF, Bian H, Gao X. NAFLD and diabetes: two sides of the same coin? Rationale for gene-based personalized NAFLD treatment. Front Pharmacol 2019;10:877.
11. Masarone M, Rosato V, Aglitti A, Bucci T, Caruso R, et al. Liver biopsy in type 2 diabetes mellitus: steatohepatitis represents the sole feature of liver damage. PLoS One 2017;12:e0178473.
12. Bril F, Cusi K. Management of nonalcoholic fatty liver disease in patients with type 2 diabetes: a call to action. Diabetes Care 2017;40:419-30.
13. Mantovani A, Targher G. Type 2 diabetes mellitus and risk of hepatocellular carcinoma: spotlight on nonalcoholic fatty liver disease. Ann Transl Med 2017;5:270.
14. Agosti P, Sabba C, Mazzocca A. Emerging metabolic risk factors in hepatocellular carcinoma and their influence on the liver microenvironment. Biochim Biophys Acta Mol Basis Dis 2018;1864:607-17.
15. Yang JD, Ahmed F, Mara KC, Addissie BD, Allen AM, et al. Diabetes is associated with increased risk of hepatocellular carcinoma in cirrhosis patients with nonalcoholic fatty liver disease. Hepatology 2019. Epub ahead of print. doi: 10.1002/hep.30858
16. Margini C, Dufour JF. The story of HCC in NAFLD: from epidemiology, across pathogenesis, to prevention and treatment. Liver Int 2016;36:317-24.
17. Younossi Z, Stepanova M, Ong JP, Jacobson IM, Bugianesi E, et al. Nonalcoholic steatohepatitis is the fastest growing cause of hepatocellular carcinoma in liver transplant candidates. Clin Gastroenterol Hepatol 2019;17:748-55.e3.
18. Reig M, Gambato M, Man NK, Roberts JP, Victor D, et al. Should patients with NAFLD/NASH be surveyed for HCC? Transplantation 2019;103:39-44.
19. Sadler EM, Mehta N, Bhat M, Ghanekar A, Greig PD, et al. Liver transplantation for NASH-related hepatocellular carcinoma versus non-NASH etiologies of hepatocellular carcinoma. Transplantation 2018;102:640-7.
20. Olofson AM, Gonzalo DH, Chang M, Liu X. Steatohepatitic variant of hepatocellular carcinoma: a focused review. Gastroenterology Res 2018;11:391-6.
21. Kanwal F, Kramer JR, Mapakshi S, Natarajan Y, Chayanupatkul M, et al. Risk of hepatocellular cancer in patients with non-alcoholic fatty liver disease. Gastroenterology 2018;155:1828-37.e2.
22. Estes C, Anstee QM, Arias-Loste MT, Bantel H, Bellentani S, et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016-2030. J Hepatol 2018;69:896-904.
23. Uygun A. Is that possible to stop or cease the NASH to turn into HCC? J Gastrointest Cancer 2017;48:250-5.
24. Yopp AC, Choti MA. Non-alcoholic steatohepatitis-related hepatocellular carcinoma: a growing epidemic? Dig Dis 2015;33:642-7.
25. Weinmann A, Alt Y, Koch S, Nelles C, Duber C, et al. Treatment and survival of non-alcoholic steatohepatitis associated hepatocellular carcinoma. BMC Cancer 2015;15:210.
26. Oda K, Uto H, Mawatari S, Ido A. Clinical features of hepatocellular carcinoma associated with nonalcoholic fatty liver disease: a review of human studies. Clin J Gastroenterol 2015;8:1-9.
28. Zen Y, Katayanagi K, Tsuneyama K, Harada K, Araki I, et al. Hepatocellular carcinoma arising in non-alcoholic steatohepatitis. Pathol Int 2001;51:127-31.
29. Gawrieh S, Dakhoul L, Miller E, Scanga A, deLemos A, et al. Characteristics, aetiologies and trends of hepatocellular carcinoma in patients without cirrhosis: a United States multicentre study. Aliment Pharmacol Ther 2019;50:809-21.
30. Grohmann M, Wiede F, Dodd GT, Gurzov EN, Ooi GJ, et al. Obesity drives STAT-1-dependent NASH and STAT-3-dependent HCC. Cell 2018;175:1289-306.e20.
31. Younossi ZM, Otgonsuren M, Henry L, Venkatesan C, Mishra A, et al. Association of nonalcoholic fatty liver disease (NAFLD) with hepatocellular carcinoma (HCC) in the United States from 2004 to 2009. Hepatology 2015;62:1723-30.
32. Piscaglia F, Svegliati-Baroni G, Barchetti A, Pecorelli A, Marinelli S, et al. Clinical patterns of hepatocellular carcinoma in nonalcoholic fatty liver disease: a multicenter prospective study. Hepatology 2016;63:827-38.
33. Yasui K, Hashimoto E, Komorizono Y, Koike K, Arii S, et al. Characteristics of patients with nonalcoholic steatohepatitis who develop hepatocellular carcinoma. Clin Gastroenterol Hepatol 2011;9:428-33.
34. Ertle J, Dechene A, Sowa JP, Penndorf V, Herzer K, et al. Non-alcoholic fatty liver disease progresses to hepatocellular carcinoma in the absence of apparent cirrhosis. Int J Cancer 2011;128:2436-43.
35. Chen K, Ma J, Jia X, Ai W, Ma Z, et al. Advancing the understanding of NAFLD to hepatocellular carcinoma development: From experimental models to humans. Biochim Biophys Acta Rev Cancer 2019;1871:117-25.
36. Anstee QM, Day CP. The genetics of nonalcoholic fatty liver disease: spotlight on PNPLA3 and TM6SF2. Semin Liver Dis 2015;35:270-90.
37. Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016;65:1038-48.
38. Bessone F, Razori MV, Roma MG. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell Mol Life Sci 2019;76:99-128.
39. Spahis S, Delvin E, Borys JM, Levy E. Oxidative stress as a critical factor in nonalcoholic fatty liver disease pathogenesis. Antioxid Redox Signal 2017;26:519-41.
40. Zhang XQ, Xu CF, Yu CH, Chen WX, Li YM. Role of endoplasmic reticulum stress in the pathogenesis of nonalcoholic fatty liver disease. World J Gastroenterol 2014;20:1768-76.
41. Gadd VL, Skoien R, Powell EE, Fagan KJ, Winterford C, et al. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014;59:1393-405.
42. Krenkel O, Puengel T, Govaere O, Abdallah AT, Mossanen JC, et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018;67:1270-83.
43. Sookoian S, Pirola CJ. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 2011;53:1883-94.
44. Singal AG, Manjunath H, Yopp AC, Beg MS, Marrero JA, et al. The effect of PNPLA3 on fibrosis progression and development of hepatocellular carcinoma: a meta-analysis. Am J Gastroenterol 2014;109:325-34.
45. Liu YL, Patman GL, Leathart JB, Piguet AC, Burt AD, et al. Carriage of the PNPLA3 rs738409 C > G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J Hepatol 2014;61:75-81.
46. Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2014;46:352-6.
47. Chen LZ, Xia HH, Xin YN, Lin ZH, Xuan SY. TM6SF2 E167K variant, a novel genetic susceptibility variant, contributing to nonalcoholic fatty liver disease. J Clin Transl Hepatol 2015;3:265-70.
48. Donati B, Dongiovanni P, Romeo S, Meroni M, McCain M, et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci Rep 2017;7:4492.
49. Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet 2015;47:505-11.
50. Tryndyak VP, Han T, Muskhelishvili L, Fuscoe JC, Ross SA, et al. Coupling global methylation and gene expression profiles reveal key pathophysiological events in liver injury induced by a methyl-deficient diet. Mol Nutr Food Res 2011;55:411-8.
51. Liu F, Li H, Chang H, Wang J, Lu J. Identification of hepatocellular carcinoma-associated hub genes and pathways by integrated microarray analysis. Tumori 2015;101:206-14.
52. Erstad DJ, Fuchs BC, Tanabe KK. Molecular signatures in hepatocellular carcinoma: a step toward rationally designed cancer therapy. Cancer 2018;124:3084-104.
53. de Conti A, Ortega JF, Tryndyak V, Dreval K, Moreno FS, et al. MicroRNA deregulation in nonalcoholic steatohepatitis-associated liver carcinogenesis. Oncotarget 2017;8:88517-28.
54. Xu Z, Hu J, Cao H, Pilo MG, Cigliano A, et al. Loss of Pten synergizes with c-Met to promote hepatocellular carcinoma development via mTORC2 pathway. Exp Mol Med 2018;50:e417.
55. Tessitore A, Cicciarelli G, Del Vecchio F, Gaggiano A, Verzella D, et al. MicroRNA expression analysis in high fat diet-induced NAFLD-NASH-HCC progression: study on C57BL/6J mice. BMC Cancer 2016;16:3.
56. Takakura K, Oikawa T, Nakano M, Saeki C, Torisu Y, et al. Recent insights into the multiple pathways driving non-alcoholic steatohepatitis-derived hepatocellular carcinoma. Front Oncol 2019;9:762.
57. Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol 2019;16:411-28.
58. Grattagliano I, de Bari O, Bernardo TC, Oliveira PJ, Wang DQ, et al. Role of mitochondria in nonalcoholic fatty liver disease - from origin to propagation. Clin Biochem 2012;45:610-8.
59. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 2005;115:1343-51.
60. Nakagawa H, Hayata Y, Kawamura S, Yamada T, Fujiwara N, et al. Lipid metabolic reprogramming in hepatocellular carcinoma. Cancers (Basel) 2018;10:E447.
61. Hirsova P, Ibrahim SH, Gores GJ, Malhi H. Lipotoxic lethal and sublethal stress signaling in hepatocytes: relevance to NASH pathogenesis. J Lipid Res 2016;57:1758-70.
62. Garcia-Ruiz C, Fernandez-Checa JC. Mitochondrial oxidative stress and antioxidants balance in fatty liver disease. Hepatol Commun 2018;2:1425-39.
63. Masarone M, Rosato V, Dallio M, Gravina AG, Aglitti A, et al. Role of oxidative stress in pathophysiology of nonalcoholic fatty liver disease. Oxid Med Cell Longev 2018;2018:9547613.
65. Nishida N, Yada N, Hagiwara S, Sakurai T, Kitano M, et al. Unique features associated with hepatic oxidative DNA damage and DNA methylation in non-alcoholic fatty liver disease. J Gastroenterol Hepatol 2016;31:1646-53.
66. Song MJ, Malhi H. The unfolded protein response and hepatic lipid metabolism in non alcoholic fatty liver disease. Pharmacol Ther 2019;203:107401.
67. Nakagawa H, Umemura A, Taniguchi K, Font-Burgada J, Dhar D, et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014;26:331-43.
68. Wu WKK, Zhang L, Chan MTV. Autophagy, NAFLD and NAFLD-related HCC. Adv Exp Med Biol 2018;1061:127-38.
69. Tian Y, Yang B, Qiu W, Hao Y, Zhang Z, et al. ER-residential Nogo-B accelerates NAFLD-associated HCC mediated by metabolic reprogramming of oxLDL lipophagy. Nat Commun 2019;10:3391.
70. Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell 2013;51:618-31.
71. Sinha RA, Singh BK, Zhou J, Wu Y, Farah BL, et al. Thyroid hormone induction of mitochondrial activity is coupled to mitophagy via ROS-AMPK-ULK1 signaling. Autophagy 2015;11:1341-57.
72. Sun K, Xu L, Jing Y, Han Z, Chen X, et al. Autophagy-deficient Kupffer cells promote tumorigenesis by enhancing mtROS-NF-kappaB-IL1alpha/beta-dependent inflammation and fibrosis during the preneoplastic stage of hepatocarcinogenesis. Cancer Lett 2017;388:198-207.
73. Kanno M, Kawaguchi K, Honda M, Horii R, Takatori H, et al. Serum aldo-keto reductase family 1 member B10 predicts advanced liver fibrosis and fatal complications of nonalcoholic steatohepatitis. J Gastroenterol 2019;54:549-57.
74. Arendt BM, Teterina A, Pettinelli P, Comelli EM, Ma DWL, et al. Cancer-related gene expression is associated with disease severity and modifiable lifestyle factors in non-alcoholic fatty liver disease. Nutrition 2019;62:100-7.
75. Torres-Mena JE, Salazar-Villegas KN, Sanchez-Rodriguez R, Lopez-Gabino B, Del Pozo-Yauner L, et al. Aldo-Keto reductases as early biomarkers of hepatocellular carcinoma: a comparison between animal models and human HCC. Dig Dis Sci 2018;63:934-44.
76. Nikolaou N, Gathercole LL, Marchand L, Althari S, Dempster NJ, et al. AKR1D1 is a novel regulator of metabolic phenotype in human hepatocytes and is dysregulated in non-alcoholic fatty liver disease. Metabolism 2019;99:67-80.
77. Marra F, Svegliati-Baroni G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J Hepatol 2018;68:280-95.
78. Iannucci LF, Sun J, Singh BK, Zhou J, Kaddai VA, et al. Short chain fatty acids induce UCP2-mediated autophagy in hepatic cells. Biochem Biophys Res Commun 2016;480:461-7.
79. Koopman N, Molinaro A, Nieuwdorp M, Holleboom AG. Review article: can bugs be drugs? The potential of probiotics and prebiotics as treatment for non-alcoholic fatty liver disease. Aliment Pharmacol Ther 2019;50:628-39.
80. Jandhyala SM, Talukdar R, Subramanyam C, Vuyyuru H, Sasikala M, et al. Role of the normal gut microbiota. World J Gastroenterol 2015;21:8787-803.
81. Mouries J, Brescia P, Silvestri A, Spadoni I, Sorribas M, et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J Hepatol 2019;71:1216-28.
82. Liu Q, Liu S, Chen L, Zhao Z, Du S, et al. Role and effective therapeutic target of gut microbiota in NAFLD/NASH. Exp Ther Med 2019;18:1935-44.
83. Kim HN, Joo EJ, Cheong HS, Kim Y, Kim HL, et al. Gut microbiota and risk of persistent nonalcoholic fatty liver diseases. J Clin Med 2019;8:E1089.
84. Jasirwan COM, Lesmana CRA, Hasan I, Sulaiman AS, Gani RA. The role of gut microbiota in non-alcoholic fatty liver disease: pathways of mechanisms. Biosci Microbiota Food Health 2019;38:81-8.
85. Leung C, Rivera L, Furness JB, Angus PW. The role of the gut microbiota in NAFLD. Nat Rev Gastroenterol Hepatol 2016;13:412-25.
86. Ezzaidi N, Zhang X, Coker OO, Yu J. New insights and therapeutic implication of gut microbiota in non-alcoholic fatty liver disease and its associated liver cancer. Cancer Lett 2019;459:186-91.
87. Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013;499:97-101.
88. Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 2012;21:504-16.
89. Chu H, Williams B, Schnabl B. Gut microbiota, fatty liver disease, and hepatocellular carcinoma. Liver Res 2018;2:43-51.
90. Nguyen J, Jiao J, Smoot K, Watt GP, Zhao C, et al. Toll-like receptor 4: a target for chemoprevention of hepatocellular carcinoma in obesity and steatohepatitis. Oncotarget 2018;9:29495-507.
91. Liu Y, Yan W, Tohme S, Chen M, Fu Y, et al. Hypoxia induced HMGB1 and mitochondrial DNA interactions mediate tumor growth in hepatocellular carcinoma through Toll-like receptor 9. J Hepatol 2015;63:114-21.
92. Brandi G, De Lorenzo S, Candela M, Pantaleo MA, Bellentani S, et al. Microbiota, NASH, HCC and the potential role of probiotics. Carcinogenesis 2017;38:231-40.
93. Takahashi S, Tanaka N, Fukami T, Xie C, Yagai T, et al. Role of farnesoid X receptor and bile acids in hepatic tumor development. Hepatol Commun 2018;2:1567-82.
94. He G, Karin M. NF-kappa B and STAT3 - key players in liver inflammation and cancer. Cell Res 2011;21:159-68.
95. Park EJ, Lee JH, Yu GY, He G, Ali SR, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010;140:197-208.
96. Min HK, Mirshahi F, Verdianelli A, Pacana T, Patel V, et al. Activation of the GP130-STAT3 axis and its potential implications in nonalcoholic fatty liver disease. Am J Physiol Gastrointest Liver Physiol 2015;308:G794-803.
97. Widjaja AA, Singh BK, Adami E, Viswanathan S, Dong J, et al. Inhibiting interleukin 11 signaling reduces hepatocyte death and liver fibrosis, inflammation, and steatosis in mouse models of nonalcoholic steatohepatitis. Gastroenterology 2019;157:777-92.e14.
98. Zheng H, Yang Y, Han J, Jiang WH, Chen C, et al. TMED3 promotes hepatocellular carcinoma progression via IL-11/STAT3 signaling. Sci Rep 2016;6:37070.
99. Gomes AL, Teijeiro A, Buren S, Tummala KS, Yilmaz M, et al. Metabolic inflammation-associated IL-17A causes non-alcoholic steatohepatitis and hepatocellular carcinoma. Cancer Cell 2016;30:161-75.
100. Syn WK, Choi SS, Liaskou E, Karaca GF, Agboola KM, et al. Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology 2011;53:106-15.
101. Chung SI, Moon H, Ju HL, Cho KJ, Kim DY, et al. Hepatic expression of sonic hedgehog induces liver fibrosis and promotes hepatocarcinogenesis in a transgenic mouse model. J Hepatol 2016;64:618-27.
102. Chen J, Gingold JA, Su X. Immunomodulatory TGF-beta signaling in hepatocellular carcinoma. Trends Mol Med 2019;25:1010-23.
103. Yang L, Roh YS, Song J, Zhang B, Liu C, et al. Transforming growth factor beta signaling in hepatocytes participates in steatohepatitis through regulation of cell death and lipid metabolism in mice. Hepatology 2014;59:483-95.
104. Jin Z, Lei L, Lin D, Liu Y, Song Y, et al. IL-33 released in the liver inhibits tumor growth via promotion of CD4(+) and CD8(+) T cell responses in hepatocellular carcinoma. J Immunol 2018;201:3770-9.
105. Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016;531:253-7.
106. Malehmir M, Pfister D, Gallage S, Szydlowska M, Inverso D, et al. Platelet GPIbalpha is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat Med 2019;25:641-55.
107. de Oliveira S, Houseright RA, Graves AL, Golenberg N, Korte BG, et al. Metformin modulates innate immune-mediated inflammation and early progression of NAFLD-associated hepatocellular carcinoma in zebrafish. J Hepatol 2019;70:710-21.
108. Bril F, Lomonaco R, Orsak B, Ortiz-Lopez C, Webb A, et al. Relationship between disease severity, hyperinsulinemia, and impaired insulin clearance in patients with nonalcoholic steatohepatitis. Hepatology 2014;59:2178-87.
109. De Minicis S, Agostinelli L, Rychlicki C, Sorice GP, Saccomanno S, et al. HCC development is associated to peripheral insulin resistance in a mouse model of NASH. PLoS One 2014;9:e97136.
110. Kudo Y, Tanaka Y, Tateishi K, Yamamoto K, Yamamoto S, et al. Altered composition of fatty acids exacerbates hepatotumorigenesis during activation of the phosphatidylinositol 3-kinase pathway. J Hepatol 2011;55:1400-8.
111. Jeong SH, Kim HB, Kim MC, Lee JM, Lee JH, et al. Hippo-mediated suppression of IRS2/AKT signaling prevents hepatic steatosis and liver cancer. J Clin Invest 2018;128:1010-25.
112. Chettouh H, Lequoy M, Fartoux L, Vigouroux C, Desbois-Mouthon C. Hyperinsulinaemia and insulin signalling in the pathogenesis and the clinical course of hepatocellular carcinoma. Liver Int 2015;35:2203-17.
113. Polyzos SA, Aronis KN, Kountouras J, Raptis DD, Vasiloglou MF, et al. Circulating leptin in non-alcoholic fatty liver disease: a systematic review and meta-analysis. Diabetologia 2016;59:30-43.
114. Jiang N, Sun R, Sun Q. Leptin signaling molecular actions and drug target in hepatocellular carcinoma. Drug Des Devel Ther 2014;8:2295-302.
115. Stefanou N, Papanikolaou V, Furukawa Y, Nakamura Y, Tsezou A. Leptin as a critical regulator of hepatocellular carcinoma development through modulation of human telomerase reverse transcriptase. BMC Cancer 2010;10:442.
116. Bruinstroop E, Dalan R, Cao Y, Bee YM, Chandran K, et al. Low-dose levothyroxine reduces intrahepatic lipid content in patients with type 2 diabetes mellitus and NAFLD. J Clin Endocrinol Metab 2018;103:2698-706.
117. Pinter M, Haupt L, Hucke F, Bota S, Bucsics T, et al. The impact of thyroid hormones on patients with hepatocellular carcinoma. PLoS One 2017;12:e0181878.
118. Lonardo A, Ballestri S, Mantovani A, Nascimbeni F, Lugari S, et al. Pathogenesis of hypothyroidism-induced NAFLD: evidence for a distinct disease entity? Dig Liver Dis 2019;51:462-70.
119. Sinha RA, Bruinstroop E, Singh BK, Yen PM. Nonalcoholic fatty liver disease and hypercholesterolemia: roles of thyroid hormones, metabolites, and agonists. Thyroid 2019;29:1173-91.
120. Ali MA, Lacin S, Abdel-Wahab R, Uemura M, Hassan M, et al. Nonalcoholic steatohepatitis-related hepatocellular carcinoma: is there a role for the androgen receptor pathway? Onco Targets Ther 2017;10:1403-12.
121. Wu EM, Wong LL, Hernandez BY, Ji JF, Jia W, et al. Gender differences in hepatocellular cancer: disparities in nonalcoholic fatty liver disease/steatohepatitis and liver transplantation. Hepatoma Res 2018;4.
122. Bosch FX, Ribes J, Diaz M, Cleries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology 2004;127:S5-16.
123. Gan L, Chitturi S, Farrell GC. Mechanisms and implications of age-related changes in the liver: nonalcoholic fatty liver disease in the elderly. Curr Gerontol Geriatr Res 2011;2011:831536.
124. Hashimoto E, Tokushige K. Prevalence, gender, ethnic variations, and prognosis of NASH. J Gastroenterol 2011;46 Suppl 1:63-9.
125. Iyer JK, Kalra M, Kaul A, Payton ME, Kaul R. Estrogen receptor expression in chronic hepatitis C and hepatocellular carcinoma pathogenesis. World J Gastroenterol 2017;23:6802-16.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Raza S, Rajak S, Anjum B, Sinha RA. Molecular links between non-alcoholic fatty liver disease and hepatocellular carcinoma. Hepatoma Res 2019;5:42. http://dx.doi.org/10.20517/2394-5079.2019.014
AMA Style
Raza S, Rajak S, Anjum B, Sinha RA. Molecular links between non-alcoholic fatty liver disease and hepatocellular carcinoma. Hepatoma Research. 2019; 5: 42. http://dx.doi.org/10.20517/2394-5079.2019.014
Chicago/Turabian Style
Raza, Sana, Sangam Rajak, Baby Anjum, Rohit A. Sinha. 2019. "Molecular links between non-alcoholic fatty liver disease and hepatocellular carcinoma" Hepatoma Research. 5: 42. http://dx.doi.org/10.20517/2394-5079.2019.014
ACS Style
Raza, S.; Rajak S.; Anjum B.; Sinha RA. Molecular links between non-alcoholic fatty liver disease and hepatocellular carcinoma. Hepatoma. Res. 2019, 5, 42. http://dx.doi.org/10.20517/2394-5079.2019.014
About This Article
Special Issue
Copyright

Data & Comments
Data



 Cite This Article 21 clicks
Cite This Article 21 clicks

 Like This Article 31
likes
Like This Article 31
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.