
Alteration in immune function in patients with fatty liver disease

Abstract
Nonalcoholic fatty liver disease (NAFLD) is a disease spectrum that spans simple steatosis, fibrosis, and ultimately cirrhosis, and is a leading cause of chronic liver disease globally. The severe variant of NAFLD, non-alcoholic steatohepatitis (NASH), is characterized by triglyceride accumulation within hepatocytes and the subsequent inflammatory pathway activation, ultimately progressing to cirrhosis in 10%-20% of patients. NASH is a known major risk factor for the development of hepatocellular carcinoma (HCC), and there is emerging data demonstrating the impact of NASH on immune subsets and the tumor microenvironment that may influence therapeutic response. This review describes the various ways in which the immune system is altered in patients with NASH. The innate immune system in NASH shows alterations in dendritic and Kupffer cells, impaired cytotoxicity of Natural Killer cells, and an accumulation of neutrophils. Additionally, there is emerging evidence emphasizing the role of the adaptive immune system in the development and progression of NASH, seen in the alteration of B-cells, T-cells, and NKT Cells. Due to the complex interplay of the immune system in NAFLD/NASH and its progression to HCC, many current treatments focus on targeting immune cells for HCC therapy. Recently, immune checkpoint inhibitors such as atezolizumab and bevacizumab have been approved as first-line therapy for unresectable HCC. Although an emerging field of research, further studies and clinical trials are needed to understand the complex interface of NASH, HCC and the immune response.
Keywords
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is a leading cause of chronic liver disease in both the Western and Eastern hemispheres, making it a significant global problem[1]. NAFLD is a disease spectrum that ranges from simple steatosis, fibrosis, and ultimately cirrhosis. The severe form of NAFLD, non-alcoholic steatohepatitis (NASH), is characterized by triglyceride accumulations within hepatocytes, and subsequent inflammatory pathway activation as hepatocytes are injured and replaced, ultimately progressing to cirrhosis in 10%-20% of patients[2-5]. In the United States, the prevalence of NASH is rapidly increasing, with an annual incident of 3.6 million cases[6]. Additionally, NASH has been predicted to increase to 56% between 2016 and 2030[7,8]. As the incidence of NASH rises, it is becoming a significant risk factor for the development of hepatocellular carcinoma (HCC), as evidenced by the cumulative HCC incidence of 2.4% to 12.8% over 2 to 3 years, respectively among patients with NASH and cirrhosis[9].
HCC is the most common primary liver cancer and the fourth leading cause of cancer-related death worldwide[10]. Liver cirrhosis is a major risk factor for the development of HCC. Etiologies of cirrhosis include NAFLD/NASH as well as viral hepatitis and alcoholic liver disease[11]. Treatment for HCC takes into consideration tumor-related factors such as tumor size and anatomic location as well as the severity of background liver disease which is a driving factor for post-operative complications. For select patients, liver resection, ablation or transplantation can be curative. However, in patients with more advanced tumors or liver disease, curative treatment options are not available and patients are treated with arterially directed therapies such as transarterial chemoembolization (TACE) or systemic therapy[12-15]. Systemic therapy options have improved with the introduction of atezolizumab [anti-programmed death-ligand 1 (PD-L1)] with bevacizumab [anti-vascular endothelial growth factor (VEGF)] as the first-line standard of care after demonstrating improved overall survival (OS) and progression-free survival (PFS) compared to sorafenib[16,17]. However, emerging evidence indicates fatty liver disease alters the tumor microenvironment and may affect response to treatment. Over the past decade, there has been immense progress in elucidating the mechanisms and immune microenvironment that factor into the development of NASH and its progression to cirrhosis and HCC. This review discusses the various ways in which the immune system is altered in patients with NAFLD/NASH.
PATHOGENESIS OF FATTY LIVER DISEASE
NAFLD and NASH can also be thought of as the manifestation of metabolic syndrome of the liver, as there is a correlation with type 2 diabetes mellitus (DM), dyslipidemia, cardiovascular disease, and obesity[18]. The risk of progression for NAFLD to NASH is directly correlated with the increasing number of risk factors in a patient[19]. Among these features, DM is shown to have the strongest association with the progression of NAFLD to NASH, and up to 75% of individuals with DM type II have NAFLD. The relationship between DM and the risk of HCC in patients with NASH is not well quantified. However, Yang et al. followed 354 patients with NASH cirrhosis, of which 253 (71%) had DM, and thirty of these patients developed HCC over a median follow-up period of 47 months.[20] Both a univariate analysis [hazard ratio (HR) = 3.6; 95% confidence interval (CI) = 1.1-11.9; P = 0.04] and multivariate analysis (HR = 4.2; 95%CI: 1.2-14.2; P = 0.02) revealed an association with DM and an increased risk of developing HCC among patients with NASH cirrhosis.
From a genetic perspective, the patatin-like phospholipase domain-contained protein (PNPLA) 3 has been identified as a risk factor for NAFLD. A loss of function mutation in the PNPLA 3 gene impairs triglyceride hydrolysis resulting in increased triglyceride accumulation within hepatocytes and lipotoxicity resulting in oxidative stress[21]. While genetics, insulin resistance, and obesity have a synergistic role in the perpetuation of hepatic steatosis, there are immune changes in response to inflammation and oxidative stress that provide additional “hits” that promote the progression of NASH[18].
TUMOR IMMUNE MICROENVIRONMENT OF HCC
HCC frequently develops in the setting of chronic liver disease, where inflammation aids in driving carcinogenesis[22]. The tumor microenvironment (TME) is now recognized as a key part of tumor growth and immune evasion. The liver is a natural immune tolerant organ; it has developed intrinsic mechanisms to promote tolerance in the innate and adaptive immune responses in order to protect itself from autoimmune damage secondary to large antigen presentation from the gastrointestinal tract[23,24]. The liver also bestows a unique proinflammatory microenvironment that consists of various immunologically active cells, such as Kupffer cells (KCs), antigen-presenting cells, T cells, and hepatic stellate cells (HSCs)[25-27]. These cells participate in a multifaceted proinflammatory response that occurs during pathologic liver injury that results in hepatocyte death and disease progression. In addition, proinflammatory cytokines, such as interleukin (IL)-6 and tumor necrosis factor (TNF)-α, as well as hyperinsulinemia, free fatty acids, and leptin, can interact with components in the liver microenvironment and create inflammation, fibrosis, and lipotoxicity[25]. Recently, literature has shown that the liver TME has been a complex interplay in the development of NAFLD/NASH and progression to HCC[1,28,29]. Thus, it is imperative to understand the hepatic microenvironment and the innate and adaptive immune system alterations that occur in the development of NASH [Figure 1].
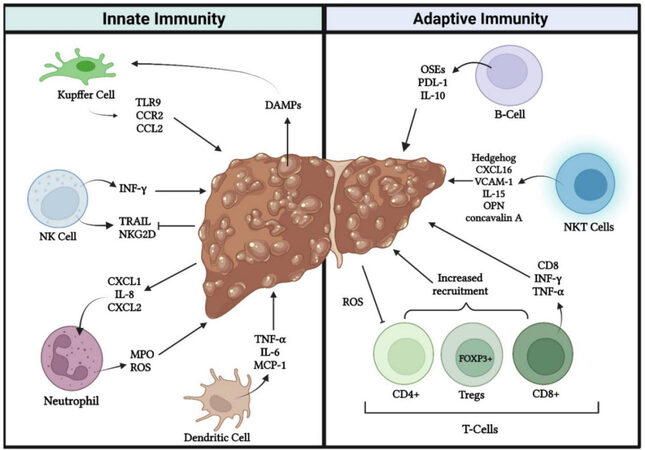
Figure 1. The Immune System. Immunopathogenesis of nonalcoholic steatohepatitis (NASH). Interactions between innate immune cells (left frame) and adaptive immune cells (right frame) on hepatocytes that promote NASH and the transition to hepatocellular carcinoma (HCC). NK cell: Natural killer cell; CD4+: CD4+ helper T cell; CD8+: Cytotoxic CD8+ T cells; Tregs: regulatory T-cells; DAMPs: damage associated molecular patterns; IFN: interferon; ROS: reactive oxygen species; MPO: myeloperoxidase; TNF: tumor necrosis factor; TLR: toll-like receptor; TRAIL: tumor necrosis factor-related apoptosis-inducing ligand; OSEs: oxidative stress-derived epitopes; OPN: Osteopontin; VCAM: vascular cell adhesion molecule; IL: interleukin; PDL: programed death ligand.
INNATE IMMUNE SYSTEM
The innate immune system is the first line of defense against pathogens in the body. The liver activates innate immunity due to its direct antigen exposure from the gastrointestinal tract. This activation has been shown to be vital in activating inflammation in NASH[30] [Table 1].
Kupffer Cells
KCs, also classified as liver resident macrophages, account for 10%-20% of total cells in the liver[31]. KCs are the liver’s first line of immune defense as they are in constant contact with antigens arriving in the liver from the gastrointestinal tract[32,33]. These antigens are presented to toll-like receptors (TLRs) on KCs that trigger mechanisms to induce immunity, which may, in turn, contribute to liver diseases such as cirrhosis and HCC[33]. For example, translocation of lipopolysaccharide (LPS), a bacterial endotoxin from the gut microbiome, is presented to TLR4 on KCs and activates transcription factor (NF)-κB. In return, this pathway generates a potent and persistent proinflammatory cascade through the generation of reactive oxygen species (ROS), especially through NADPH oxidase 2 (NOX2)[34,35]. During ROS-induced lipotoxicity, stressed or dying hepatocytes release intracellular molecules called damage-associated molecular patterns (DAMPs). Under physiologic conditions, DAMPs act on a variety of immune cells in the liver to activate a homeostatic wound-healing response to repair injured tissue[36,37]. However, if these signals are prolonged in the setting of tissue inflammation, DAMPs can induce an excessive inflammatory response through direct activation of TLR9 in KCs that can lead to advanced fibrosis and progression to cirrhosis[30,38,39]. Particularly, NOX2-induced ROS has been associated with premature senescence of liver sinusoidal endothelial cells which are associated with fibrogenesis[40,41]. Furthermore, oxidative stress in the setting of NAFLD has been demonstrated through increased levels of soluble NOX2-derived peptide (sNOX2-dp) as well as urinary 8-iso-prostagladin F2α (8-iso-PGF2α), which is a commonly accepted biomarker of oxidative stress produced by NOX2[42]. While KCs aim to protect the liver from endotoxins and antigens, the resulting persistent oxidative stress can be further damaging.
Alongside liver resident KCs, an abundant population of peripheral macrophages are recruited into the liver in both NAFLD and NASH. These infiltrative macrophages are differentiated from KCs through the expression of different surface markers. Peripheral infiltrating macrophages have high levels of CD11b and low F4/80 expression, whereas KCs were noted to have low levels of CD11b and high F4/80 expression[43]. CCR2 was another marker shown to have increased expression in infiltrated macrophages compared to KCs. Recruitment of the CCR2 macrophages into the liver was seen in parallel with increased levels of CCL2 found in steatotic hepatocytes[44,45]. In fact, murine models using drugs targeting the CCL2/CCR2 axis to impair macrophage recruitment to the liver revealed a reduction of hepatosteatosis, inflammation, and fibrosis[46,47]. This murine model demonstrates the importance of both the KCs as well as the infiltration of additional macrophages for the promotion of NASH.
Natural killer cells
Natural killer (NK) cells are another subset of the innate immune system that contribute to NASH. NK cells are a lineage of immune cells related to lymphocytes that have cytolytic activity against stressed cells, virus-infected cells, and malignantly transformed cells[48,49]. NK cells also aid in the resolution of hepatic fibrosis through the elimination of hepatic stellate cells (HSCs)[50]. NK cells derive their name from their ability to kill targeted cells without a need for secondary activation, unlike CD8+ T-cells. NK cells originate in the bone marrow, but after maturation, they disperse widely to both lymphoid and non-lymphoid tissue. NK cells are found most frequently in the lung, then liver, peripheral blood, and spleen, respectively[51]. In addition, NK cells are recognized for their ability to activate macrophages through the release of INF-γ, a proinflammatory cytokine[49].
Regarding NASH, liver fibrosis is a key component of activation for NK Cells. In comparison to patients with low fibrosis scores, patients with high fibrosis scores were shown to have a significant reduction in the tumor-cell-killing capacity of CD56dim NK cells[52]. For NK cells to repair liver fibrosis and kill activated HSCs, NK cell tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and NKG2D are critical components[53]. In a murine model utilizing DDC-induced liver fibrosis, NK cells were activated by polyinosinic-polycytidylic acid, a Toll-like Receptor (TLR)-3 ligand, or INF-γ, to then activate HSCs and replicate the severity of liver fibrosis seen in NASH. This study demonstrated that activation of the NK cells with either polyionosinic-polycytidylic acid or INF-γ improved the cytotoxicity of the NK cells towards HSCs and amplified expression of NKG2D and TRAILs on the liver resident NK cells, resulting in overall improvement of liver fibrosis. In contrast, inhibiting NKG2D or TRAIL in the mice was shown to markedly diminish the cytotoxicity of NK cells on HSCs, resulting in a decrease in protection against fibrosis[53]. This study implies that the impaired cytotoxicity of NK cell activity seen in fatty liver may contribute to fibrosis formation and progression in patients with NASH. Additionally, NK cells are reduced in number and impaired in the generation of IFN-y and cytotoxic functions in HCC conditions[54].
Neutrophils
Neutrophils are a copious population of circulating white blood cells and activate the early phases of the inflammatory response in the innate immune system[49]. In the event of injury or pathogen invasion, neutrophils are recruited to the site through various chemokines. Activated neutrophils then attack the foreign pathogen by the generation of reactive oxygen species (ROS) through a process called the respiratory burst. In order to generate this process, the enzyme myeloperoxidase (MPO) is used to create ROS to kill microbes. The large influx of neutrophils is a usual phenomenon seen in NASH[55]. Neutrophils are recruited to the liver in the early stages of hepatic injury by chemokines such as CXCL1, IL-8, and CXCL2[56]. In patients with NASH, an increase in MPO in the blood plasma has been found, suggesting that MPO accumulation and its oxidative products may contribute to liver inflammation and the promotion of NASH[57]. Further, neutrophils exacerbate the ongoing inflammatory state and hepatocyte damage through macrophage recruitment and interaction with antigen-presenting cells[30]. In addition, neutrophils release chromatin-associated traps called neutrophil extracellular traps (NETs)[58,59]. NETs are long chromatin fibers embedded with inflammatory proteins and neutrophil proteases[58]. NETs were initially observed during the microbial invasion; however, recent studies revealed NET formation occurs in the early stages of NAFLD and persists in livers with advancement to NASH[59]. Van der Windt et al. revealed that livers from mice with NASH induced by high-fat diet and neonatal streptozotocin had early neutrophil infiltration and NET formation, followed by an influx of monocyte-derived macrophages, inflammatory cytokine production (IL-6, TNF-α), and progression from NASH to HCC[58]. Inhibition of the NET formations caused a reduction of monocyte infiltration, inflammatory cytokine production, reduction of NASH through improvements in hepatocyte ballooning, and decreased tumor growth[58]. Overall, the chronic inflammatory hepatic microenvironment promotes the persistent formation of NETs, resulting in a continued proinflammatory state and, ultimately, liver damage.
Dendritic cells
Dendritic cells (DCs), antigen-presenting cells, are innate immune cells that initiate the adaptive immune system by presenting antigens to naïve T-cells. DCs ingest tumor cells or virus-infected cells and present antigens to T-cells on class I MHC molecules through cross-presentation[49]. DC cells originate in the bone marrow, but a small percentage represent the non-parenchymal liver cells inhabiting the central and portal veins[30]. While there is a paucity of literature on the role of DCs in NASH, a study by Henning et al. revealed a rapid infiltration of phenotypically active DCs into the liver of experimentally induced NASH in mice that were expressing increased levels of TNF-α, IL-6, and MCP-1. In addition, depletion of DCs in murine models was shown to delay the resolution of intrahepatic inflammation and fibrosis, thus promoting NASH[60].
ADAPTIVE IMMUNE SYSTEM
Although the innate immune system has routinely been recognized to play an integral part in the pathogenesis of NASH, emerging evidence has recently emphasized the role of the adaptive immune system in the development and progression of NASH in the alteration of B-cells, T-cells, and natural killer-T (NKT) cells [Table 2].
Cells of the adaptive immune system
| Adaptive Cell type | Activity-pro-fibrosis | Activity-anti-fibrosis |
| B cells | Accumulation in clusters within liver Presentation of OSEs to CD4+ helper T-cells IgA expression of PD-L1 and IL-10 to suppress CD8+ T-cells | Plasma cell differentiation Anti-OSE IgG |
| CD4+ helper T (Th) cells | TRM cell secretion of IFN-γ | Inhibit tumor growth and promote tumor clearance |
| CD8+ cytotoxic T cells | Increased IFN-γ and TNF-α production | |
| Tregs | Increased accumulation in liver evasion of immunosurveillance | |
| NKT cells | Production of IL-4 and INF-y in response to lipids Increased levels of CXCL16, VCAM-1, IL-15 Overactive Hedgehog pathway Secretion of OPN |
B cells
B cells are a subset of the adaptive immune system and influence immune-mediated inflammatory responses. For instance, B cells are known for their ability to present antigens, secrete cytokines upon TLR-mediated activation by pathogen-associated molecular patterns, and produce immunoglobulins[61-63]. B-cells are suspected of contributing to the pathogenesis of NASH, as an accumulation of B-cells in the liver is seen in parallel with high levels of lobular inflammation and fibrosis[64]. In fact, liver biopsies in both human and experimental NASH models show increased infiltration of B- and T- lymphocyte clusters[65]. These clusters correlate with increased levels of oxidative stress-derived epitopes (OSEs), which are released from damaged hepatocytes[66]. A study by Bruzzi et al. revealed that B-cells were able to present OSEs to CD4+ T helper (Th1) cells allowing them to release proinflammatory cytokines, thus inducing an inflammatory state[64]. On the contrary, B-cells can also differentiate into plasma cells and produce anti-OSE immunoglobulins (IgG), thus counteracting an inflammatory state[64]. Similarly, Shalapour et al. revealed that liver-resident IgA cell accumulation was associated with chronic inflammation and fibrosis in both humans and mice with NAFLD[67]. These IgA cells were able to express programmed death ligand 1 (PD-L1) and IL-10, which were found to directly suppress hepatic cytotoxic CD8+ T cells. In addition, inhibition of the IgA cells resulted in reactivation of cytotoxic CD8+ T cells and regression of established HCC[67].
T-cells
Conventional T-cells are abundant in healthy livers and are characterized into several subsets: CD4+ helper T (Th) cells, CD8+ cytotoxic T cells, and regulatory T-cells (Tregs)[68]. CD4+ T cells are required for tumor control by inhibiting tumor initiation as well as facilitating clearance of malignant cells[69-71]. Analysis has ascertained CD4+ T cells as provokers of anti-tumor response and has linked CD4+ T cells to favorable responses to immunotherapy. In the setting of NASH, anti-tumor surveillance is impaired through the promotion of ROS-dependent cell death of hepatic CD4+ T cells. In fact, disruption of the loss of CD4+ T-cells in association with NASH phenotype suggests immunotherapy may be impaired in the setting of NASH-related hepatic tumors[72,73]. In one study, steatohepatitis induced through a methionine-choline deficient diet in mice was shown to reduce the effect of immunotherapeutic agents such as M30, an mRNA vaccine, and aOX40, an antibody against OX40, on inhibiting hepatic tumor growth through reduction of tumor infiltration by CD4+ T cells and effector memory cells[69]. Another experimental NASH model showed worsening of steatohepatitis in relation to the increased recruitment of CD4+ and CD8+ T-lymphocytes within the liver[74]. Similarly, Rag1-/- mice, which lack lymphocyte populations, lead to the reversal of NASH phenotype, irrespective of metabolic alterations such as obesity[68].
CD8+ cytotoxic T-cells are the main effector cells of the cellular immune system, and yield cytotoxic processes to destroy infected or malignant cells through recognition of antigens presented on MHC I molecules[75]. In healthy livers, there are populations of non-recirculating CD8+ tissue-resident memory (TRM) cells that act as local immune sentinels[76,77]. In inflammation or pathogen invasion situations, hepatic TRMs secrete IFN-γ as an immune response[78]. In the early stages of NASH, an accumulation of intrahepatic cytotoxic CD8 T-cells is associated with an obesity-induced hepatic type I interferon (INF-1) response[79]. INF-1 response induces increased CD8+ T-cell production of proinflammatory cytokine IFN-γ and TNF- α, which promotes continued metabolic dysfunction and, ultimately, hepatocyte damage[79]. In fact, deletion of CD8+ T cells in experimental animal models resulted in amelioration of NASH and improvements such as restored hepatic insulin sensitivity and reduced fibrosis[75,79]. The resolution of NASH through depletion of CD8+ T-cells suggests a direct impact on NASH pathogenesis and subsequent transition to HCC.
Tregs, immunosuppressive subset of CD4+ T-cells, are essential for maintaining homeostasis and immune tolerance. Tregs are distinguished from other T-cells through their expression of master transcription factor forkhead box P3 (FoxP3)[59,80]. In an experimental NASH murine model, Tregs were found to be increased in NASH livers, whereas CD4+ T cells were found in lower concentrations. When Tregs were then depleted using FoxP3-DTR mice, HCC initiation and progression of NASH were drastically inhibited[59]. Therefore, the accumulation of Tregs allows evasion of immunosurveillance in the NASH liver and plays a critical role in hepatocarcinogenesis.
NKT cells
NKT cells are considered a bridge between innate and adaptive immunity and are mostly located in the hepatic sinusoids to provide intravascular immune surveillance[49,81]. NKT cells are a small subset of nonconventional T-cells that express surface markers of NK cells as well as an antigen receptor (TCR) characteristic of a T-cell, allowing effector functions similar to helper T-cells[30,49]. NKT cell TCRs recognize lipids that are bound to class I MHC-like molecules called CD1 molecules. When triggered by lipids bound to CD1 molecules, NKT cells can rapidly produce cytokines such as IL-4 and INF-γ[49]. NKT cells are both proinflammatory through type I NKT cells and protect against liver injury through NKT Type II cells[82]. Syn et al. reported that hedgehog (Hh)-mediated accumulation of NKT cells contributed to both the formation and progression of NASH in both mice and humans[83]. Overly active Hh pathway in mice resulted in increased levels of NKT cell chemokine, CXCL16, vascular cell adhesion molecule 1 (VCAM-1), and IL-15, a proinflammatory cytokine. These mice were shown to accumulate more NKT cells in their livers and develop worse NASH fibrosis. In addition, NASH cirrhosis was found to have four times as many NKT cells as individuals with healthy livers[84]. Osteopontin (OPN), a matricellular protein and cytokine secreted by NKT cells, has also been shown to promote NAFLD progression through exacerbation of concanavalin A-induced hepatitis[85,86]. Feeding mice with methionine-choline-deficient diets resulted in activated hepatic NKT cells that generated Hh and OPN and thus activated HSCs to differentiate into myofibroblasts and induce fibrosis. In contrast, Ja18-/- mice, which lack NKT cells, showed decreased Hh and OPN expression as well as decreased fibrosis[84]. In humans, plasma levels of OPN and Hh expression directly correlated with the severity of fibrosis in patients, concluding that hepatic NKT cells promote fibrogenesis in NASH through their production of Hh and OPN[84]. Lastly, Wolf et al. discovered a cross-talk between CD8 T-cells, NKT cells, and hepatocytes in NASH development and progression to HCC. In patients with NASH, NKT cells and CD8 T-cells were found to be significantly elevated[87]. Using choline-deficient high-fat diet mouse models, depletion of CD8 T-cells revealed the reversal of liver damage; however, upon reduction of NKT cells despite elevated levels of CD8 T-cells, liver damage was shown to be prevented[87]. This result suggests that CD8 T-cells on their own are not enough to cause liver damage in the absence of NKT cells, thus providing evidence that NKT cells are needed to induce liver damage.
IMPACT ON THERAPY
NASH is a known major risk factor for developing HCC and data is emerging as to the impact of NASH on immune subsets and the potential ability to influence the effectiveness of systemic therapies[88]. Due to the crucial role immune cells play in NAFLD/NASH and its progression to HCC, many current treatments focus on targeting immune cells for HCC therapy[Table 3]. Historically, sorafenib, an oral multi-kinase inhibitor, was the gold standard therapy for advanced HCC for decades with a minimal prolonged survival of approximately 3 months vs. placebo[89]. Several years later, Lenvatinib, an oral tyrosine kinase inhibitor, was granted approval for treatment of chemotherapy-naïve patients with HCC after an open-label phase III trial noted non-inferiority to sorafenib[90-92]. More recently, immune checkpoint inhibitors (ICIs) have become an emerging therapy for HCC after success in the treatment of patients with other malignancies[93,94]. Atezolizumab, in combination with bevacizumab, is now the new standard of care for patients with advanced HCC as a result of the IMbrave-150 trial. The IMbrave-150 trial was a randomized, phase III trial evaluating the efficacy and safety of atezo + bev compared with sorafenib in unresectable HCC patients who were systemic therapy naive[95,96]. Compared to sorafenib, atezo + bev was shown to have statistically significant improvement in overall survival (OS) [hazard ratio (HR), 0.58; 95%CI: 0.42-0.79] and progression-free survival (PFS) (HR: 0.59; 95%CI: 0.47-0.76). As such, atezo + bev is now approved for first-line therapy for unresectable HCC[95]. However, a subgroup analysis of patients from IMBrave150 revealed the ORR for patients with NASH-related HCC was 27% versus 35% among patients with HCC due to other etiologies[97]. In addition, a phase III clinical trial evaluating the efficacy and safety of lenvatinib versus atezo + bev as first-line therapy for the treatment of unresectable HCC was conducted on 232 patients in Korea. There were no statistical differences in OS, objective response rates, or PFS with lenvatinib vs atezo + bev, resulting in either drug as the first-line option[98]. In contrast, a meta-analysis by Pfister et al. that examined the outcomes of PD-L1 or programmed cell death protein 1 (PD-1) inhibitors as monotherapy or in combination with bevacizumab for the treatment of HCC revealed no significant difference in overall survival with immune therapy vs the control group for nonviral HCC[99,100]. While atezo + bev is now a promising first-line therapy, there is still a need for future studies to further understand the complex interface of NASH, hepatocellular carcinoma, and the immune response.
Recruiting and upcoming clinical trials utilizing immunotherapy for the treatment of HCC
| Trial | Treatment | Phase | Setting | Estimated enrollment (n) | Primary outcomes | Secondary outcomes |
| NCT04721132 | Atezolizumab + Bevacizumab | II | Neoadjuvant | 30 | pCR AEs | ORR DOR RFS OS |
| NCT04442581 | Cabozantinib + Pembrolizumab | II | First-line | 29 | OR | OR PFS OS AEs DC |
| NCT05359861 | Atezolizumab + Bevacizumab + SRF388 | II | First-line | 110 | PFS | ORR DPR DC TTP OS Tmax T1/2 |
| NCT05199285 | Nivolumab + Ipilumumab | II | Second-line | 40 | ORR | OS PFS DC AEs |
| NCT03680508 | TSR-022 (Cobolimab) + TSR-042 (Dostarlimab) | II | First-line | 42 | ORR | DOR TTP PFS OS |
CONCLUSIONS
NASH is a multifactorial, complex, immunologic disease that results in severe liver alterations. A vast network of immune cells is mobilized and functionally changed in patients with NASH, resulting in a persistent driving force for the progression of cirrhosis and HCC. While we have described a few of the immune alterations above in both the innate and adaptive immune systems, there are still many mechanisms responsible for steatohepatitis that are incompletely understood. At this time, further studies are needed to elicit a more robust understanding of the complex immune pathophysiology of this disease.
DECLARATIONS
Authors contributionsSubstantial contributions to conception and design of the study, as well as provided material and editorial support: Gregory SN, Perati SR, and Brown ZJ
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interested.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
2. Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018;67:328-57.
3. Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol 2019;16:411-28.
4. Machado MV, Diehl AM. Pathogenesis of nonalcoholic steatohepatitis. Gastroenterology 2016;150:1769-77.
5. Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol 2013;10:656-65.
7. Huang DQ, El-Serag HB, Loomba R. Global epidemiology of NAFLD-related HCC: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 2021;18:223-38.
8. Estes C, Anstee QM, Arias-Loste MT, et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016-2030. J Hepatol 2018;69:896-904.
9. White DL, Kanwal F, El-Serag HB. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin Gastroenterol Hepatol 2012;10:1342-1359.e2.
11. Llovet JM, Zucman-Rossi J, Pikarsky E, et al. Hepatocellular carcinoma. Nat Rev Dis Primers 2016;2:16018.
12. Maluccio MA, Covey AM, Porat LB, et al. Transcatheter arterial embolization with only particles for the treatment of unresectable hepatocellular carcinoma. J Vasc Interv Radiol 2008;19:862-9.
13. Llovet JM, Real MI, Montaña X, et al. Arterial embolisation or chemoembolisation versus symptomatic treatment in patients with unresectable hepatocellular carcinoma: a randomised controlled trial. Lancet 2002;359:1734-9.
14. Maluccio M, Covey AM, Gandhi R, et al. Comparison of survival rates after bland arterial embolization and ablation versus surgical resection for treating solitary hepatocellular carcinoma up to 7 cm. J Vasc Interv Radiol 2005;16:955-61.
15. Raoul JL, Forner A, Bolondi L, Cheung TT, Kloeckner R, de Baere T. Updated use of TACE for hepatocellular carcinoma treatment: how and when to use it based on clinical evidence. Cancer Treat Rev 2019;72:28-36.
16. Finn RS, Ryoo BY, Merle P, et al. KEYNOTE-240 investigators. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: a randomized, double-blind, phase III trial. J Clin Oncol 2020;38:193-202.
17. Greten TF, Abou-Alfa GK, Cheng AL, et al. Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immunotherapy for the treatment of hepatocellular carcinoma. J Immunother Cancer 2021;9:e002794.
18. Cusi K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: pathophysiology and clinical implications. Gastroenterology 2012;142:711-725.e6.
19. Dixon JB, Bhathal PS, Hughes NR, O'Brien PE. Nonalcoholic fatty liver disease: improvement in liver histological analysis with weight loss. Hepatology 2004;39:1647-54.
20. Yang JD, Ahmed F, Mara KC, et al. Diabetes is associated with increased risk of hepatocellular carcinoma in patients with cirrhosis from nonalcoholic fatty liver disease. Hepatology 2020;71:907-16.
21. Rotman Y, Koh C, Zmuda JM, Kleiner DE, Liang TJ. NASH CRN. The association of genetic variability in patatin-like phospholipase domain-containing protein 3 (PNPLA3) with histological severity of nonalcoholic fatty liver disease. Hepatology 2010;52:894-903.
22. Prieto J, Melero I, Sangro B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 2015;12:681-700.
24. Brown ZJ, Greten TF, Heinrich B. Adjuvant treatment of hepatocellular carcinoma: prospect of immunotherapy. Hepatology 2019;70:1437-42.
25. Koo SY, Park EJ, Lee CW. Immunological distinctions between nonalcoholic steatohepatitis and hepatocellular carcinoma. Exp Mol Med 2020;52:1209-19.
26. Agosti P, Sabbà C, Mazzocca A. Emerging metabolic risk factors in hepatocellular carcinoma and their influence on the liver microenvironment. Biochim Biophys Acta Mol Basis Dis 2018;1864:607-17.
27. Stauffer JK, Scarzello AJ, Jiang Q, Wiltrout RH. Chronic inflammation, immune escape, and oncogenesis in the liver: a unique neighborhood for novel intersections. Hepatology 2012;56:1567-74.
28. Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology 2004;127:S35-50.
29. Hernandez-Gea V, Toffanin S, Friedman SL, Llovet JM. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology 2013;144:512-27.
30. Arrese M, Cabrera D, Kalergis AM, Feldstein AE. Innate immunity and inflammation in NAFLD/NASH. Dig Dis Sci 2016;61:1294-303.
33. Nakamoto N, Kanai T. Role of toll-like receptors in immune activation and tolerance in the liver. Front Immunol 2014;5:221.
34. Soares JB, Pimentel-Nunes P, Roncon-Albuquerque R, Leite-Moreira A. The role of lipopolysaccharide/toll-like receptor 4 signaling in chronic liver diseases. Hepatol Int 2010;4:659-72.
35. Kim SY, Jeong JM, Kim SJ, et al. Pro-inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4-MD2 complex. Nat Commun 2017;8:2247.
36. Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology 2015;61:1066-79.
37. Feldstein AE, Canbay A, Angulo P, et al. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003;125:437-43.
38. Mridha AR, Haczeyni F, Yeh MM, et al. TLR9 is up-regulated in human and murine NASH: pivotal role in inflammatory recruitment and cell survival. Clin Sci 2017;131:2145-59.
39. Garcia-Martinez I, Santoro N, Chen Y, et al. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J Clin Invest 2016;126:859-64.
40. Sarkar S, Saha P, Seth RK, et al. Higher intestinal and circulatory lactate associated NOX2 activation leads to an ectopic fibrotic pathology following microcystin co-exposure in murine fatty liver disease. Comp Biochem Physiol C Toxicol Pharmacol 2020;238:108854.
41. Gabbia D, Cannella L, De Martin S. The Role of Oxidative Stress in NAFLD-NASH-HCC Transition-Focus on NADPH Oxidases. Biomedicines 2021;9:687.
42. Polimeni L, Del Ben M, Baratta F, et al. Oxidative stress: new insights on the association of non-alcoholic fatty liver disease and atherosclerosis. World J Hepatol 2015;7:1325-36.
43. Holt MP, Cheng L, Ju C. Identification and characterization of infiltrating macrophages in acetaminophen-induced liver injury. J Leukoc Biol 2008;84:1410-21.
44. Stienstra R, Saudale F, Duval C, et al. Kupffer cells promote hepatic steatosis via interleukin-1beta-dependent suppression of peroxisome proliferator-activated receptor alpha activity. Hepatology 2010;51:511-22.
45. Obstfeld AE, Sugaru E, Thearle M, et al. C-C chemokine receptor 2 (CCR2) regulates the hepatic recruitment of myeloid cells that promote obesity-induced hepatic steatosis. Diabetes 2010;59:916-25.
46. Baeck C, Wehr A, Karlmark KR, et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 2012;61:416-26.
47. Lefebvre E, Moyle G, Reshef R, et al. Antifibrotic effects of the dual CCR2/CCR5 antagonist cenicriviroc in animal models of liver and kidney fibrosis. PLoS One 2016;11:e0158156.
48. Kahraman A, Schlattjan M, Kocabayoglu P, et al. Major histocompatibility complex class I-related chains A and B (MIC A/B): a novel role in nonalcoholic steatohepatitis. Hepatology 2010;51:92-102.
50. Male V, Stegmann KA, Easom NJ, Maini MK. Natural killer cells in liver disease. Semin Liver Dis 2017;37:198-209.
51. Martínez-Chantar ML, Delgado TC, Beraza N. Revisiting the role of natural killer cells in non-alcoholic fatty liver disease. Front Immunol 2021;12:640869.
52. Amer J, Salhab A, Noureddin M, et al. Insulin signaling as a potential natural killer cell checkpoint in fatty liver disease. Hepatol Commun 2018;2:285-98.
53. Radaeva S, Sun R, Jaruga B, Nguyen VT, Tian Z, Gao B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology 2006;130:435-52.
54. Cai L, Zhang Z, Zhou L, et al. Functional impairment in circulating and intrahepatic NK cells and relative mechanism in hepatocellular carcinoma patients. Clin Immunol 2008;129:428-37.
55. Feng M, Zhang J, Anver M, Hassan R, Ho M. In vivo imaging of human malignant mesothelioma grown orthotopically in the peritoneal cavity of nude mice. J Cancer 2011;2:123-31.
56. Lalor PF, Faint J, Aarbodem Y, Hubscher SG, Adams DH. The role of cytokines and chemokines in the development of steatohepatitis. Semin Liver Dis 2007;27:173-93.
57. Rensen SS, Slaats Y, Nijhuis J, et al. Increased hepatic myeloperoxidase activity in obese subjects with nonalcoholic steatohepatitis. Am J Pathol 2009;175:1473-82.
58. van der Windt DJ, Sud V, Zhang H, et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology 2018;68:1347-60.
59. Wang H, Zhang H, Wang Y, et al. Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J Hepatol 2021;75:1271-83.
60. Henning JR, Graffeo CS, Rehman A, et al. Dendritic cells limit fibroinflammatory injury in nonalcoholic steatohepatitis in mice. Hepatology 2013;58:589-602.
61. Huby T, Gautier EL. Immune cell-mediated features of non-alcoholic steatohepatitis. Nat Rev Immunol 2022;22:429-43.
63. Fillatreau S. B cells and their cytokine activities implications in human diseases. Clin Immunol 2018;186:26-31.
64. Bruzzì S, Sutti S, Giudici G, et al. B2-Lymphocyte responses to oxidative stress-derived antigens contribute to the evolution of nonalcoholic fatty liver disease (NAFLD). Free Radic Biol Med 2018;124:249-59.
65. Garnelo M, Tan A, Her Z, et al. Interaction between tumour-infiltrating B cells and T cells controls the progression of hepatocellular carcinoma. Gut 2017;66:342-51.
66. Sutti S, Albano E. Adaptive immunity: an emerging player in the progression of NAFLD. Nat Rev Gastroenterol Hepatol 2020;17:81-92.
67. Shalapour S, Lin X, Bastian IN, et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 2017;551:340-5.
68. Ramadori P, Kam S, Heikenwalder M. T cells: friends and foes in NASH pathogenesis and hepatocarcinogenesis. Hepatology 2022;75:1038-49.
69. Heinrich B, Brown ZJ, Diggs LP, et al. Steatohepatitis impairs T-cell-directed immunotherapies against liver tumors in mice. Gastroenterology 2021;160:331-345.e6.
70. Kang TW, Yevsa T, Woller N, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011;479:547-51.
71. Rakhra K, Bachireddy P, Zabuawala T, et al. CD4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell 2010;18:485-98.
72. Ma C, Kesarwala AH, Eggert T, et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016;531:253-7.
73. Brown ZJ, Fu Q, Ma C, et al. Carnitine palmitoyltransferase gene upregulation by linoleic acid induces CD4+ T cell apoptosis promoting HCC development. Cell Death Dis 2018;9:620.
74. Sutti S, Jindal A, Locatelli I, et al. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology 2014;59:886-97.
75. Van Herck MA, Weyler J, Kwanten WJ, et al. The differential roles of T cells in non-alcoholic fatty liver disease and obesity. Front Immunol 2019;10:82.
76. Hirsova P, Bamidele AO, Wang H, Povero D, Revelo XS. Emerging roles of T cells in the pathogenesis of nonalcoholic steatohepatitis and hepatocellular carcinoma. Front Endocrinol (Lausanne) 2021;12:760860.
77. MacParland SA, Liu JC, Ma XZ, et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun 2018;9:4383.
79. Ghazarian M, Revelo XS, Nøhr MK, et al. Type I interferon responses drive intrahepatic T cells to promote metabolic syndrome. Sci Immunol 2017;2:eaai7616.
80. Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol 2019;16:356-71.
81. Geissmann F, Cameron TO, Sidobre S, et al. Intravascular immune surveillance by CXCR6+ NKT cells patrolling liver sinusoids. PLoS Biol 2005;3:e113.
82. Kumar V. NKT-cell subsets: promoters and protectors in inflammatory liver disease. J Hepatol 2013;59:618-20.
83. Syn WK, Oo YH, Pereira TA, et al. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology 2010;51:1998-2007.
84. Syn WK, Agboola KM, Swiderska M, et al. NKT-associated hedgehog and osteopontin drive fibrogenesis in non-alcoholic fatty liver disease. Gut 2012;61:1323-9.
85. Syn WK, Choi SS, Liaskou E, et al. Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology 2011;53:106-15.
86. Diao H, Kon S, Iwabuchi K, et al. Osteopontin as a mediator of NKT cell function in T cell-mediated liver diseases. Immunity 2004;21:539-50.
87. Wolf MJ, Adili A, Piotrowitz K, et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 2014;26:549-64.
88. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med 2018;24:908-22.
89. Llovet JM, Ricci S, Mazzaferro V, et al. SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359:378-90.
90. Greten TF, Lai CW, Li G, Staveley-O’Carroll KF. Targeted and immune-based therapies for hepatocellular carcinoma. Gastroenterology 2019;156:510-24.
91. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. The Lancet 2018;391:1163-1173.
92. Brown ZJ, Gregory S, Hewitt DB, et al. Safety, efficacy, and tolerability of immune checkpoint inhibitors in the treatment of hepatocellular carcinoma. Surg Oncol 2022;42:101748.
93. Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med 2015;373:1627-39.
94. Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320-30.
95. Finn RS, Qin S, Ikeda M, et al. IMbrave150 Investigators. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med 2020;382:1894-905.
96. Lee MS, Ryoo B, Hsu C, et al. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (GO30140): an open-label, multicentre, phase 1b study. The Lancet Oncology 2020;21:808-20.
97. Ducreux M, Zhu AX, Cheng A, et al. IMbrave150: exploratory analysis to examine the association between treatment response and overall survival (OS) in patients (pts) with unresectable hepatocellular carcinoma (HCC) treated with atezolizumab (atezo) + bevacizumab (bev) versus sorafenib (sor). JCO 2021;39:4071-4071.
98. Kim BK, Cheon J, Kim H, et al. Atezolizumab/Bevacizumab vs. Lenvatinib as first-line therapy for unresectable hepatocellular carcinoma: a real-world, multi-center study. Cancers 2022;14:1747.
99. Pfister D, Núñez NG, Pinyol R, et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021;592:450-6.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Gregory SN, Perati SR, Brown ZJ. Alteration in immune function in patients with fatty liver disease. Hepatoma Res 2022;8:31. http://dx.doi.org/10.20517/2394-5079.2022.34
AMA Style
Gregory SN, Perati SR, Brown ZJ. Alteration in immune function in patients with fatty liver disease. Hepatoma Research. 2022; 8: 31. http://dx.doi.org/10.20517/2394-5079.2022.34
Chicago/Turabian Style
Gregory, Stephanie N., Shruthi R. Perati, Zachary J. Brown. 2022. "Alteration in immune function in patients with fatty liver disease" Hepatoma Research. 8: 31. http://dx.doi.org/10.20517/2394-5079.2022.34
ACS Style
Gregory, SN.; Perati SR.; Brown ZJ. Alteration in immune function in patients with fatty liver disease. Hepatoma. Res. 2022, 8, 31. http://dx.doi.org/10.20517/2394-5079.2022.34
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 29 clicks
Cite This Article 29 clicks

 Like This Article 32
likes
Like This Article 32
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.