
Immunological characterization of hepatocellular carcinoma

 , ...
, ... Abstract
Hepatocellular carcinoma (HCC) is the most common type of primary liver malignancy and the fourth leading cause of cancer-related deaths globally. HCC is often diagnosed in late stage, difficult to treat, and has poor prognosis with a median survival of 6-20 months. Innate and adaptive immunity play a pivotal role in determining tumor control versus progression. Genomic instability and abnormal signaling in the setting of chronic liver inflammation lead to tumorigenesis. Tumor progression occurs due to a sustained inflammatory response that promotes fibrogenesis and angiogenesis. This review discusses the key innate and adaptive cellular players that mediate the anti-tumor response. This review explores the complex interactions that occur within the tumor microenvironment and their clinical implications. HCC is a fastidious malignancy that is able to evade and downregulate the host immune response. Mechanisms of how this occurs are discussed, along with how they may be exploited in the development of novel therapeutics. From our research, it appears that striking a balance between immunotolerance and a robust immune response may yield the best prognosis. This review assesses major and recent developments in HCC immunotherapy, including adoptive cell therapy, cancer vaccines, and targeted therapy such as checkpoint inhibitors. Overall, the importance of the immune response in determining outcomes for HCC cannot be understated. Improved animal models and better characterization of the tumor microenvironment are needed. We determine that a better understanding of the HCC immune profile would facilitate advancements in diagnosis, monitoring, and ultimately treatment.
Keywords
Introduction
Hepatocellular carcinoma (HCC) is a primary malignancy of the liver that often develops in the setting of chronic liver disease. Worldwide, HCC is the second leading cause of lost life-years and fourth leading cause of cancer-related deaths[1]. Major risk factors for HCC include chronic hepatitis B and C viral infection and alcohol use, although patients with liver cirrhosis of any etiology are at risk. Other risk factors include exposure to environmental toxins such as aflatoxin, metabolic disease such as diabetes mellitus, and genetic predisposition such as alpha-1 antitrypsin deficiency. The global disease burden of liver cancers, the majority of which are caused by HCC, is substantial and increasing, with an estimated 42,810 new cases and 30,160 deaths expected to occur in the year 2020[2,3]. The mainstay of treatment involves surgical resection or liver transplantation. However, the prognosis for HCC remains poor because a significant proportion of patients are diagnosed at advanced stage and/or are unsuitable surgical candidates.
The pathophysiological mechanisms of HCC are incompletely understood. However, HCC is believed to arise from aberrant molecular signaling, genomic instability, and chronic inflammation[4]. The role of the immune system in HCC is a trending topic of research, primarily due to the investigation of potential immunotherapies. The liver itself is an immunological organ. Housing the body’s largest population of phagocytes such as macrophages, the liver acts as a filter that detects and removes exogenous molecules[5]. Portal drainage from the gut continuously supplies an abundance of food antigens and microbe-associated molecular patterns (MAMPs) to the liver via the portal vein[6]. In response, the liver must exert the dual functions of immune activation against harmful antigens and immunotolerance for inert compounds. Balanced immunoregulation is key in response to HCC, as it can mean the difference between tumor progression and control [Figure 1][7]. A better understanding of the immune landscape of HCC would be valuable in improving future clinical outcomes. The purpose of this review is to discuss the immune response to hepatocellular carcinoma, to highlight the importance of the tumor immune microenvironment, and to identify tumor mechanisms of immune evasion and suppression. Previous and current investigations on HCC immunotherapy including checkpoint inhibitors are examined. Furthermore, this review identifies gaps in current knowledge and directions for future research on HCC.
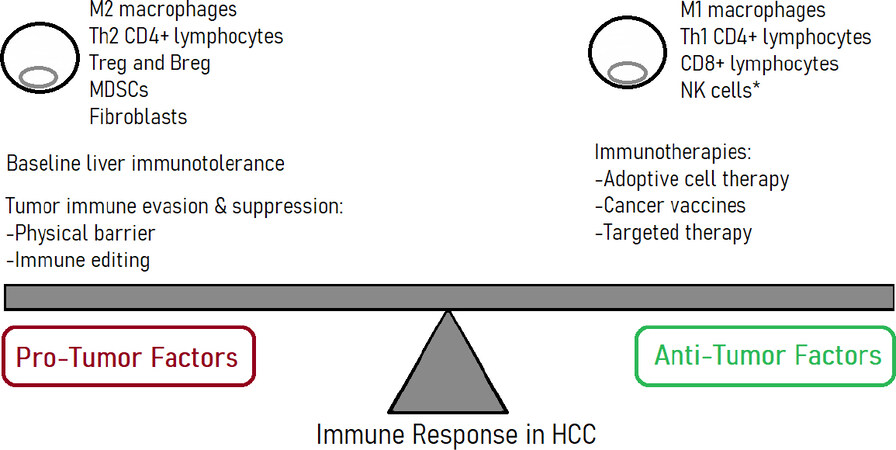
Figure 1. Balance in the immune response to HCC. *NK cells are known to have both pro-tumor and anti-tumor features. HCC: hepatocellular carcinoma; Treg: regulatory T lymphocytes; Breg: regulatory B lymphocytes; MDSC: myeloid-derived stellate cells; NK cells: natural killer cells.
Immune response to HCC
Innate immune response
The innate immune response is the body’s first line of defense against invading pathogens. In the context of cancer, effector cells of innate immunity include macrophages, natural killer (NK) cells, and dendritic cells (DCs)[8]. Innate immunity is important in initial tumor detection and control, as well as presenting antigens to cells involved in adaptive immunity[9]. This section describes the roles of innate effector cells and their dynamic relationship with tumor cells. In addition, we discuss the importance of deranged physiologic function and mechanisms by which HCC subverts innate immunity. For more detailed information, Roderburg et al.[10] expertly summarized the relationship between innate immunity in HCC in a recent review. Please refer to Table 1 for a comparison between innate and adaptive immune response to HCC.
Innate and adaptive immune response to HCC
| Innate Immune Response | Adaptive Immune Response | |
|---|---|---|
| Role | First line of defense | More pathogen-specific response |
| Relative speed of onset | Quicker, almost immediate | Slower, usually days |
| Specificity | Limited specificity | Highly specific |
| Memory | Absent | Present |
| Cellular component and major function | Macrophages
KCs – phagocytose pathogens TAMs – promote (M2) or control (M1) tumor progression NK cells – direct tumor cytotoxicity DCs – antigen presentation, phagocytosis pDCs – immune tolerance cDCs – immune suppression | T cells
CD4/Th1 – promote inflammation CD4/Th2 – promote tumor growth CD4/Th17/Th22 - angiogenesis CD8/Tc – direct tumor cytotoxicity Treg – immune suppression B cells – antibody production Breg – immune suppression CD5 – promote tumor growth CD20 – direct tumor cytotoxicity |
Macrophages
Kupffer cells (KCs) are macrophages that reside in sinusoids of the liver. The major function of KCs is to detect and phagocytose bloodborne pathogens that have been marked as foreign through complement-mediated opsonization. While other cell types use complement receptors such as CR1 to tether opsonized pathogens, they may be inadequate under shear forces of blood flow[11]. In contrast, KCs can tightly capture pathogens despite shear forces through specialized receptors called VSIG4 (V-set immunoglobulin-domain-containing 4), which belong in the complement receptor of the immunoglobulin superfamily (CRIg)[11]. CRIg is also expressed by macrophages in the peritoneum and spleen[12]. It has been shown that higher CRIg expression is associated with reduced disease severity among cirrhotic patients, due to enhanced phagocytic activity[13]. As cirrhosis is a major risk factor for the development of HCC, increased CRIg expression by KCs may potentially have a similar positive effect on the clinical course of HCC as well.
However, not all macrophages within the liver are equal. Macrophages that live within or surrounding the tumor microenvironment are called tumor-associated macrophages (TAMs). TAMs can be classified into two subsets: the anti-tumorigenic M1 phenotype and the pro-tumorigenic M2 phenotype[14]. TAMs have been shown to promote tumor progression through the production of growth factors and inflammatory cytokines/chemokines[14]. In HCC and other tumors, TAMs predominantly differentiate towards the M2 subset in a process called macrophage polarization, facilitated in part by colony stimulating factor 1 (CSF-1) and C-C motif ligand 2 (CCL2)[14,15]. M2 macrophages produce cytokines that promote HCC tumor growth (IL-5 and TGF-β), invasion (IL-6), and metastasis (SDF-1)[14]. In contrast, M1 macrophages secrete a different set of cytokines that aid in tumor control, including IL-12[8]. Several studies have shown that a greater proportion of M2 TAMs in the immune response results in worse clinical outcomes, not only in HCC but also in other cancers such as breast and lymphoma[16]. Further investigation into TAM subtypes and their crosstalk is under way, in an effort to discover potential targets for HCC immunotherapy. Mechanisms being explored include depletion of M2 TAMs, inhibition of TAM recruitment, and shifting macrophage polarization to a more favorable/antitumor profile[16].
Natural killer (NK) cells
Similar to macrophages, NK cells are found in the greatest concentration within the liver. NK cells are unique in that they target cells with absence of self. In an effort to evade the immune response, tumor cells downregulate antigen-presenting molecules such as MHC-I; this absence of self is recognized by NK cells that then eliminate the suspect cell[5]. NK cells exert their cytotoxic effect through secretion of granules containing perforin, granzymes (proteases), and IFN-γ that result in cell lysis. NK cells can protect against insults that lead to HCC through various mechanisms, such as clearance of hepatitis virus-infected cells and exerting antifibrotic properties in alcoholic liver disease[17]. Recent research on tissue-resident NK cells (trNK) has provided new knowledge on this cell population. Unlike their counterparts that circulate in peripheral blood (cNK), trNK differ in the profile of MHC-recognizing receptors and cytokines produced[18]. Hepatic trNKs maintain hepatic homeostasis by reacting to liver insults such as acute viral infection and hepatoma, the latter of which elicits a clonal-like expansion of trNKs that enable a memory-like, more robust immune response[19]. Indeed, a greater proportion of NK cells in the tumor microenvironment is correlated with improved survival among HCC patients[17]. Conversely, reduced NK cell frequency and activity are associated with HCC progression[20]. Impaired NK cell function is found in cirrhosis and HCC. The exact mechanism is not well understood but several hypotheses have been proposed, including phagocytosis of NK cells by hepatic stellate cells and downregulation of NK receptors[15,17]. Furthermore, alterations in the cytokine profile within the tumor microenvironment may explain failure of NK cells to control HCC development[21].
Myeloid-derived suppressor cells (MDSCs), a subset of inflammatory monocytes, are also thought to play a role in suppressing the HCC immune response. MDSCs in HCC have been shown to directly suppress the cytotoxic function of NK cells in a contact-dependent manner involving ARG1 and iNOS[22]. MDSCs are known to promote tumor angiogenesis and are upregulated in HCC, partly due to the hypoxic tumor microenvironment[23,24]. Immunotherapies that enhance NK function in the killing of HCC tumor cells are being developed, including stimulation of NK cell growth through cytokine infusions and adoptive NK cell transfer[17].
Dendritic cells
DCs bridge innate and adaptive immunity through a spectrum of functions. Not only can DCs phagocytose tumor cells, but they also act as professional antigen-presenting cells (APCs) that elicit a robust T cell response[9]. DC subsets have an important role in balancing immune activation and tolerance in the liver. A cytotoxic subset of DCs (NK1.1+ DCs) stimulate an inflammatory Th1-predominant response that helps with tumor control[5]. Conversely, other DC populations in the liver have immunosuppressive properties. It has been shown that activated DCs exist in fewer numbers among HCC patients; even fewer DCs are found in the tumor nodule itself due to blunted recruitment[15]. Furthermore, DCs that are present in the liver elicit a weaker T cell response compared to those in other tissues[5].
The majority of hepatic DCs can be categorized into two classes: plasmacytoid DCs (pDCs) and conventional/myeloid DCs (cDCs)[8]. The cDC subset dampens T cell activation through secretion of IL-10 and IDO, while pDCs induce immune tolerance through expression of PD-L1[25]. A subset of cDCs (LAMP3+ DCs) identified in HCC and other cancers such as lung is found in much greater numbers in lymph nodes than in the tumor[26]. Analysis of HCC patients has shown that reduced LAMP3+ expression led to worse survival, adding evidence to the anti-tumor function of certain DC subsets[27]. While LAMP3+ DCs can facilitate T cell migration to the tumor site, they have also been associated with increased Treg population and T cell dysfunction[26,27]. Taken together, DCs appear to have numerous immunoregulatory properties in HCC. Further investigation into the mechanisms and different subsets of DCs would allow better understanding of how this specialized cell population affects HCC prognosis.
Adaptive immune response
Whereas the innate immune response is almost immediate, the adaptive immune response takes at least a few days to mount[28]. The tradeoff is that the adaptive immune system can exert a highly specific and amplifiable response against antigens. The hallmark of adaptive immunity is immunological memory, whereby future exposure to the same or similar antigens will trigger a quicker response. This section discusses the role of T and B lymphocytes in the adaptive immune response against HCC. Interestingly, adaptive immune cells appear to have both pro- and anti-tumor features[29].
T cells
The adaptive immune system recognizes tumor cells through tumor-associated antigens (TAAs). HCC has multiple TAAs, with alpha-fetoprotein (AFP) being one of the most common, specific, and immunogenic[28]. TAAs are recognized, processed, and presented by innate immune cells to T and B lymphocytes, which then exert effector functions.
T lymphocytes can broadly be placed into three categories: CD4+ T cells (Th “helper” subtypes), CD8+ T cells (Tc “cytotoxic” and Treg “regulatory” subtypes), and γδ-T cells[30]. Shuai et al.[28] summarized well in table format the various roles of T cell subsets in combating HCC. In general, it is accepted that CD8+ T cells play the most essential role in tumor control[29,31]. CD8+ T cells are capable of exerting direct tumor-killing effect through the secretion of perforins and granzymes[28]. CD8+ T cells also release IL-33, a pro-inflammatory cytokine that activates anti-tumor effector cells including CD4+ T, CD8+ T, and NK cells[32,33]. These mechanisms are of clinical significance as multiple studies have illustrated that greater intratumoral CD8+ T cell density is associated with lower rate of HCC recurrence[34-36]. A recent metanalysis by Xu et al.[37] involving more than 3,500 patients showed that high levels of CD8+ tumor-infiltrating lymphocytes were correlated with both improved overall and disease-free survival in HCC. Although these researchers suggested that greater CD8+ T cell density is a positive prognostic factor, other studies have found the reverse. Tissue analysis has shown that higher density of CD8+ T cells was associated with higher recurrence and lower five-year survival[38]. Mice models have shown that depletion of CD8+ T cells actually delayed HCC tumor development in the setting of chronic liver disease[29]. Thus, the role of CD8+ T cells in modulating HCC remains elusive.
CD4+ T helper cells are similar in this regard. Different Th subtypes appear to exert opposing effects. Current research suggests that a Th1-dominant response leads to more favorable outcomes in HCC than a Th2-dominant response[39-41]. Th1 cells secrete pro-inflammatory cytokines, including IL-2 and IFN-γ, that have potent anti-tumor effects[41]. A Th2-shifted immune response, on the contrary, is associated with venous metastasis in HCC[15]. Interestingly, a study of patients with chronic hepatitis C virus (HCV) showed that Th2 dominance may induce hepatocarcinogenesis in the first place[42]. Other helper T cell subtypes worth mentioning include Th17 and Th22, both of which have been shown to have pro-tumor effects[15,43]. Th22 cells secrete IL-22, which, with the assistance of Th17 cells, led to angiogenesis to the tumor site[43].
Arguably the most important T cell subtype in determining clinical outcomes in HCC is the regulatory T cell (Treg). Tregs are important in preventing an exaggerated immune response, however excess numbers lead to an immunosuppressive state. Treg is known to suppress anti-tumor CD4+ and CD8+ T cell function, along with NK-mediated tumor destruction[44]. Although intratumoral lymphocyte penetration is generally poor in HCC, studies show that Treg populations are paradoxically upregulated[28,45,46]. This is in part due to Treg-attracting CCL22 and CCL1 chemokines[47]. Increased intratumoral Treg density is associated with greater HCC invasion and tumor recurrence[48]. A study of patients who underwent HCC resection showed that higher preoperative Treg levels was associated with significantly higher recurrence rate at one-year follow-up[49]. Together, these findings suggest that increased Treg density portends a poorer prognosis in HCC.
The myriad T cell subsets and functions present multiple potential targets for immunotherapy. Adoptive cell therapy involving T lymphocytes, genetically-engineered TAA-specific T cell receptors (TCR), and molecules that engage TAA with TCR (bispecific T cell engagers/BiTEs) are only some examples[50]. In addition, many checkpoint inhibitors involved in the T cell response are under investigation. These are discussed further in the section titled “HCC Immunotherapy” to follow.
B cells
B lymphocytes have several essential functions in adaptive immunity. Similar to dendritic cells and macrophages, B cells are professional APCs that can alert T cells to novel pathogens. B cells are most notable for the production and secretion of antibodies (i.e., humoral immunity). Antibodies have anti-tumor properties through a number of mechanisms: mediating tumor cell phagocytosis, activating complement, and facilitating antibody-dependent cellular cytotoxicity (ADCC)[51]. Although B cells comprise a significant portion of the tumor microenvironment, their role in modulating HCC has not been clearly elucidated[52]. Fewer studies have examined B lymphocytes as compared to T cells.
Existing research suggests that B lymphocytes have both anti- and pro-tumor effects[51]. Analysis of HCC tissue databases showed that overexpression of immunoglobulins including IgM by B cells correlated with improved survival[53]. Mature CD20+ B cells in the HCC tumor margin are known to exert direct anti-tumor effects by releasing granzymes and inducing tumor cell death through TRAIL[54]. Depletion of B cells in mice models led to decreased tumor control[31]. Clinically, it has been shown that increased density of tumor-infiltrating T and B lymphocytes is associated with improved survival among HCC patients[31]. These and other studies have illustrated the favorable effect of B cells in fighting not only HCC, but other cancers such as lung and breast[55,56].
Many B cell subsets exist with distinctly different functions. Regulatory B cells (Breg) in particular are known to suppress the immune response through the production of IL-10 and TGF-β cytokines[57]. Furthermore, mice models have shown the propensity of Breg to migrate from peripheral circulation into HCC tumor tissue, further amplifying the pro-tumor profile[58]. Tumor-infiltrating B cells have been shown to promote tumor angiogenesis/growth through lymphotoxin production[59]. Notably, B cells that express high levels of CD5+ have been associated with poorer clinical outcomes in HCC[60,61]. In a tumor microenvironment rich in pro-inflammatory cytokines such as IL-6, CD5+ B cells promote cancer growth through STAT3[62]. Under physiologic conditions, STAT3 is a key element of wound healing. However, the excess STAT3 activation found in HCC creates favorable conditions for tumor growth, for instance augmented vascularization and production of extracellular matrix[63]. Certain B cell subsets are also known to induce polarization of macrophages to the M2 phenotype which, as discussed above, is associated with HCC invasion/metastasis and worse survival[52,64].
Few studies have examined the possibility of B lymphocyte-based immunotherapy. A recent review by Wennhold et al.[65] suggests that CD40L/CD40 signaling may be exploited to develop B cell-based therapy. Specifically, B cells would be isolated from the patient, activated and expanded with CD40L solution, incubated with HCC tumor antigen, and then infused back into the patient with the goal of eliciting a powerful anti-tumor immune response. Other proposed methods include B cell depletion therapy (specifically of Breg subset), stimulation of plasma B cells to increase antibody production, etc.[66]. In summary, B lymphocytes appear to be an under-studied immune cell type with good therapeutic potential.
Tumor immune microenvironment
Tumorigenesis
Hepatocellular carcinoma develops in the setting of chronic liver disease in at least 80% of cases[67]. Although normal hepatocytes have the remarkable ability to self-repair and self-regenerate, repeated insults over time can cause irreparable damage[30]. Cycles of injury and compensatory regeneration lead to dysregulated hepatocyte growth, fibrosis, and formation of dysplastic nodules[67,68]. This creates an inflammatory microenvironment favorable for carcinogenesis. The process of tumorigenesis can be divided into three phases: initiation, progression, and metastasis[69].
As in other cancers, genomic instability and epigenetic changes are attributed to the initiation of hepatocellular carcinoma[4]. Copy number alterations (deletion or amplification of large genomic sections) and driver mutations in tumor suppressor genes such as TP53 are examples of DNA-level changes that occur in HCC[70]. Additionally, epigenetic changes including transcription regulation and production of microRNAs can promote HCC growth and invasion[70,71]. Aberrant signaling that results from genomic changes also contributes to tumor initiation. For example, the STAT3 cascade is thought to be central in HCC development due to the anti-apoptotic effects and angiogenesis that it affords tumor cells[72]. Indeed, it has been shown that constitutively active STAT3 alone can drive development of HCC even in non-cirrhotic livers, a further testament to the importance of this signaling cascade[73].
Tumor progression is driven by the inflammatory and hypoxic environment of cirrhosis. A study that highlights the importance of cirrhosis in tumorigenesis was carried out by He et al.[74], who discovered that transplantation of HCC progenitor cells only led to cancer formation when injected into cirrhotic livers. Key in promoting cirrhosis are the reactive oxygen species (ROS) produced by injured hepatocytes. ROS can mediate DNA damage and inhibit adaptive immunosurveillance[75]. ROS also promote fibrosis by activating hepatic stellate cells (HSC) and myofibroblasts, which produce excess collagen and extracellular matrix (ECM), respectively[76]. The effect of fibrosis is localized acute hypoxia within the liver parenchyma that, in turn, triggers the production of vascular endothelial growth factor (VEGF) and other pro-angiogenic molecules[76]. Most cells found in the liver also produce cytokines, including platelet-derived growth factor (PDGF) and TGF-β, which amplify fibrogenesis[77,78]. Fibrosis drives tumor progression and metastasis. A fibrotic environment promotes epithelial proliferation, differentiation, and epithelial–mesenchymal transition (EMT)[79]. EMT is the process by which epithelial cells gain features of mesenchymal cells, including invasion of the basement membrane and enhanced migration. It is well established that EMT is a major contributor of both tumor progression and metastasis[80].
Tumor metastasis occurs in an established sequence that involves the following: (1) invasion of normal tissue; (2) intravasation into surrounding tissue and vasculature; (3) propagation through the circulatory system; (4) extravasation out of capillaries; and (5) seeding and growth in new sites of metastasis[14]. HCC most commonly metastasizes to the lung and lymph nodes. Although extrahepatic metastasis is only estimated to occur in 10% of HCC cases, it is associated with worse one- and three-year survival compared to patients without metastasis[81]. Researchers found that the site of metastasis did not have an impact on patient survival[81]. However, HCC metastasis plus vascular invasion was associated with worse prognosis than either metastasis alone or vascular invasion alone. Overall, it is clear that tumorigenesis in HCC is directly influenced by the tumor microenvironment. Components and features of the tumor microenvironment are discussed in the following section.
Components of the tumor microenvironment
Cellular players
The HCC tumor microenvironment (TME) is complex and composed of both normal and neoplastic cells[82]. Single cell sequencing technology has allowed for better characterization of the tumor landscape in primary liver cancers[83]. Aside from HCC parenchymal cells, major cell types found in the TME include cancer-associated fibroblasts (CAF), immune cells such as TAMs and T cells, hepatic stellate cells (HSC), and endothelial cells[68]. CAFs and HSCs appear to be of special importance in forming a pro-tumor microenvironment. Overall, it appears that greater density of these stromal components predicts poor prognosis in HCC, specifically reduced overall and disease-free survival[84]. The role of these cellular components and their clinical significance are summarized in Table 2.
Function of cellular components of the HCC TME and their clinical implications
| Cell type | Function in the tumor microenvironment | Clinical implications & findings |
|---|---|---|
| CAF | Produce growth factors such as EGF[68] Increase collagen production[85] Favor tumor encapsulation[86] | Greater tumor cell proliferation
Reduced overall survival[87] Promote hepatocarcinogenesis |
| HSC | Reduce lymphocyte infiltration of tumor[88] Shift monocytes towards immunosuppression[89] Inhibit lymphocyte infiltration in tumors[90] Enhance tumor angiogenesis[90] | Increased hepatic fibrosis[91] Greater HCC recurrence[89] Reduced immune surveillance Sustains tumor metabolism |
| TAM | Release growth factors and cytokines
Suppress immunity by recruiting Tregs[92] | Larger tumor size[85] Reduced overall survival[93] |
| MDSC | Suppress non-regulatory T cell response[94] | Marker of advanced disease[95] |
| Treg | Suppress the overall immune response[68] | Worse prognosis post-resection[48] |
| Endothelial cells | Express pro-angiogenic receptors[85] Exhibit rapid cell turnover[68] | Quicker tumor growth[96] Increased mortality[96] |
| Kupffer Cells | Detect and phagocytose foreign pathogens | Tumor surveillance |
Non-cellular players
Non-cellular components of the tumor microenvironment also influence HCC progression and prognosis[68]. Such components include enzymes, growth factors, cytokines/chemokines, and signaling pathways; their functions are well summarized in a recent review by Novikova et al.[68]. Non-cellular players of particular importance that are discussed here include pro-inflammatory cytokines (specifically IL-6 and TGF-β) and MAPK signaling.
The pro-inflammatory cytokines IL-6 and TGF-β are well-known acute phase reactants that are implicated in many cancers. IL-6 production by cells of the TME (most notably Kupffer cells) enhances HCC tumor growth through several mechanisms. Firstly, IL-6 triggers tumor proliferation in a STAT3 signaling-dependent manner[97]. Secondly, IL-6 inhibits apoptosis that normally occurs in response to DNA damage, thereby facilitating tumor progression[98]. Thirdly, IL-6 promotes angiogenesis and tumor invasion[98,99]. A study involving patients with mostly TNM stage I HCC, post-resection, found that high tissue expression of IL-6/IL-6R was significantly associated with earlier tumor recurrence[100]. Similar to IL-6, TGF-β is also a master regulator of cell growth and apoptosis. TGF-β has innumerable roles in modulating HCC and appears to be an essential component of tumorigenesis[101]. Interestingly, TGF-β has different roles in the early versus advanced stages of HCC[102]. TGF-β induces apoptosis of neoplastic cells in the early phase of tumor development. However, late-stage tumor cells appear to benefit from TGF-β-induced cellular proliferation[102]. In addition, TGF-β has immunosuppressive properties that assist tumor cells in escaping immune surveillance[103]. Exact mechanisms of how this occurs is not well understood. However, it has been shown that activation of CAFs by TGF-β leads to dysregulated extracellular matrix (ECM) architecture and excess fibrosis, both of which favor tumor progression[104]. The clinical significance of TGF-β is that it assists tumor cells with immune evasion and causes immunotherapy failure[101,104]. Evidently, greater concentrations of IL-6 and TGF-β in the tumor microenvironment may be predictors of worse prognosis in HCC.
Although studies have shown that IL-6 deletion reduces spontaneous carcinogenesis and delays HCC tumorigenesis, IL-6 has not yet been exploited as a target for immunotherapy[105,106]. However, it is worth mentioning that clinical trials examining anti-hepatitis virus drugs such as telbivudine and sofosbuvir use IL-6 as a marker of therapeutic response[107,108]. More experiments have examined TGF-β as a potential target for HCC therapy. Galunisertib, a TGF-β receptor 1 inhibitor, has been demonstrated to enhance T cell penetration into the tumor microenvironment[109]. An alkaloid compound called sanguinarine has been shown to inhibit TGF-β-induced EMT and tumor cell migration in HCC cell lines[110]. Antibodies against TGF-β have been shown to have anti-tumor effects in HCC and other cancers[111]. Thus, TGF-β appears to be a promising target for the development of novel HCC immunotherapy.
Several signaling pathways involved in HCC have been studied, including STAT3-NFκB, JAK-STAT, RAS-MAPK, PI3K-AKT-mTOR, and Wnt-β-catenin[112]. Of these, cascades involving mitogen-activated protein kinase (MAPK) emerge as key regulators of HCC. MAPK is a tyrosine kinase that regulates essential cellular processes, including cell proliferation and apoptosis[68]. Overexpression of MAPK signaling results in uncontrolled tumor growth. For example, activation of the EGFR-RAS-MAPK pathway leads to dysregulated cell proliferation in HCC[113]. The MAPK/p38 signaling has been shown to promote angiogenesis and drive tumor cell metastasis (migration and invasion)[114,115]. In addition, activation of the MAPK/p38 endows resistance against apoptosis to HCC cells[116]. Experiments involving inhibitors of MAPK signaling demonstrate reduced proliferation, invasion, epidermal–mesenchymal transition, and metastasis in HCC[117-119]. Erlotinib, for example, is a monoclonal antibody against MAPK under current investigation in clinical trials. Studies have shown that erlotinib is an effective adjunctive therapy to bevacizumab for the treatment of advanced-stage HCC, extending the progression-free survival with fewer side effects than the current first-line drug sorafenib[120]. More recently, the cGAS-STING (cyclic GMP-AMP synthase-stimulator of interferon genes) pathway has emerged in the field of HCC research for its potential as a therapeutic target. The cGAS-STING pathway activates the innate immune response in response to DNA products, from both foreign pathogens and self-DNA released due to cell damage[121]. It has been shown that low cGAS-STING activation is associated with reduced survival in HCC, lung adenocarcinoma, and gastric cancers[122]. In a study involving HCC mice models, activation of cGAS-STING using cyclic dinucleotide (a STING agonist) resulted in tumor shrinkage and earlier tumor detection[123].
Intercellular communication and exosomes
Aside from the aforementioned cytokines/chemokines and signaling pathways, exosomes represent another mechanism of intercellular communication. Exosomes are membrane-coated extracellular vesicles that play a key role in intercellular communication within the tumor microenvironment[124]. Exosomes contain nucleic acids, lipids, proteins, and signaling molecules. It has been shown that exosomes are involved in the spread of hepatitis virus and progression of fibrosis in alcoholic liver disease, both of which are major contributors of HCC[125]. Exosomes promote the proliferation and migration of HCC through multiple mechanisms, including induction of the EMT that is key in tumor metastasis[126]. HCC cells also use exosomes in immune evasion. For instance, transformed hepatocytes secrete exosomes that help to upregulate Breg cell populations in the TME, which results in reduced CD8+ T cell activity[61]. Tumor-derived exosomes have been found to expand immunosuppressive M2 macrophages and downregulate the anti-tumor activity of NK and T cells[127]. Tumor-derived exosomes have also been found to contain high levels of 14-3-3ζ, a key regulatory molecule used in class-switching[128]. The effect of this is T cell exhaustion. Indeed, it has been shown that overexpression of 14-3-3ζ is associated with worse survival in HCC[129].
Therefore, exosomes present an opportunity for the development of immunotherapy. For example, exosomes may be used as delivery vehicles for tumor suppressive molecules and nucleic acids[130]. It has been shown in HCC mice models that the infusion of DCs impregnated with tumor-derived exosomes (called DC-TEX) restored exhausted T cells; the combination of DC-TEX plus PD-1 antibodies appeared to enhance the efficacy of the front-line HCC drug sorafenib[131]. Current experiments suggest that exosomes may have useful applications in HCC, either as tumor biomarkers or as a therapeutic target[132].
Tumor metabolism and classification
The metabolism of HCC cells is an important aspect of the tumor microenvironment. Tumor cells preferentially use aerobic glycolysis for energy production as opposed to oxidative phosphorylation, in a type of modified metabolism called the Warburg effect[133]. The resulting lactate undergoes fermentation, which activates hypoxia-inducible factor 1 (HIF-1). HIF-1 is a well-known transcription factor that activates a host of genes contributing to the tenacity of tumors; these genes are involved in angiogenesis, cell survival, and cell invasion[134]. In fact, mouse hepatoma cell lines were used in some of the first studies examining the function of HIF-1. These early experiments found that HIF-1-rich hepatoma cells had greater vascularity and rate of cell growth compared to HIF-1-deficient cell lines[135]. Little research has since been conducted on the metabolism of HCC tumors. One study found that high lipid concentration in the tumor microenvironment sustained the increased metabolic demands of myeloid-derived suppressor cells (MDSC), which have potent pro-tumor effects[136]. Further investigation into the metabolic profile and derangements of the TME may guide specific, even personalized, treatment of HCC by way of genome-scale metabolic models[137].
Tumors are often described using criteria that involve size and spread, most notably TNM staging. Although TNM classification can be applied to hepatocellular carcinoma, other staging schemes that consider liver function are also used. Among these, the Barcelona Clinic Liver Cancer (BCLC) system has been widely adopted in clinical practice[138,139]. BCLC stratifies patients according to factors such as radiologic tumor characteristics, Child–Pugh score, and patient performance status; based on the stage, BCLC suggests next steps in management[140]. Despite its popularity, some studies have questioned the validity and prognostic utility of BCLC staging[141,142]. Other staging systems for HCC include the French classification, Hong Kong Liver Cancer (HKLC) staging system, Cancer of the Liver Italian Program (CLIP) score, Okuda classification, etc.[143,144]. Increasingly, there has been recognition of the importance of the tumor microenvironment on cancer prognosis. In an attempt to describe intratumoral cell diversity and the tumor microenvironment, a panel of experts has developed the Evo-index and Eco-index, respectively[145]. The Evo-index reflects the heterogeneity of the tumor composition and how it evolves over time, due to spontaneous mutations and selective pressures. The Eco-index describes the hazards and resources that tumor cells are exposed to. By proposing these additional classification schemes, Maley et al.[145] characterized tumor behavior and response to interventions in order to inform clinical decision-making. Lee et al.[146] showed that fluorodeoxyglucose (18F-FDG) PET/CT scan can predict HCC tumor aggressiveness. Furthermore, the researchers showed that PET/CT more sensitively detected extrahepatic metastasis of HCC that other imaging modalities missed. Since 18F-FDG is a marker of metabolism, specifically of cellular glucose uptake, this study is further testament to the interplay between HCC progression and the TME.
In summary, the tumor microenvironment clearly plays an important role in shaping the immune response to HCC. From cancer initiation to metastasis, elements of the tumor milieu exhibit an overall pro-HCC profile. A better understanding of the HCC TME is vital as it not only identifies potential targets for therapy, but also ultimately determines clinical outcomes.
Immune evasion and suppression by HCC
The fact that tumors are not always eradicated by adaptive immunity indicates that HCC cells can develop “resistance”. Several mechanisms allow HCC to suppress and/or evade the anti-tumor response[128]. The liver itself generally leans towards immunotolerance. Tumors create physical protective barriers as they grow[147]. Tumors can downregulate expression of MHC molecules, becoming undetectable by immune surveillance. Tumor cells can produce immunosuppressive compounds such as cytokines that blunt the immune response. These mechanisms are discussed in the following sections.
Promotion of immune tolerance
The liver is tasked with maintaining a basal level of tolerance for foreign antigens. What is known about factors that promote a tolerogenic microenvironment comes from studies involving liver transplant models, HCC mice models, and tumor tissue from patients with HCC[7]. The tolerogenic tendency of the liver is, in part, established by naturally high concentrations of endotoxins in portal blood that blunt antigen processing/presentation and the CD4+ T cell response in the liver[148]. Immune tolerance is mediated by T lymphocyte exhaustion, particularly by TGF-β1[149]. Staining of tumor tissue has revealed a microenvironment rich in PD-L1 expression[150]. PD-L1 is a known immune suppressor that mediates tolerance in pregnancy and transplant[151,152]. The PD-1/PD-L1 interaction is an important immune checkpoint that can inhibit the co-stimulation required to elicit a robust T cell response[153]. Clinically, higher expression of PD-L1 in tumor tissue manifests as reduced overall survival in HCC[154]. The PD-1/PD-L1 pathway’s role in mediating tolerance has been exploited in the development of immunotherapies such as nivolumab[155]. Tolerance in the liver is also governed by AFP. Although AFP has previously been interpreted as a simple biomarker of HCC, it appears to have many more functions than that[156]. AFP levels are used in diagnosing and prognosticating HCC[157]. AFP exerts tolerogenic and immunosuppressive mechanisms that enable tumor progression. For instance, AFP can induce apoptosis in antigen-presenting cells and inhibit apoptosis of HCC cells[156,158]. Recruitment of Tregs, Bregs, and pDCs, as discussed above, plays additional roles in establishing a tolerogenic TME. Overall, the upregulation of PD-L1 and AFP among other factors favor immune evasion in HCC.
Formation of a physical barrier
HCC can protect itself by increasing the degree of fibrosis in the peritumoral environment[159]. Okrah et al.[160] conducted a study that compared genome-wide RNA profiles between tumor and non-tumor liver tissue in patients with HCC[160]. They found that peri-tumoral tissue had upregulated expression of 23 genes involved in the production of extracellular matrix and collagen, which create a fibrotic barrier that inhibits penetration of tumor-killing CD8+ T lymphocytes. Hepatic stellate cells and myofibroblasts are responsible for the production of ECM and related proteins[85]. Recent research suggests that fibrosis may have different roles in early- versus late-stage liver disease. In response to hepatocyte damage, regenerative nodules form in order to “fence off” potential spread of tumor cells[161]. Advanced cirrhosis, however, lays the groundwork for HCC tumor formation. The subsequent encapsulation of tumors by ECM and collagen serve as a protective barrier against immune cells[161]. The degree of liver fibrosis can be estimated using the fibrosis-4 (FIB-4) index, which takes into account the patient’s age, AST, ALT, and platelet count[162]. Several studies have demonstrated that higher FIB-4 score is associated with worse outcomes, including increased risk of developing HCC in setting of chronic HCV infection and increased persistence of HCC post-hepatectomy[163-165]. Fibrosis evidently influences clinical outcomes in HCC. Thus, reduction of fibrosis particularly in late-stage HCC may be a potential anti-tumor strategy[160].
Immune editing
The immune response is robust in the early stage of tumor growth. Although some cancer cells are eliminated in this fashion, less immunogenic tumor cells go undetected and multiply in number. The phenomenon whereby the immune response inadvertently places a selective pressure on the tumor microenvironment is called immune editing[166]. Immune editing allows tumors to enter dormancy, escaping immune surveillance only to recur years later after many more rounds of cell division[167]. This concept can be compared to the development of antibiotic resistance in bacteria, whereby only select cells survive and go on to seed stronger populations. Immune editing in cancer is governed by many of the mechanisms mentioned above, such as downregulation of antigen presentation (loss of MHC I)[168]. In breast cancer, methylation of promoters in MHC genes have effectively cloaked tumors from immune surveillance[169]. HCC cells evade the immune response through reduced antigen processing/presentation as well. The exact mechanisms of how this occurs, whether epigenetic and/or molecular, have not been described but is an active area of research[166,170].
HCC immunotherapy
Current approaches to treating HCC with immunotherapy amplify the anti-tumor immune response and/or block mechanisms of immune evasion by tumor cells[171]. This section provides an overview of the three main strategies under current investigation: adoptive cell therapy, cancer vaccines, and checkpoint inhibitors[172]. Please see Figure 2 for a summary of the different approaches in treating HCC with immunotherapy.

Figure 2. Approaches to immunotherapy in treating HCC: (A) adoptive cell therapy demonstrating leukopheresis, lymphocyte engineering, and subsequent reintroduction of modified lymphocytes to the patient; (B) cancer vaccine development demonstrating the activation of T cells by mature DCs in vitro and/or purification of TAAs from excised tumor tissue; and (C) checkpoint inhibitor therapy demonstrating the interaction between APCs, tumor cells, and T lymphocytes along with their associated receptors. HCC: hepatocellular carcinoma; DCs: dendritic cells; TAAs: tumor-associated antigens; APCs: antigen-presenting cells
Adoptive cell therapy
At its most basic level, adoptive cell therapy involves the introduction of autologous anti-tumor immune cells. Notable cell types that have been studied include NK cells, cytokine-induced killer (CIK) cells, and chimeric antigen receptor T (CAR-T) cells[173]. In adoptive cell therapy, the desired immune cells are removed from the patient, modified and/or expanded, and then infused back into the patient. The goal of therapy is to amplify the strength of the body’s natural anti-tumor immune response.
NK cells were one of the first cell lines explored in adoptive cell therapy due to their tumor-killing effects. As discussed above, NK cells exert direct cytotoxicity through the release of granzymes. It has been shown that increased NK cell density within the tumor microenvironment is correlated with improved prognosis in HCC[17]. However, it appears that intratumoral NK cells are functionally impaired by other immune cells and/or immune suppressor signals[17]. NK cell-based immunotherapy aims to overcome these immunosuppressive barriers. For example, NK cell activity can be enhanced through the infusion of stimulating cytokines and/or blockade of NK cell inhibitory receptors with monoclonal antibodies[174]. A major limitation of this approach is that cytokine infusions often lead to global, non-specific activation of the innate inflammatory response. Therefore, adoptive NK cell therapy is an increasingly attractive alternative. NK cells from HCC patients can be extracted and modified in vitro, and then re-introduced back into circulation. Genetic modification of NK cells has aimed to boost expression of activating receptors that enable tumor-killing[175]. This would, in theory, enhance tumor destruction or at least prevent tumor progression. There are two challenges in studying NK cell therapy. Firstly, studies have shown that autologous NK cells do not appear to change clinical outcomes in several solid tumors, including melanoma, possibly due to their aberrant function[176]. Secondly, infused NK cells have weak penetration into the tumor microenvironment so that, while their circulating numbers may be impressive, the actual intratumoral density of transferred cells remains subtherapeutic[175]. However, adoptive NK cell therapy is an active area of investigation as researchers explore ways to fine tune NK cell targeting, activity, and specificity.
CIK cells are a heterogenous group of cells composed mainly of CD3+CD56+ T lymphocytes with an NK-like phenotype[177]. In addition to their tumor-killing activity, CIK cells appear to play a role in the downregulation of immunosuppressive cells such as Treg[178]. A recent meta-analysis showed that CIK therapy has a positive influence on HCC prognosis[179]. Specifically, these researchers found that CIK cell transfer was correlated with improvement in overall survival and reduction of tumor recurrence in the first three years. The long-term effects of CIK therapy are, however, not well-characterized. A study involving >160 HCC patients found that autologous CIK cell transfer may be an effective adjunct to surgery and/or transcatheter arterial chemoembolization (TACE)[177]. Jia et al.[177] found that the addition of CIK therapy improved HCC prognosis compared to either surgery-alone or TACE-alone. Interestingly, the researchers found that patients receiving CIK therapy who developed post-infusion fever had better overall survival than those who did not. This suggests that fever may be used as a predictor of therapeutic response in future experiments involving CIK cell transfer.
Chimeric antigen receptor T (CAR-T) cells represent one of the most promising areas of adoptive cell therapy research. CAR-T cells are created by removing a patient’s T lymphocytes and inducing the expression of synthetic receptors (CARs) through in vitro viral transduction. CARs are genetically engineered to be able to recognize specific tumor antigens[180]. A significant advantage of CAR-T cells is that, unlike regular T lymphocytes, they do not require the expression of MHC in order to recognize and destroy tumor cells[181]. As discussed above, tumor can evade the immune response by downregulating self-MHC. Therefore, CAR-T cells circumvent the need for MHC expression and are able to independently mount an anti-tumor response. CAR-T cells have proved to be effective in treating hematologic cancers such as acute myelogenous leukemia (AML), however their efficacy for solid tumors has not been thoroughly studied[182]. This may be in part due to the neurotoxicity imparted by CAR-T therapy that halted early clinical trials[183]. CAR-T therapy is complicated by the need to identify reliable tumor markers that can be used as antigenic targets. Furthermore, the resource-intensive nature of CAR-T trials presents a practical barrier to research. Regarding hepatocellular carcinoma, one promising antigen appears to be glypican-3 (GPC3)[184]. Healthy adult hepatocytes do not express GPC3. In contrast, GPC3 is overexpressed by HCC tumor cells and has been associated with worse disease-free survival and overall survival[185,186]. GPC3 is involved in many signaling cascades that promote cell growth and invasion, including the Wnt pathway that is well-known for its role in embryogenesis[187]. Experiments involving monoclonal antibodies targeting GPC3, such as codrituzumab, have successfully induced antibody-dependent cellular cytotoxicity in solid tumors[184]. This became the basis for exploring CAR-T therapy in treating HCC. There are currently > 20 clinical trials involving GPC3-targeted CAR-T therapy, mostly occurring in China where there is a disproportionately high number of HCC cases[188]. However, none of these trials have entered Phase III and/or have not published preliminary results. Nevertheless, CAR-T therapy for the treatment of HCC is a promising area of research. Advantages of CAR-T transfer include its ability to circumvent tumor immune evasion (through MHC-independent recognition) and specificity in the anti-tumor response[181]. Current barriers of CAR-T therapy include its high production cost and need to identify validated extracellular HCC-specific antigens[189,190].
Cancer vaccines
While antiviral vaccines are widely accepted and adopted in clinical practice, cancer vaccines represent a much newer area of research. The core concept of cancer vaccines is that they are designed to induce an amplified immune response against tumor-specific antigens[191]. To date, there are no cancer vaccines known to prevent the initial development of tumors. However, some vaccines have demonstrated a positive therapeutic effect in a limited subset of cancers, such as those related to chronic human papillomavirus (HPV) infection[192]. A major hurdle in cancer vaccine development is the need to identify tumor-specific antigens. The ideal antigen should be foreign to the body (non-self), elicit a robust immune response, and have restricted expression on tumor cells only. Two types of cancer vaccine targets exist: tumor-associated antigens (TAA) and tumor neoantigens.
TAAs are peptides overexpressed by cancer cells that may be expressed at low levels by normal cells[193]. Important TAAs in hepatocellular carcinoma include AFP and glypican-3 (GPC3)[194]. Such TAAs can be delivered in the form of peptide vaccines. For example, clinical trials involving cancer vaccines against GPC3 epitopes have shown increased cytotoxic T lymphocyte recruitment and activity in the HCC tumor microenvironment[195]. Several of these Phase I/II trials have demonstrated that the GPC3 vaccine is well-tolerated without major adverse effects. However, whether these findings translate to clinical significance will remain unknown until progression to Phase III/IV trials. Another approach to inducing a TAA-specific response involves oncolytic virus vaccines[171]. In oncolytic immunotherapy, viruses are designed to lyse tumor cells through replication and to induce an adaptive immune response. For example, JX-594 (Pexa-Vec) is a poxvirus that has been studied in HCC clinical trials. JX-594 is genetically engineered with insertion of granulocyte-macrophage colony-stimulating factor (GM-CSF) and deletion of thymidine kinase, features that allow this modified virus to selectively replicate in HCC cells and elicit a tumor-specific immune response[196]. Phase II trials in 2013 showed promising results, where the high-dose vaccine group had a median survival of 14.1 months compared to 6.7 months for the low-dose group[196]. However, Phase III trials were terminated in 2019 as interim results did not show a significant survival benefit of JX-594 plus standard-of-care (sorafenib) when compared to sorafenib alone[197]. Although this may be a disappointing early setback, there remains several active clinical trials involving oncolytic cancer vaccines due to their overall anti-tumor potential[188].
Tumor neoantigens represent another target for cancer vaccine development. Genomic instability is the cornerstone of malignant cell transformation. DNA mutations in cancer cells produce new molecules that are unique to the tumor, called tumor neoantigens[198]. Whereas TAAs have overlapping expression in both transformed and healthy cells, tumor neoantigens represent a true molecular fingerprint[199]. Tumor neoantigens are entirely non-self and thus easily recognized by the immune system. The advent of high-throughput genomics sequencing has allowed for the identification of tumor neoantigens in a variety of cancers, including HCC[200]. The rationale for developing tumor neoantigen vaccines is the high degree of specificity that it affords. Beyond being tumor-specific, these neoantigens may be patient-specific owing to the fact that no two driver mutations are alike. Early clinical trials have demonstrated that neoantigen vaccines are able to elicit robust tumor-specific T lymphocyte response in cancers such as glioblastoma and melanoma[200-202]. Despite all its apparent advantages, few clinical trials currently examine neoantigen-based vaccines. One barrier is the resource-intensive nature of experiments, which must be catered to each individual patient. Another challenge is the new nature of this technology, which has resulted in slow uptake in the scientific community and insufficient patient recruitment. These practical roadblocks have resulted in the early terminal of trials, such as the Phase II trial (NCT03480152) led by Rosenberg et al.[188]at the National Cancer Institute examining an mRNA-based neoantigen vaccine for solid tumors. A third, more concerning challenge is that tumor neoantigens may undergo immune evasion once it is familiar to the adaptive immune system[200]. A study investigating the effect of tumor vaccine in colon cancer patients showed that those with later-stage disease had poorer survival than those with early-stage cancer[203]. This suggests that tumor vaccine may not be an effective or appropriate therapy for advanced cancers. Overall, neoantigen vaccines represent an emerging area of research as scientists hope that it may be the next frontier in personalized tumor immunotherapy.
There are other key factors to consider in cancer vaccine development, including vaccine formulation, method of delivery, and immunoadjuvants[191]. A concern regarding cancer vaccines is their questionable efficacy among patients with advanced age. Natural aging is associated with reduced lymphocyte populations and a less robust immune response to vaccination[204]. Immunosuppressed patients also present a challenging group to evaluate and treat. As current clinical trials exclude these populations, the current understanding of cancer vaccines for HCC have limited generalizability.
Targeted therapy
Enzyme inhibition
Kinase inhibition appears to be the most adopted strategy in targeting HCC. Currently, three oral mulikinase inhibitors are approved for HCC treatment. These include sorafenib, regorafenib, and lenvatinib[205]. Sorafenib is a protein kinase inhibitor that has been considered the drug of choice for unresectable HCC since its FDA approval in 2007[206]. Sorafenib prevents tumor progression by inhibiting multiple tyrosine kinase enzymes, notably VEGF, PDGFR, and Raf kinases[207]. Sorafenib granted a modest survival advantage in phase III clinical trials by a median of 2.5 months, compared to placebo[208]. However, there are several issues with sorafenib. There is no validated biomarker that can measure response to sorafenib therapy[207]. The toxicity that sorafenib causes leads to undesirable adverse effects, including weight loss, nausea, diarrhea, and rash. One of the biggest limitations of sorafenib is that fewer than 5% of patients respond to treatment[208]. Therefore, many ongoing trials are exploring the efficacy of sorafenib as part of combination therapy with novel agents.
Checkpoint inhibition
Immune checkpoint inhibitors have emerged as part of standard-of-care treatment for various cancers, such as melanoma and Hodgkin lymphoma[209]. Chief immune checkpoint targets for HCC are cytotoxic T lymphocyte-associated protein (CTLA-4) and programmed cell death protein/ligand (PD-1/PD-L1). CTLA-4 and PD-1 are expressed by immunosuppressive cell types, such as Tregs, to downregulate the immune response[210]. Thus, tumors exploit these natural host mechanisms in order to evade immunity.
Ipilimumab and tremelimumab are anti-CTLA-4 antibodies under active investigation. CTLA-4 mediates immune suppression by blocking B7/CD28 costimulation. Early trials have demonstrated acceptable safety profile of CTLA-4 blockers for HCC, with clinical response rate ranging 18%–26%[211]. Currently, both ipilimumab and tremelimumab are involved as combination therapy in a few phase III clinical trials, the results of which have not been released[212].
PD-L1 expression is common in HCC tumor tissue and, more importantly, PD-L1 positivity is associated with a greater therapeutic response[213]. Nivolumab was the first, and currently only, anti-PD-1 antibody that is FDA-approved for the treatment of hepatocellular carcinoma[205,214]. In 2017, promising results from a phase II clinical trial involving nivolumab led to its accelerated approval[215]. Phase III clinical trials thus began examining PD-1 inhibitors nivolumab (CheckMate-459 trial) and pembrolizumab (KEYNOTE-240 trial). The KEYNOTE-240 trial found that there was no statistically significant survival advantage to pembrolizumab treatment, and this also applied to the preliminary results for the CheckMate-459 trial[216]. However, trials thereafter have demonstrated success with these PD-1 inhibitors. For example, pembrolizumab in the phase II KEYNOTE-224 trial showed durable response in 17% of patients[216]. Nivolumab in the CheckMate-040 trial demonstrated up to 20% tumor shrinkage, according to Huppert et al.[217] who conducted a review of checkpoint inhibitors under investigation for HCC.
There are clinical differences between CTLA-4 and PD-1 inhibitors. In general, toxicity associated with CTLA-4 inhibitors are more severe than those of PD-1 inhibitors[218]. For example, Chae et al.[219] conducted a review of checkpoint inhibitor therapy among transplant patients and determined that CTLA-4 inhibitors may be preferable to PD-1 inhibitors, which were associated with increased risk of graft rejection. Other potential targets for checkpoint inhibition are Tim-3, LAG-3, and galectin-9, all of which are involved in mediating immunotolerance[220]. Immune checkpoint inhibitors have certain limitations. Adverse effects of checkpoint inhibitors tend to be immune-related, as is consistent with their mechanism of action. Toxic adverse effects of CTLA-4 and PD-1 inhibitors include dermatitis, pneumonitis, enterocolitis, elevated AST/ALT, and hypothyroidism[215]. While these effects were relatively uncommon in trials, they dictate the need for close monitoring should checkpoint therapy be adopted in clinical use. Overall, immune checkpoint inhibitors have great potential and are worth further exploration.
Conclusion
Hepatocellular carcinoma is a difficult-to-treat cancer associated with high degree of morbidity and mortality. The complex interactions between HCC cells and immune cells determine clinical outcomes. Crosstalk between signaling cascades and cytokines within the tumor microenvironment present challenges in elucidating the mechanisms of tumor progression. Interestingly, when it comes to the immune response against HCC, it appears that “less is more”. We hypothesize that excessive immune reaction against tumor cells activate pro-inflammatory cycles of wound healing and fibrosis that are ultimately maladaptive. However, we recognize that immunotolerance cannot dominate the immune response. Several challenges exist in the field of HCC research. These include the lack of an animal model that closely mimics the human tumor microenvironment and the abandonment of clinical trials due to high rate of attrition and/or resource demand. Furthermore, genomic changes in HCC and the tumor’s ability to evade host immunity mean that researchers are always playing catch up, so to speak. From our review, we note that certain patients, such as transplant recipients, are often neglected while they paradoxically belong in some of the most at-risk groups.
Several solutions and directions for future research seem to arise from our review. Firstly, a standardized method of HCC staging should be developed, and it must take into account the nature of the TME. Secondly, a greater effort should be made to identify HCC tumor markers; this would potentially allow researchers to identify targets for immunotherapy and enable clinicians earlier to make diagnosis or to better monitor disease progression. Thirdly, combination therapies involving sorafenib and novel biologics may be the next frontier in HCC treatment. An example of this is multiple antigen stimulating cellular therapy (MASCT), which uses a combination of adoptive cell transfer and DC-based cancer vaccine therapy. In a small set of HCC patients, MASCT was shown to induce a robust and highly specific anti-tumor response[221]. No two tumors are the same, therefore personalized medicine that considers the immunological profile unique to each patient may be the best approach to treatment. Lastly, novel biologics that fail to demonstrate a dramatic clinical response in clinical trials should be abandoned. The rationale is that the tumor milieu is a dynamic environment. Compounds must be given at the appropriate stage of disease, in the adequate dose, and through the appropriate mode of delivery. It is challenging to delineate what combination of these factors would be most efficacious, especially given the small number of HCC patients enrolled in trials. Until more is known about the immune profile of HCC, primary prevention should be emphasized to reduce global disease burden. Clinicians may consider more rigorous screening methods, particularly among high-risk groups. It is important to recognize that well-known disease markers such as AFP have limited sensitivity. Thus, physicians may wish to consider adopting other modes of HCC detection such as through PET/CT, for cases in which the hepatic panels are unremarkable but there is strong clinician suspicion[146]. In conclusion, a deeper understanding of the immune response to HCC would enable better tumor characterization, identification of potential immunotherapy targets, and personalization of therapy.
Declarations
Authors’ contributionsAuthored the majority of this review: Wang SZ, Lee SD
Provided proofreading and editorial assistance: Sarkar D, Lee HM, Khan A, Bhati C, Sharma A, Kumaran V, Bruno D, Cotterell A, Levy MF
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol 2019;16:589-604.
2. SEER*Explorer: An interactive website for SEER cancer statistics [Internet]. Surveillance Research Program, National Cancer Institute. Available from https://seer.cancer.gov/explorer/. [Last accessed on 14 Dec 2020].
3. American Cancer Society. Cancer Facts & Figures 2020. Atlanta: American Cancer Society; 2020. Available from: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2020.html.[Last accessed on 14 Dec 2020].
4. Singh AK, Kumar R, Pandey AK. Hepatocellular carcinoma: causes, mechanism of progression and biomarkers. Curr Chem Genom Transl Med 2018;12:9-26.
6. Hilmi M, Vienot A, Rousseau B, Neuzillet C. Immune therapy for liver cancers. Cancers (Basel) 2019;12:77.
7. Makarova-Rusher OV, Medina-Echeverz J, Duffy AG, Greten TF. The yin and yang of evasion and immune activation in HCC. J Hepatol 2015;62:1420-9.
8. Woo SR, Corrales L, Gajewski TF. Innate immune recognition of cancer. Annu Rev Immunol 2015;33:445-74.
9. Liu Y, Zeng G. Cancer and innate immune system interactions: translational potentials for cancer immunotherapy. J Immunother 2012;35:299-308.
10. Roderburg C, Wree A, Demir M, Schmelzle M, Tacke F. The role of the innate immune system in the development and treatment of hepatocellular carcinoma. Hepat Oncol 2020;7:HEP17.
11. van Lookeren Campagne M, Verschoor A. Pathogen clearance and immune adherence “revisited”: Immuno-regulatory roles for CRIg. Semin Immunol 2018;37:4-11.
12. Lu C, Rong D, Zhang B, et al. Current perspectives on the immunosuppressive tumor microenvironment in hepatocellular carcinoma: challenges and opportunities. Mol Cancer 2019;18:130.
13. Irvine KM, Banh X, Gadd VL, et al. CRIg-expressing peritoneal macrophages are associated with disease severity in patients with cirrhosis and ascites. JCI Insight 2016;1:e86914.
14. Lin Y, Xu J, Lan H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J Hematol Oncol 2019;12:76.
15. Li S, Yang F, Ren X. Immunotherapy for hepatocellular carcinoma. Drug Discov Ther 2015;9:363-71.
16. Degroote H, Van Dierendonck A, Geerts A, Van Vlierberghe H, Devisscher L. Preclinical and clinical therapeutic strategies affecting tumor-associated macrophages in hepatocellular carcinoma. J Immunol Res 2018;2018:7819520.
17. Liu P, Chen L, Zhang H. Natural killer cells in liver disease and hepatocellular carcinoma and the NK cell-based immunotherapy. J Immunol Res 2018;2018:1206737.
18. Hashemi E, Malarkannan S. Tissue-resident NK cells: development, maturation, and clinical relevance. Cancers (Basel) 2020;12:1553.
19. Sung PS, Jang JW. Natural killer cell dysfunction in hepatocellular carcinoma: pathogenesis and clinical implications. Int J Mol Sci 2018;19:3648.
20. Juengpanich S, Shi L, Iranmanesh Y, et al. The role of natural killer cells in hepatocellular carcinoma development and treatment: A narrative review. Transl Oncol 2019;12:1092-107.
21. Mikulak J, Bruni E, Oriolo F, Di Vito C, Mavilio D. Hepatic natural killer cells: organ-specific sentinels of liver immune homeostasis and physiopathology. Front Immunol 2019;10:946.
22. Yu SJ, Ma C, Heinrich B, et al. Targeting the crosstalk between cytokine-induced killer cells and myeloid-derived suppressor cells in hepatocellular carcinoma. J Hepatol 2019;70:449-57.
23. Chiu DK, Tse AP, Xu IM, et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun 2017;8:517.
24. Prieto J, Melero I, Sangro B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 2015;12:681-700.
25. Doherty DG. Immunity, tolerance and autoimmunity in the liver: A comprehensive review. J Autoimmun 2016;66:60-75.
26. Zhang Q, He Y, Luo N, et al. Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell 2019;179:829-45.e20.
27. Gui Y, Liu WB, Chen H, Ma JL, Li JS. Expression of LAMP3 and its correlation with clinicopathologic characteristics and prognosis in hepatocellular carcinoma. Int J Clin Exp Pathol 2018;11:367-74.
28. Shuai Z, Leung MW, He X, et al. Adaptive immunity in the liver. Cell Mol Immunol 2016;13:354-68.
29. Endig J, Buitrago-Molina LE, Marhenke S, et al. Dual role of the adaptive immune system in liver injury and hepatocellular carcinoma development. Cancer Cell 2016;30:308-23.
30. Ringelhan M, Pfister D, O’Connor T, Pikarsky E, Heikenwalder M. The immunology of hepatocellular carcinoma. Nat Immunol 2018;19:222-32.
31. Garnelo M, Tan A, Her Z, et al. Interaction between tumour-infiltrating B cells and T cells controls the progression of hepatocellular carcinoma. Gut 2017;66:342-51.
32. Fournié JJ, Poupot M. The pro-tumorigenic IL-33 involved in antitumor immunity: a yin and yang cytokine. Front Immunol 2018;9:2506.
33. Jin Z, Lei L, Lin D, et al. IL-33 Released in the liver inhibits tumor growth via promotion of CD4+ and CD8+ T cell responses in hepatocellular carcinoma. J Immunol 2018;201:3770-9.
34. Gabrielson A, Wu Y, Wang H, et al. Intratumoral CD3 and CD8 T-cell densities associated with relapse-free survival in HCC. Cancer Immunol Res 2016;4:419-30.
35. Kalathil SG, Hutson A, Barbi J, Iyer R, Thanavala Y. Augmentation of IFN-γ+ CD8+ T cell responses correlates with survival of HCC patients on sorafenib therapy. JCI Insight 2019;4:130116.
36. Zhang M, Pang HJ, Zhao W, et al. VISTA expression associated with CD8 confers a favorable immune microenvironment and better overall survival in hepatocellular carcinoma. BMC Cancer 2018;18:511.
37. Xu X, Tan Y, Qian Y, et al. Clinicopathologic and prognostic significance of tumor-infiltrating CD8+ T cells in patients with hepatocellular carcinoma: A meta-analysis. Medicine (Baltimore) 2019;98:e13923.
38. Ramzan M, Sturm N, Decaens T, et al. Liver-infiltrating CD8(+) lymphocytes as prognostic factor for tumour recurrence in hepatitis C virus-related hepatocellular carcinoma. Liver Int 2016;36:434-44.
39. Ji L, Gu J, Chen L, Miao D. Changes of Th1/Th2 cytokines in patients with primary hepatocellular carcinoma after ultrasound-guided ablation. Int J Clin Exp Pathol 2017;10:8715-20.
40. Lee HL, Jang JW, Lee SW, et al. Inflammatory cytokines and change of Th1/Th2 balance as prognostic indicators for hepatocellular carcinoma in patients treated with transarterial chemoembolization. Sci Rep 2019;9:3260.
41. Saxena R, Kaur J. Th1/Th2 cytokines and their genotypes as predictors of hepatitis B virus related hepatocellular carcinoma. World J Hepatol 2015;7:1572-80.
42. Kogame M, Nagai H, Shinohara M, Igarashi Y, Sumino Y, Ishii K. Th2 dominance might induce carcinogenesis in patients with HCV-related liver cirrhosis. Anticancer Res 2016;36:4529-36.
43. Kuang DM, Xiao X, Zhao Q, et al. B7-H1-expressing antigen-presenting cells mediate polarization of protumorigenic Th22 subsets. J Clin Invest 2014;124:4657-67.
44. Keenan BP, Fong L, Kelley RK. Immunotherapy in hepatocellular carcinoma: the complex interface between inflammation, fibrosis, and the immune response. J Immunother Cancer 2019;7:267.
45. Shen Y, Wei Y, Wang Z, et al. TGF-β regulates hepatocellular carcinoma progression by inducing Treg cell polarization. Cell Physiol Biochem 2015;35:1623-32.
46. Lan YT, Fan XP, Fan YC, Zhao J, Wang K. Change in the Treg/Th17 cell imbalance in hepatocellular carcinoma patients and its clinical value. Medicine (Baltimore) 2017;96:e7704.
47. Wiedemann GM, Röhrle N, Makeschin MC, et al. Peritumoural CCL1 and CCL22 expressing cells in hepatocellular carcinomas shape the tumour immune infiltrate. Pathology 2019;51:586-92.
48. Gao Q, Qiu SJ, Fan J, et al. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol 2007;25:2586-93.
49. Zhou Y, Wang B, Wu J, et al. Association of preoperative EpCAM circulating tumor cells and peripheral treg cell levels with early recurrence of hepatocellular carcinoma following radical hepatic resection. BMC Cancer 2016;16:506.
50. Ma W, Wu L, Zhou F, Hong Z, Yuan Y, Liu Z. T cell-associated immunotherapy for hepatocellular carcinoma. Cell Physiol Biochem 2017;41:609-22.
51. Sharonov GV, Serebrovskaya EO, Yuzhakova DV, Britanova OV, Chudakov DM. B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat Rev Immunol 2020;20:294-307.
52. Zhang Z, Ma L, Goswami S, et al. Landscape of infiltrating B cells and their clinical significance in human hepatocellular carcinoma. Oncoimmunology 2019;8:e1571388.
53. Brunner SM, Itzel T, Rubner C, et al. Tumor-infiltrating B cells producing antitumor active immunoglobulins in resected HCC prolong patient survival. Oncotarget 2017;8:71002-11.
54. Shi JY, Gao Q, Wang ZC, et al. Margin-infiltrating CD20(+) B cells display an atypical memory phenotype and correlate with favorable prognosis in hepatocellular carcinoma. Clin Cancer Res 2013;19:5994-6005.
55. Garaud S, Buisseret L, Solinas C, et al. Tumor infiltrating B-cells signal functional humoral immune responses in breast cancer. JCI Insight 2019;5:129641.
56. Wang SS, Liu W, Ly D, Xu H, Qu L, Zhang L. Tumor-infiltrating B cells: their role and application in anti-tumor immunity in lung cancer. Cell Mol Immunol 2019;16:6-18.
57. Tsou P, Katayama H, Ostrin EJ, Hanash SM. The emerging role of B cells in tumor immunity. Cancer Res 2016;76:5597-601.
58. Shao Y, Lo CM, Ling CC, et al. Regulatory B cells accelerate hepatocellular carcinoma progression via CD40/CD154 signaling pathway. Cancer Lett 2014;355:264-72.
59. Yuen GJ, Demissie E, Pillai S. B lymphocytes and cancer: a love-hate relationship. Trends Cancer 2016;2:747-57.
60. He H, Wu J, Zang M, et al. CCR6+ B lymphocytes responding to tumor cell-derived CCL20 support hepatocellular carcinoma progression via enhancing angiogenesis. Am J Cancer Res 2017;7:1151-63.
61. Ye L, Zhang Q, Cheng Y, et al. Tumor-derived exosomal HMGB1 fosters hepatocellular carcinoma immune evasion by promoting TIM-1+ regulatory B cell expansion. J Immunother Cancer 2018;6:145.
62. Zhang C, Xin H, Zhang W, et al. CD5 binds to interleukin-6 and induces a feed-forward loop with the transcription factor STAT3 in B cells to promote cancer. Immunity 2016;44:913-23.
63. Huynh J, Chand A, Gough D, Ernst M. Therapeutically exploiting STAT3 activity in cancer - using tissue repair as a road map. Nat Rev Cancer 2019;19:82-96.
64. Liu RX, Wei Y, Zeng QH, et al. Chemokine (C-X-C motif) receptor 3-positive B cells link interleukin-17 inflammation to protumorigenic macrophage polarization in human hepatocellular carcinoma. Hepatology 2015;62:1779-90.
65. Wennhold K, Shimabukuro-Vornhagen A, von Bergwelt-Baildon M. B Cell-Based Cancer Immunotherapy. Transfus Med Hemother 2019;46:36-46.
66. Guo FF, Cui JW. The role of tumor-infiltrating B cells in tumor immunity. J Oncol 2019;2019:2592419.
67. Marquardt JU, Andersen JB, Thorgeirsson SS. Functional and genetic deconstruction of the cellular origin in liver cancer. Nat Rev Cancer 2015;15:653-67.
68. Novikova MV, Khromova NV, Kopnin PB. Components of the hepatocellular carcinoma microenvironment and their role in tumor progression. Biochemistry (Mosc) 2017;82:861-73.
69. Wang M, Zhao J, Zhang L, et al. Role of tumor microenvironment in tumorigenesis. J Cancer 2017;8:761-73.
70. Ho DW, Lo RC, Chan LK, Ng IO. Molecular pathogenesis of hepatocellular carcinoma. Liver Cancer 2016;5:290-302.
71. Liu M, Jiang L, Guan XY. The genetic and epigenetic alterations in human hepatocellular carcinoma: a recent update. Protein Cell 2014;5:673-91.
72. Subramaniam A, Shanmugam MK, Perumal E, et al. Potential role of signal transducer and activator of transcription (STAT)3 signaling pathway in inflammation, survival, proliferation and invasion of hepatocellular carcinoma. Biochim Biophys Acta 2013;1835:46-60.
73. Grohmann M, Wiede F, Dodd GT, et al. Obesity drives STAT-1-dependent NASH and STAT-3-dependent HCC. Cell 2018;175:1289-1306.e20.
74. He G, Dhar D, Nakagawa H, et al. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell 2013;155:384-96.
75. Ma C, Kesarwala AH, Eggert T, et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016;531:253-7.
76. O’Rourke JM, Sagar VM, Shah T, Shetty S. Carcinogenesis on the background of liver fibrosis: implications for the management of hepatocellular cancer. World J Gastroenterol 2018;24:4436-47.
77. Yang R, Gao N, Chang Q, Meng X, Wang W. The role of IDO, IL-10, and TGF-β in the HCV-associated chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma. J Med Virol 2019;91:265-71.
78. Ying HZ, Chen Q, Zhang WY, et al. PDGF signaling pathway in hepatic fibrosis pathogenesis and therapeutics (Review). Mol Med Rep 2017;16:7879-89.
79. Chandler C, Liu T, Buckanovich R, Coffman LG. The double edge sword of fibrosis in cancer. Transl Res 2019;209:55-67.
80. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009;119:1420-8.
81. Hsu CY, Liu PH, Ho SY, et al. Metastasis in patients with hepatocellular carcinoma: prevalence, determinants, prognostic impact and ability to improve the barcelona clinic liver cancer system. Liver Int 2018;38:1803-11.
82. Hou J, Zhang H, Sun B, Karin M. The immunobiology of hepatocellular carcinoma in humans and mice: Basic concepts and therapeutic implications. J Hepatol 2020;72:167-82.
83. Zheng C, Zheng L, Yoo JK, et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 2017;169:1342-56.e16.
84. Fang M, Yuan J, Chen M, et al. The heterogenic tumor microenvironment of hepatocellular carcinoma and prognostic analysis based on tumor neo-vessels, macrophages and α-SMA. Oncol Lett 2018;15:4805-12.
85. Affo S, Yu LX, Schwabe RF. The role of cancer-associated fibroblasts and fibrosis in liver cancer. Annu Rev Pathol 2017;12:153-86.
86. Zhao G, Cui J, Qin Q, et al. Mechanical stiffness of liver tissues in relation to integrin β1 expression may influence the development of hepatic cirrhosis and hepatocellular carcinoma. J Surg Oncol 2010;102:482-9.
87. Ju MJ, Qiu SJ, Fan J, et al. Peritumoral activated hepatic stellate cells predict poor clinical outcome in hepatocellular carcinoma after curative resection. Am J Clin Pathol 2009;131:498-510.
88. Amann T, Bataille F, Spruss T, et al. Activated hepatic stellate cells promote tumorigenicity of hepatocellular carcinoma. Cancer Sci 2009;100:646-53.
89. Ji J, Eggert T, Budhu A, et al. Hepatic stellate cell and monocyte interaction contributes to poor prognosis in hepatocellular carcinoma. Hepatology 2015;62:481-95.
90. Zhao W, Zhang L, Yin Z, et al. Activated hepatic stellate cells promote hepatocellular carcinoma development in immunocompetent mice. Int J Cancer 2011;129:2651-61.
91. Barry AE, Baldeosingh R, Lamm R, et al. Hepatic stellate cells and hepatocarcinogenesis. Front Cell Dev Biol 2020;8:709.
92. Li K, Liu H, Guo T. Th17/Treg imbalance is an indicator of liver cirrhosis process and a risk factor for HCC occurrence in HBV patients. Clin Res Hepatol Gastroenterol 2017;41:399-407.
93. Li JQ, Yu XJ, Wang YC, et al. Distinct patterns and prognostic values of tumor-infiltrating macrophages in hepatocellular carcinoma and gastric cancer. J Transl Med 2017;15:37.
94. Kalathil S, Lugade AA, Miller A, Iyer R, Thanavala Y. Higher frequencies of GARP(+)CTLA-4(+)Foxp3(+) T regulatory cells and myeloid-derived suppressor cells in hepatocellular carcinoma patients are associated with impaired T-cell functionality. Cancer Res 2013;73:2435-44.
95. Kapanadze T, Gamrekelashvili J, Ma C, et al. Regulation of accumulation and function of myeloid derived suppressor cells in different murine models of hepatocellular carcinoma. J Hepatol 2013;59:1007-13.
96. Villa E, Critelli R, Lei B, et al. Neoangiogenesis-related genes are hallmarks of fast-growing hepatocellular carcinomas and worst survival. Results from a prospective study. Gut 2016;65:861-9.
97. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 2009;9:798-809.
98. Bergmann J, Müller M, Baumann N, et al. IL-6 trans-signaling is essential for the development of hepatocellular carcinoma in mice. Hepatology 2017;65:89-103.
99. Duan XH, Li H, Han XW, et al. Upregulation of IL-6 is involved in moderate hyperthermia induced proliferation and invasion of hepatocellular carcinoma cells. Eur J Pharmacol 2018;833:230-6.
100. Zhang M, Zhang S, Yang Z, et al. Association between the expression levels of IL-6 and IL-6R in the hepatocellular carcinoma microenvironment and postoperative recurrence. Oncol Lett 2018;16:7158-65.
101. Chen J, Gingold JA, Su X. Immunomodulatory TGF-β signaling in hepatocellular carcinoma. Trends Mol Med 2019;25:1010-23.
102. Arrese M, Hernandez A, Astete L, Estrada L, Cabello-Verrugio C, Cabrera D. TGF-β and hepatocellular carcinoma: when a friend becomes an enemy. Curr Protein Pept Sci 2018;19:1172-9.
103. Dituri F, Mancarella S, Cigliano A, Chieti A, Giannelli G. TGF-β as multifaceted orchestrator in HCC progression: signaling, EMT, immune microenvironment, and novel therapeutic perspectives. Semin Liver Dis 2019;39:53-69.
104. Chakravarthy A, Khan L, Bensler NP, Bose P, De Carvalho DD. TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat Commun 2018;9:4692.
105. Shakiba E, Ramezani M, Sadeghi M. Evaluation of serum interleukin-6 levels in hepatocellular carcinoma patients: a systematic review and meta-analysis. Clin Exp Hepatol 2018;4:182-90.
106. Kong L, Zhou Y, Bu H, Lv T, Shi Y, Yang J. Deletion of interleukin-6 in monocytes/macrophages suppresses the initiation of hepatocellular carcinoma in mice. J Exp Clin Cancer Res 2016;35:131.
107. Tavakolpour S, Mirsafaei HS, Elkaei Behjati S, Ghasemiadl M, Akhlaghdoust M, Sali S. Toward cure chronic hepatitis B infection and hepatocellular carcinoma prevention: Lessons learned from nucleos(t)ide analogues therapy. Immunol Lett 2017;190:206-12.
108. Hamdy H. Levels of interleukin-6 and transforming growth factor beta in HCV patients sera. 2020, US National Library of Medicine. Available from: https://clinicaltrials.gov/ct2/show/NCT03882307[Last accessed on 14 Dec 2020].
109. Mariathasan S, Turley SJ, Nickles D, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554:544-8.
110. Su Q, Fan M, Wang J, et al. Sanguinarine inhibits epithelial-mesenchymal transition via targeting HIF-1α/TGF-β feed-forward loop in hepatocellular carcinoma. Cell Death Dis 2019;10:939.
112. Saviano A, Roehlen N, Virzì A, et al. Stromal and immune drivers of hepatocarcinogenesis. In: Hoshida Y, editor. Hepatocellular carcinoma. Cham: springer international publishing; 2019. pp. 317-31.
113. Kamil F, Rowe JH. How does the tumor microenvironment play a role in hepatobiliary tumors? J Gastrointest Oncol 2018;9:180-95.
114. Feng PC, Ke XF, Kuang HL, Pan LL, Ye Q, Wu JB. BMP2 secretion from hepatocellular carcinoma cell HepG2 enhances angiogenesis and tumor growth in endothelial cells via activation of the MAPK/p38 signaling pathway. Stem Cell Res Ther 2019;10:237.
115. Zhu B, Shi S, Ma YG, Fan F, Yao ZZ. Lysophosphatidic acid enhances human hepatocellular carcinoma cell migration, invasion and adhesion through P38 MAPK pathway. Hepatogastroenterology 2012;59:785-9.
116. Huang JL, Ren TY, Cao SW, et al. HBx-related long non-coding RNA DBH-AS1 promotes cell proliferation and survival by activating MAPK signaling in hepatocellular carcinoma. Oncotarget 2015;6:33791-804.
117. Deng L, Yang J, Chen H, et al. Knockdown of TMEM16A suppressed MAPK and inhibited cell proliferation and migration in hepatocellular carcinoma. Onco Targets Ther 2016;9:325-33.
118. Zhao Y, Yang Q, Wang X, et al. AnnexinA7 down-regulation might suppress the proliferation and metastasis of human hepatocellular carcinoma cells via MAPK/ ERK pathway. Cancer Biomark 2018;23:527-37.
119. Jiang Y, Fang Y, Ye Y, et al. Anti-Cancer Effects of 3, 3’-Diindolylmethane on human hepatocellular carcinoma cells is enhanced by calcium ionophore: the role of cytosolic Ca2+ and p38 MAPK. Front Pharmacol 2019;10:1167.
120. He L, Deng H, Lei J, et al. Efficacy of bevacizumab combined with erlotinib for advanced hepatocellular carcinoma: a single-arm meta-analysis based on prospective studies. BMC Cancer 2019;19:276.
121. Qi Z, Yan F, Chen D, et al. Identification of prognostic biomarkers and correlations with immune infiltrates among cGAS-STING in hepatocellular carcinoma. Biosci Rep 2020;40:BSR20202603.
122. Yum S, Li M, Frankel AE, Chen ZJ. Roles of the cGAS-STING Pathway in Cancer Immunosurveillance and Immunotherapy. Annu Rev Cancer Biol 2019;3:323-44.
123. Thomsen MK, Skouboe MK, Boularan C, et al. The cGAS-STING pathway is a therapeutic target in a preclinical model of hepatocellular carcinoma. Oncogene 2020;39:1652-64.
124. Wu Q, Zhou L, Lv D, Zhu X, Tang H. Exosome-mediated communication in the tumor microenvironment contributes to hepatocellular carcinoma development and progression. J Hematol Oncol 2019;12:53.
125. Wang H, Lu Z, Zhao X. Tumorigenesis, diagnosis, and therapeutic potential of exosomes in liver cancer. J Hematol Oncol 2019;12:133.
126. Yu LX, Zhang BL, Yang Y, et al. Exosomal microRNAs as potential biomarkers for cancer cell migration and prognosis in hepatocellular carcinoma patient-derived cell models. Oncol Rep 2019;41:257-69.
127. Han Q, Zhao H, Jiang Y, Yin C, Zhang J. HCC-derived exosomes: critical player and target for cancer immune escape. Cells 2019;8:558.
128. Sasaki R, Kanda T, Yokosuka O, Kato N, Matsuoka S, Moriyama M. Exosomes and hepatocellular carcinoma: from bench to bedside. Int J Mol Sci 2019;20:1406.
129. Huang XY, Ke AW, Shi GM, et al. αB-crystallin complexes with 14-3-3ζ to induce epithelial-mesenchymal transition and resistance to sorafenib in hepatocellular carcinoma. Hepatology 2013;57:2235-47.
130. Pan JH, Zhou H, Zhao XX, et al. Role of exosomes and exosomal microRNAs in hepatocellular carcinoma: potential in diagnosis and antitumour treatments (Review). Int J Mol Med 2018;41:1809-16.
131. Shi S, Rao Q, Zhang C, Zhang X, Qin Y, Niu Z. Dendritic cells pulsed with exosomes in combination with PD-1 antibody increase the efficacy of sorafenib in hepatocellular carcinoma model. Transl Oncol 2018;11:250-8.
132. Tai YL, Chen KC, Hsieh JT, Shen TL. Exosomes in cancer development and clinical applications. Cancer Sci 2018;109:2364-74.
133. Liberti MV, Locasale JW. The warburg effect: how does it benefit cancer cells? Trends Biochem Sci 2016;41:211-8.
135. Maxwell PH, Dachs GU, Gleadle JM, et al. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc Natl Acad Sci U S A 1997;94:8104-9.
136. Al-Khami AA, Zheng L, Del Valle L, et al. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology 2017;6:e1344804.
137. Agren R, Mardinoglu A, Asplund A, Kampf C, Uhlen M, Nielsen J. Identification of anticancer drugs for hepatocellular carcinoma through personalized genome-scale metabolic modeling. Mol Syst Biol 2014;10:721.
138. Li JW, Goh BG, Chang PE, Tan CK. Barcelona clinic liver cancer outperforms Hong Kong liver cancer staging of hepatocellular carcinoma in multiethnic Asians: real-world perspective. World J Gastroenterol 2017;23:4054-63.
139. Ayuso C, Rimola J, Vilana R, et al. Diagnosis and staging of hepatocellular carcinoma (HCC): current guidelines. Eur J Radiol 2018;101:72-81.
140. Jun CH, Yoon JH, Cho E, et al. Barcelona clinic liver cancer-stage C hepatocellular carcinoma: A novel approach to subclassification and treatment. Medicine (Baltimore) 2017;96:e6745.
141. Tsilimigras DI, Bagante F, Sahara K, et al. Prognosis after resection of barcelona clinic liver cancer (BCLC) stage 0, A, and B hepatocellular carcinoma: a comprehensive assessment of the current BCLC classification. Ann Surg Oncol 2019;26:3693-700.
142. Barman PM, Su GL. Limitations of the barcelona clinic liver cancer staging system with a focus on transarterial chemoembolization as a key modality for treatment of hepatocellular carcinoma. Clin Liver Dis (Hoboken) 2016;7:32-5.
143. Pons F, Varela M, Llovet JM. Staging systems in hepatocellular carcinoma. HPB (Oxford) 2005;7:35-41.
144. Tellapuri S, Sutphin PD, Beg MS, Singal AG, Kalva SP. Staging systems of hepatocellular carcinoma: a review. Indian J Gastroenterol 2018;37:481-91.
145. Maley CC, Aktipis A, Graham TA, et al. Classifying the evolutionary and ecological features of neoplasms. Nat Rev Cancer 2017;17:605-19.
146. Lee SD, Kim SH. Role of positron emission tomography/computed tomography in living donor liver transplantation for hepatocellular carcinoma. Hepatobiliary Surg Nutr 2016;5:408-14.
147. Spurrell EL, Lockley M. Adaptive immunity in cancer immunology and therapeutics. Ecancermedicalscience 2014;8:441.
148. Knolle PA, Germann T, Treichel U, Uhrig A, Schmitt E, Hegenbarth S, Lohse AW, Gerken G. Endotoxin down-regulates T cell activation by antigen-presenting liver sinusoidal endothelial cells. J Immunol 1999;162:1401-7.
149. Sia D, Jiao Y, Martinez-Quetglas I, et al. Identification of an immune-specific class of hepatocellular carcinoma, based on molecular features. Gastroenterology 2017;153:812-26.
150. Yarchoan M, Xing D, Luan L, et al. Characterization of the immune microenvironment in hepatocellular carcinoma. Clin Cancer Res 2017;23:7333-9.
151. Okuyama M, Mezawa H, Kawai T, Urashima M. Elevated soluble PD-L1 in pregnant women’s serum suppresses the immune reaction. Front Immunol 2019;10:86.
152. Lanza R, Russell DW, Nagy A. Engineering universal cells that evade immune detection. Nat Rev Immunol 2019;19:723-33.
153. Macek Jilkova Z, Aspord C, Decaens T. Predictive factors for response to PD-1/PD-L1 checkpoint inhibition in the field of hepatocellular carcinoma: current status and challenges. Cancers (Basel) 2019;11:1554.
154. Dai X, Pi G, Yang SL, Chen GG, Liu LP, Dong HH. Association of PD-L1 and HIF-1α coexpression with poor prognosis in hepatocellular carcinoma. Transl Oncol 2018;11:559-66.
155. Xu W, Liu K, Chen M, et al. Immunotherapy for hepatocellular carcinoma: recent advances and future perspectives. Ther Adv Med Oncol 2019;11:1758835919862692.
156. Meng W, Bai B, Bai Z, Li Y, Yue P, Li X, Qiao L. The immunosuppression role of alpha-fetoprotein in human hepatocellular carcinoma. Discov Med 2016;21:489-94.
157. Özdemir F, Baskiran A. The importance of AFP in liver transplantation for HCC. J Gastrointest Cancer 2020;51:1127-32.
158. Wang S, Zhu M, Wang Q, et al. Alpha-fetoprotein inhibits autophagy to promote malignant behaviour in hepatocellular carcinoma cells by activating PI3K/AKT/mTOR signalling. Cell Death Dis 2018;9:1027.
159. Buonaguro L, Mauriello A, Cavalluzzo B, Petrizzo A, Tagliamonte M. Immunotherapy in hepatocellular carcinoma. Ann Hepatol 2019;18:291-7.
160. Okrah K, Tarighat S, Liu B, et al. Transcriptomic analysis of hepatocellular carcinoma reveals molecular features of disease progression and tumor immune biology. NPJ Precis Oncol 2018;2:25.
161. Garrido A, Djouder N. Cirrhosis: a questioned risk factor for hepatocellular carcinoma. Trends Cancer 2020:S2405-8033(20)30237-5.
162. Kim JH, Lee M, Park SW, et al. Validation of modified fibrosis-4 index for predicting hepatocellular carcinoma in patients with compensated alcoholic liver cirrhosis. Medicine (Baltimore) 2018;97:e13438.
163. Lee YB, Nam JY, Lee JH, et al. Differential Effect of HCV eradication and fibrosis grade on hepatocellular carcinoma and all-cause mortality. Sci Rep 2018;8:13651.
164. Wang H, Liu A, Bo W, et al. Fibrosis-4 model influences results of patients with hepatocellular carcinoma undergoing hepatectomy. Biomed Res Int 2018;2018:4305408.
165. Ioannou GN, Beste LA, Green PK, et al. Increased risk for hepatocellular carcinoma persists up to 10 years after HCV eradication in patients with baseline cirrhosis or high FIB-4 scores. Gastroenterology 2019;157:1264-78.e4.
166. Nishida N, Kudo M. Oncogenic signal and tumor microenvironment in hepatocellular carcinoma. Oncology 2017;93:160-4.
167. Vinay DS, Ryan EP, Pawelec G, et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin Cancer Biol 2015;35:S185-98.
168. Gomez S, Tabernacki T, Kobyra J, Roberts P, Chiappinelli KB. Combining epigenetic and immune therapy to overcome cancer resistance. Semin Cancer Biol 2020;65:99-113.
169. Luo N, Nixon MJ, Gonzalez-Ericsson PI, et al. DNA methyltransferase inhibition upregulates MHC-I to potentiate cytotoxic T lymphocyte responses in breast cancer. Nat Commun 2018;9:248.
170. Zhang C, Peng L, Zhang Y, et al. The identification of key genes and pathways in hepatocellular carcinoma by bioinformatics analysis of high-throughput data. Med Oncol 2017;34:101.
171. Johnston MP, Khakoo SI. Immunotherapy for hepatocellular carcinoma: Current and future. World J Gastroenterol 2019;25:2977-89.
172. Sim HW, Knox J. Hepatocellular carcinoma in the era of immunotherapy. Curr Probl Cancer 2018;42:40-8.
173. Zhang R, Zhang Z, Liu Z, et al. Adoptive cell transfer therapy for hepatocellular carcinoma. Front Med 2019;13:3-11.
174. Sun C, Sun HY, Xiao WH, Zhang C, Tian ZG. Natural killer cell dysfunction in hepatocellular carcinoma and NK cell-based immunotherapy. Acta Pharmacol Sin 2015;36:1191-9.
175. Rezvani K. Adoptive cell therapy using engineered natural killer cells. Bone Marrow Transplant 2019;54:785-8.
176. Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin Cancer Res 2011;17:6287-97.
177. Jia CC, Chen YH, Cai XR, et al. Efficacy of cytokine-induced killer cell-based immunotherapy for hepatocellular carcinoma. Am J Cancer Res 2019;9:1254-65.
178. Gao X, Mi Y, Guo N, et al. Cytokine-induced killer cells as pharmacological tools for cancer immunotherapy. Front Immunol 2017;8:774.
179. Wang J, Shen T, Wang Q, et al. The long-term efficacy of cytokine-induced killer cellular therapy for hepatocellular carcinoma: a meta-analysis. Immunotherapy 2019;11:1325-35.
180. Karschnia P, Jordan JT, Forst DA, et al. Clinical presentation, management, and biomarkers of neurotoxicity after adoptive immunotherapy with CAR T cells. Blood 2019;133:2212-21.
181. June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science 2018;359:1361-5.
182. Miliotou AN, Papadopoulou LC. CAR T-cell therapy: a new era in cancer immunotherapy. Curr Pharm Biotechnol 2018;19:5-18.
184. Nishida T, Kataoka H. Glypican 3-targeted therapy in hepatocellular carcinoma. Cancers (Basel) 2019;11:1339.
185. Wu Y, Liu H, Ding H. GPC-3 in hepatocellular carcinoma: current perspectives. J Hepatocell Carcinoma 2016;3:63-7.
186. Zhang J, Zhang M, Ma H, et al. Overexpression of glypican-3 is a predictor of poor prognosis in hepatocellular carcinoma: An updated meta-analysis. Medicine (Baltimore) 2018;97:e11130.
187. Zhou F, Shang W, Yu X, Tian J. Glypican-3: a promising biomarker for hepatocellular carcinoma diagnosis and treatment. Med Res Rev 2018;38:741-67.
188. Clinical Trials. ClinicalTrials.gov 2020. Available from: https://clinicaltrials.gov/ct2/search [Last accessed on 14 Dec 2020].
189. Vormittag P, Gunn R, Ghorashian S, Veraitch FS. A guide to manufacturing CAR T cell therapies. Curr Opin Biotechnol 2018;53:164-81.
190. Guo M, Zhang H, Zheng J, Liu Y. Glypican-3: a new target for diagnosis and treatment of hepatocellular carcinoma. J Cancer 2020;11:2008-21.
191. Hu Z, Ott PA, Wu CJ. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat Rev Immunol 2018;18:168-82.
192. Yarchoan M, Johnson BA 3rd, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer 2017;17:209-22.
193. Jiang T, Shi T, Zhang H, et al. Tumor neoantigens: from basic research to clinical applications. J Hematol Oncol 2019;12:93.
194. Wang X, Wang Q. Alpha-fetoprotein and hepatocellular carcinoma immunity. Can J Gastroenterol Hepatol 2018;2018:9049252.
195. Shimizu Y, Suzuki T, Yoshikawa T, Endo I, Nakatsura T. Next-generation cancer immunotherapy targeting glypican-3. Front Oncol 2019;9:248.
196. Heo J, Reid T, Ruo L, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med 2013;19:329-36.
197. Gilchrist VH, Jémus-Gonzalez E, Said A, Alain T. Kinase inhibitors with viral oncolysis: unmasking pharmacoviral approaches for cancer therapy. Cytokine Growth Factor Rev 2020:83-93.
198. Petrizzo A, Tagliamonte M, Mauriello A, et al. Unique true predicted neoantigens (TPNAs) correlates with anti-tumor immune control in HCC patients. J Transl Med 2018;16:286.
199. Lu L, Jiang J, Zhan M, et al. Targeting tumor-associated antigens in hepatocellular carcinoma for immunotherapy: past pitfalls and future strategies. Hepatology 2020; doi: 10.1002/hep.31502.
200. Peng M, Mo Y, Wang Y, et al. Neoantigen vaccine: an emerging tumor immunotherapy. Mol Cancer 2019;18:128.
201. Keskin DB, Anandappa AJ, Sun J, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019;565:234-9.
202. Ott PA, Hu Z, Keskin DB, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017;547:217-21.
203. Hanna MG, Hoover HC, Vermorken JB, Harris JE, Pinedo HM. Adjuvant active specific immunotherapy of stage II and stage III colon cancer with an autologous tumor cell vaccine: first randomized phase III trials show promise. Vaccine 2001;19:2576-82.
204. Varadé J, Magadán S, González-Fernández Á. Human immunology and immunotherapy: main achievements and challenges. Cell Mol Immunol 2020; doi: 10.1038/s41423-020-00530-6.
206. Llovet JM, Zucman-Rossi J, Pikarsky E, et al. Hepatocellular carcinoma. Nat Rev Dis Primers 2016;2:16018.
207. Marisi G, Cucchetti A, Ulivi P, et al. Ten years of sorafenib in hepatocellular carcinoma: are there any predictive and/or prognostic markers? World J Gastroenterol 2018;24:4152-63.
208. Llovet JM, Ricci S, Mazzaferro V, et al; SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359:378-90.
209. Murciano-Goroff YR, Warner AB, Wolchok JD. The future of cancer immunotherapy: microenvironment-targeting combinations. Cell Res 2020;30:507-19.
210. Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol 2018;15:599-616.
211. Cheng H, Sun G, Chen H, et al. Trends in the treatment of advanced hepatocellular carcinoma: immune checkpoint blockade immunotherapy and related combination therapies. Am J Cancer Res 2019;9:1536-45.
212. Lee HW, Cho KJ, Park JY. Current status and future direction of immunotherapy in hepatocellular carcinoma: what do the data suggest? Immune Netw 2020;20:e11.
213. Khemlina G, Ikeda S, Kurzrock R. The biology of hepatocellular carcinoma: implications for genomic and immune therapies. Mol Cancer 2017;16:149.
214. Waidmann O. Recent developments with immunotherapy for hepatocellular carcinoma. Expert Opin Biol Ther 2018;18:905-10.
215. Finkelmeier F, Waidmann O, Trojan J. Nivolumab for the treatment of hepatocellular carcinoma. Expert Rev Anticancer Ther 2018;18:1169-75.
216. Finn RS, Ryoo BY, Merle P, et al; KEYNOTE-240 investigators. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: a randomized, double-blind, phase III trial. J Clin Oncol 2020;38:193-202.
217. Huppert LA, Gordan JD, Kelley RK. Checkpoint inhibitors for the treatment of advanced hepatocellular carcinoma. Clin Liver Dis (Hoboken) 2020;15:53-8.
218. Heinrich B, Czauderna C, Marquardt JU. Immunotherapy of hepatocellular carcinoma. Oncol Res Treat 2018;41:292-7.
219. Chae YK, Galvez C, Anker JF, Iams WT, Bhave M. Cancer immunotherapy in a neglected population: the current use and future of T-cell-mediated checkpoint inhibitors in organ transplant patients. Cancer Treat Rev 2018;63:116-21.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Wang SZ, Lee SD, Sarkar D, Lee HM, Khan A, Bhati C, Sharma A, Kumaran V, Bruno D, Cotterell A, Levy MF. Immunological characterization of hepatocellular carcinoma. Hepatoma Res 2021;7:6. http://dx.doi.org/10.20517/2394-5079.2020.113
AMA Style
Wang SZ, Lee SD, Sarkar D, Lee HM, Khan A, Bhati C, Sharma A, Kumaran V, Bruno D, Cotterell A, Levy MF. Immunological characterization of hepatocellular carcinoma. Hepatoma Research. 2021; 7: 6. http://dx.doi.org/10.20517/2394-5079.2020.113
Chicago/Turabian Style
Wang, Sarah Ziqi, Seung Duk Lee, Devanand Sarkar, Hannah M. Lee, Aamir Khan, Chandra Bhati, Amit Sharma, Vinay Kumaran, David Bruno, Adrian Cotterell, Marlon F. Levy. 2021. "Immunological characterization of hepatocellular carcinoma" Hepatoma Research. 7: 6. http://dx.doi.org/10.20517/2394-5079.2020.113
ACS Style
Wang, SZ.; Lee SD.; Sarkar D.; Lee HM.; Khan A.; Bhati C.; Sharma A.; Kumaran V.; Bruno D.; Cotterell A.; Levy MF. Immunological characterization of hepatocellular carcinoma. Hepatoma. Res. 2021, 7, 6. http://dx.doi.org/10.20517/2394-5079.2020.113
About This Article
Copyright

Data & Comments
Data



 Cite This Article 39 clicks
Cite This Article 39 clicks

 Like This Article 31
likes
Like This Article 31
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.