
Viral hepatitis as a risk factor for the development of hepatocellular carcinoma

Abstract
Hepatocellular carcinoma (HCC) is the fourth leading global cause of tumor-related mortality. HCC has a high prevalence in patients with chronic liver diseases, and it mostly results from cirrhosis caused by infection with blood-borne viruses. Despite the implementation of various diagnostic and prevention strategies, the rates of new HCC cases and mortality are increasing globally due to the aging and growth of the world population as well as their increased exposure to dominant risk factors like alcohol, hepatitis B and C, and clinical correlates of metabolic syndrome. Modeling studies indicate that sanitation practices, implementation of vaccination programs against hepatitis B, and expanded recognition and treatment of patients with chronic hepatitis B and C could greatly contribute to the eradication of viral hepatitis B and C. While the availability of generic antiviral drugs could partially overcome the bottleneck represented by the lack of resources in low and middle-income countries, where viral hepatitis is the leading cause of liver cancer, the enthusiasm for the prevention of liver cancer through antiviral therapy is mitigated by the risk of cancer in many patients who are treated late in the hepatitis course. The present work aimed to review in detail the various types, epidemiology, and carcinogenesis mechanisms of viral infections that are associated with a significantly increased risk for the development of HCC.
Keywords
Introduction
Chronic infections with blood-borne hepatitis B (HBV), hepatitis C (HCV), and hepatitis D (HDV) viruses are the dominant causes of hepatocellular carcinoma (HCC) worldwide. In 2018, the death toll of HCC was 810,000 persons, and the attributable fractions of HCC due to HBV and HCV were 33% and 21%, respectively[1,2]. In selected regions of Eurasia, the Far East, and Africa, HDV stands as a significant risk factor for HCC and liver-related mortality[3]. While this cancer is on the rise globally, reflecting the continuing growth of the world population, the threat is not annulled by the lifestyle changes of people at risk, and many hopes are posed on the delivery of effective sanitation interventions. Mirroring the frequency and geographical distribution of blood-borne viral hepatitis, the prevalence of HCC has long been lower in developed regions than in developing regions. Yet, more recently, some peculiar changes in disease trends have emerged. Based on the 2012 data of the World Health Organization (WHO), HCC mortality was on the rise in northern Europe, North America, and some parts of Asia (China, India, and Korea), mainly as a consequence of epidemics of blood-borne viral hepatitis due to such parenteral risk behaviors, such as drug injections, tattoos, and unsafe sex. Conversely, HCC is declining in traditionally high-risk countries, including the Mediterranean European nations, Japan, and Hong Kong, as a consequence of improved sanitation, screening of blood donors, and mass vaccination of newborns against HBV. Of note, the latter also prevents the spread of HDV, another important player in the arena of HCC known to enhance cancer risk in HBV carriers[4-6]. While projections have predicted a decline of HCC mortality following massive access of infected patients to antiviral therapy against HBV and HCV, currently, only a minority of individuals with chronic hepatitis B (CHB) or C have been diagnosed, and an even smaller percentage of them has received effective antiviral therapy. The global goal of eliminating viral hepatitis as a public health threat by 2030 is expected to prevent HCC-related mortality by 65%. It would require a 7% annual decline of the global burden of viral hepatitis, a goal that has been reached by only a dozen countries to date[7].
Hepatitis B
HBV is a small, partially double-stranded DNA virus and a major contributor to chronic liver disease. The virus has a specific predilection for the liver, where it persists in hepatocyte nuclei in the form of chromosomal insertions of HBV DNA sequences and episomal covalently-closed circular DNA (cccDNA)[8]. Approximately 15%-40% of HBV carriers develop CHB[9]. The 5-year cumulative incidence of cirrhosis in untreated CHB is 8%-20%, with an annual risk of HCC in cirrhotic patients of 2%-5%[10]. At the beginning of the 1980s, a highly effective HBV vaccine was developed, and it proved to be very successful in reducing the disease burden. Nevertheless, the global number of HBV infections remains to be high, in part due to ineffective vaccination implementation programs in many less-developed countries and a high rate of perinatal transmission in certain parts of the world[11].
HBV is generally considered to be the strongest epidemiologic factor associated with HCC. Worldwide, CHB is responsible for almost half of all HCC cases, but the importance of this risk factor varies significantly between regions (e.g., critical in East Asia, but less so in Europe)[12]. Many studies have revealed that HBV-infected patients have a 15- to 20-fold increased risk for the development of HCC compared to non-infected individuals[13,14]. However, several effective antiviral therapies (e.g., nucleoside/nucleotide analogs (NAs) have been developed for patients with HBV over the last decade, and these agents were shown to reduce the rate of HCC occurrence in cirrhotic HBV patients[15].
Risk factors for HCC in HBV patients
A long list of risk factors for disease progression to HCC in CHB patients have been described. Firstly, several host-related factors have been shown to influence the HCC risk, with a higher risk in older patients and HBV carriers of African American origin[16-19]. Additionally, HCC is known to have a male preponderance, and several single-nucleotide polymorphisms have been identified to be associated with a higher genetic susceptibility for HCC[16,18-22]. Also, the lifestyle of HBV carriers can have a profound influence on HCC risk. For instance, heavy alcohol use was found to accelerate the development of cirrhosis in HBV patients, ultimately resulting in a 1.3- to 8.4-fold increase in HCC risk[23]. Similarly, tobacco smoking in HBV carriers was described to be directly correlated with the development of liver cancer. In this respect, a meta-analysis from 2010 reported a synergistic effect in HCC risk for individuals who smoke and have HBV infection. Compared to HBV-negative nonsmokers, the risk of HCC was 1.87 times greater for HBV-negative smokers, 15.8 for HBV-positive nonsmokers, and 21.6 for HBV-positive smokers[24]. More recently, this finding was confirmed in a large Chinese population-based case-control study[25]. There is an increasing body of evidence indicating an important role of metabolic risk factors in the disease process of CHB. For example, a high body mass index (BMI) has been shown to worsen the disease outcomes of HBV carriers. In a large Korean population-based cohort study, a strong association was revealed between high BMI and a higher risk for HCC among patients with CHB infection[26]. Also, diabetes mellitus (DM) was shown to have a synergistic impact on the HBV disease course, as amply illustrated by a large meta-analysis, including almost 22,000 patients with CHB[27]. In this analysis, HBV patients with type 2 DM were found to have a significantly increased risk of HCC (pooled HR = 1.77, 95%CI: 1.28-2.47) and worse overall mortality (pooled RR = 1.93, 95%CI: 1.64-2.27) compared to CHB patients without DM[27]. That being said, the relationship between nonalcoholic fatty liver disease (NAFLD) and hepatitis is complex and requires further clarification. Interestingly, HBV infection seems to protect patients from the development of steatosis, metabolic syndrome, and insulin resistance[28], whereas the presence of NAFLD-related steatosis impacts on the replication of HBV. The results of a large case-control study revealed that treatment-naïve CHB patients with NAFLD had significantly lower levels of serum HBV DNA compared to CHB without steatosis[29]. This, however, does not protect against liver damage, as the presence of steatosis was found to be associated with a higher rate of liver fibrosis and subsequent progression to HCC in HBV-infected patients, independent of antiviral therapy[30,31].
Also, specific virus-related features have been shown to impact the HCC risk, including HBV DNA levels, viral genotype, hepatitis B e-antigen/surface antigen (HBeAg/HBsAg) levels, mutations in the HBV genome, and coinfections with other hepatitis viruses or human immunodeficiency virus (HIV)[19,20,32-34]. With respect to viral factors, a high viral load has proved to be a strong predictor of the HCC risk, independent of whether or not the patient has cirrhosis, or displays high levels of serum HBsAg levels[18,20,32]. In relation to the HBV genotype, a large meta-analysis, which included more than 14,500 patients, demonstrated that genotype C was associated with a higher risk of HCC compared to the other major genotypes[35]. In the past, several large-scale studies have established baseline HBV DNA levels as a prognostic indicator in CHB patients. However, given the fact that the new effective antiviral therapies can induce a complete viral response in the majority of patients, the prognostic significance of serum HBV DNA levels has substantially diminished[36]. Finally, studies have also identified double mutations in the basal core promoter of the HBV genome as an independent predictor for an increased risk of HCC[37].
Accumulating evidence indicates that an occult hepatitis B infection (OBI) may be a risk factor for HCC. OBI refers to a condition in which HBV DNA persists in the liver tissue (and the serum in some cases) in the absence of circulating HBsAg[38]. A long list of studies has demonstrated the persistence of HBV infection in a large proportion of HBsAg-negative HCC patients[39]. While the exact relationship between OBI and HCC remains to be elucidated[40], the available data suggest that OBI is not carcinogenic per se, but that the minimal lesions produced by the presence of the occult virus might induce a worse liver disease course in the presence of co-existing causative agents of liver injury (e.g., HCV and alcohol abuse)[39]. This hypothesis is supported by studies indicating a higher prevalence of OBI in HCV-infected patients with HCC compared to HCV carriers who do not develop HCC[41-43]. Other studies, however, failed to show a correlation between serum anti-HBV detectability and HCC risk in HCV-infected patients[44,45]. Therefore, the most recent report of the Taormina occult HBV expert panel concluded that further studies on molecular epidemiology and carcinogenesis are required to confirm the role of OBI in HCC development[40].
As such, both viral- and host-related features were shown to profoundly impact HCC development in patients with HBV. However, the most important variables with respect to the HCC risk relate to the stage of liver disease. Historically, assessing the fibrosis status of the liver required a liver biopsy. However, due to the invasive nature of liver biopsy and its potential complications, this cannot be performed routinely in all CHB patients. To address this, several noninvasive methods have been validated to assess fibrosis in patients with chronic liver disease, of which transient elastography using the FibroScan® device is the most popular[46-48].
In order to help clinicians predict the risk of HCC in patients with CHB, several risk scores have been designed that incorporate host, viral, and liver characteristics. An overview of the most frequently used HCC risk prediction models is depicted in Table 1[16,18,49-51]. Of note, most of these scoring systems were validated before the availability of effective direct-acting antiviral (DAA) therapies. To assess the performance of the different risk scores in a contemporary setting, these conventional HCC prediction models were compared to the “modified risk estimation for HCC in CHB” (mREACH-B) score[52]. After a median follow-up of 75.3 months, 125 of the 1,308 subjects (9.6%) enrolled in this study developed HCC. Interestingly, the mREACH-B score proved to be associated with a significantly higher area under the receiver operating characteristic curve (AUROC) for the prediction of HCC development at 3 and 5 years (AUROC: 0.828 and 0.806, respectively), compared to the “liver stiffness measurement-HCC” (LSM-HCC) (AUROC: 0.777 and 0.759, respectively), “guide with age, gender, HBV DNA, core promoter mutations and cirrhosis-HCC” (GAG-HCC) (AUROC: 0.751 and 0.757, respectively), REACH-B (AUROC: 0.717 and 0.699, respectively), and “Chinese university-HCC” (CU-HCC) (AUROC: 0.698 and 0.700, respectively) scores (P < 0.05)[52]. As such, the prognostic performance of the mREACH-B score seems to be superior to that of the more conventional risk models.
HCC prediction models for HBV-infected patients
| CU-HCC[18] | GAG-HCC[49] | REACH-B[16] | mREACH-B[52] | LSM-HCC[50] | PAGE-B[51] | |
|---|---|---|---|---|---|---|
| Components | Age
Albumin Bilirubin Cirrhosis HBV DNA | Age
Gender BCP mutation Cirrhosis HBV DNA | Age
Gender ALT level HBeAG status HBV DNA | Age
Gender ALT level HBeAG status LS value | Age
Albumin HBV DNA Liver stiffness | Age
Gender Platelet level |
| Risk score | Low: < 5 | Low: < 100 | Low: ≤ 5 | Low: < 10 | Low: < 11 | Low: ≤ 9 |
| Medium: 5-20 | Medium: 6-11 | Medium: 10-17 | ||||
| High: > 20 | High: ≥ 100 | High: ≥ 12 | High ≥ 10 | High: ≥ 11 | High: ≥ 18 | |
| Negative predictive value | 97% at
10 years | 99% at
10 years | 98% at
10 years | 96.8% at
5 years | 99.4% at
10 years | 100% at
5 years |
Carcinogenic mechanisms
A potential carcinogenic mechanism that is mediated through HBV consists of HBV genome integration. In the vast majority of HCC cases (80%-90%), HBV DNA was found to be integrated into the host hepatocyte genome[53]. Cancer-related DNA integrations do not occur randomly; interestingly, they seem to be an early event that occurs before the development of HCC. Large-scale sequencing studies revealed recurrent HBV DNA integration sites at genetic loci that encode for proteins with a potential role in the initiation of hepatocellular carcinogenesis (e.g., CCNE1, TERT, and MLL4)[53,54]. Immune-mediated processes ultimately play a dominant role in the development of HCC in HBV-infected patients. In this light, elevated serum levels of proinflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukin (IL)-1β are typical hallmarks of HBV-infected patients[55]. The long-lasting hepatic inflammation caused by the host’s immune defense in response to CHB infection accelerates hepatocyte turnover, leading to increased mutations. As such, the proinflammatory environment in the liver of HBV patients indirectly contributes to the development of liver fibrosis, cirrhosis, and HCC progression[56].
In recent years, several studies have revealed an important role for micro-RNAs (miRNAs) in the HBV-related tumorigenesis. The miRNAs are small noncoding RNAs of 20-25 nucleotides in length that regulate the expression of certain target genes. For instance, HBeAg induces the expression of macrophage miRNA-155, which leads to an accelerated liver injury through the increased production of inflammatory cytokines (mediated by the targeting of BCL-6, SHIP-1, and SOCS-1)[57]. A deregulated miRNA expression (e.g., downregulation of miRNA-145 and upregulation of miRNA-224) occurs early and accumulates overtime in the stages of HBV-associated multistep hepatocarcinogenesis[58]. Apart from the deregulated miRNA expression, HBV was also shown to cause other epigenetic changes and regulate the expression of cellular oncogenes and tumor suppressor genes through a process of promotor hypo- or hypermethylation[59].
HBV-encoded proteins also play a role in the inflammation processes that lead to the development of HCC. In this respect, the HBV core protein and its splice variant HBeAg stimulate an immune response in the host, resulting in an increased production of proinflammatory cytokines[60]. A special role in hepatocarcinogenesis has also been described for the HBx protein, per the model of transgenic mice expressing HBx protein published in 1991[61]. Further research into the role of HBx revealed that this protein transactivates binding sites for the transcription factors AP-1 and NF-κB, and it activates the p53 and β-catenin signaling pathways involved in chromatin remodeling. All these signaling cascades were shown to be involved in the development of HCC (recently reviewed by Kanda et al.[62]). As such, HBx seems to play an essential role in the transcriptional modulation that contributes to hepatocarcinogenesis[62].
Prevention of HBV-related HCC
In 2016, the WHO, along with other health authorities, launched a campaign for expanding the recognition and treatment of HBV with the goal of eliminating viral hepatitis by 2030[63]. A package of high impact interventions of high impact was designed, following modeling studies of hepatitis epidemiology, with the anticipation of benefits conferred by articulated interventions like sanitation and even mass vaccination against HBV of newborns. Indeed, early-in-life infection with HBV is the major risk factor fueling the global reservoir of CHB, and it predisposes those individuals to HCC development. In WHO’s vision, vaccination of all newborns is linked with the implementation of screening of blood donors and harm reduction policies for people who inject drugs (i.e., exchange of sterile syringes and needles coupled with opioid substitution therapies)[63]. Strategies to interrupt vertical transmission of HBV through vaccination are in place in almost all WHO member countries, yet only 39% of newborns received the HBV vaccine globally[64]. By 2015, however, 84% of all infants globally received the vaccine (WHO target for 2020 was set at 90%). Over the last few years, the accumulated evidence showed that vaccination had substantially contributed to shrinking the burden of HBV by over 30%. In China, campaigns of mass vaccination led to a decline of HBsAg carrier state from approximately 90% in the eighties to 1% nowadays. In turn, this averted 2.8-3.5 million future HBV-related deaths, of which the majority is associated with HCC[64]. The immune prophylaxis against HBV is long-lasting, although multiple long-term studies have demonstrated a decline of protective serum anti-HBs levels (10 IU/mL) over time. While 60% of vaccinated people are anti-HBs serum positive at year 20, more than 95% retain the ability to mount an anamnestic response after a challenge dose at that time point[65]. However, to achieve optimal rates of immunization (95%), all neonates need to receive their first dose of the HBV vaccine as soon as possible after birth, preferably within 24 h. In contrast, neonates born to HBV-infected mothers should receive the vaccine along with hepatitis B immunoglobulins within 12 h[66]. Suboptimal immunoprophylaxis in neonates can occur and is related to mothers with positive serum HBeAg or high viral load. Suboptimal immunoprophylaxis can also be due to the delivery of less than the recommended three doses of vaccine, an event which occurs in 60% of HBV vaccine recipients. In adulthood, failure to achieve 95% immunization rates is mostly seen in persons older than 60 years of age and patients with morbidities like cancer, immunosuppression, renal failure, HIV, and organ transplantation. Mammalian cell-derived recombinant vaccines incorporating preS1 and preS2 antigens have shown enhanced immunogenicity and might be used to overcome non-response to second-generation recombinant vaccines.
While HBV vaccination stands as the only pragmatic approach to prevent mortality from the HDV, a recent report from WHO sheds a dark light on the other pillar of the WHO campaign of viral hepatitis elimination, being the prolonged treatment of the HBV infected population with NAs. Currently, less than 10% of all patients chronically infected with HBV have been identified and successfully linked to care with anti-HBV antivirals. This constraint might have important consequences as chemoprevention of HCC is more likely to be successful when antiviral therapy is started before the development of cirrhosis[67]. The advent of a safe, effective, and user-friendly third-generation NAs, such as tenofovir disoproxil fumarate and entecavir, overrode the constraints represented by first- and second-generation anti-HBV NAs that caused studies to be flawed by referral biases, high rates of treatment failures, and ultimately by suboptimal percent suppression of HBV. Collectively, both population and cohort studies that have been carried out in both hemispheres of the globe showed that the incidence and mortality of HCC could have been prevented in a majority of patients if they received NAs for more than five years[68,69]. Given the differences in patient access (entecavir is contraindicated in lamivudine-experienced patients), market distribution, and genetic sequences of HBV polymerase targeted by the two NAs, non-randomized studies comparing the HCC risk reduction following HBV suppression were unable to conclusively demonstrate the superiority of one regimen over the other[70]. Both NAs fail to clear the nuclei of infected hepatocytes from HBV DNA sequences integrated into chromosomes and from free viral cccDNA, two events that are known to play a role in the neoplastic transformation of the liver in HBV carriers[67]. More recently, both cohort and population studies provided some evidence that statins and aspirin may confer protection against HCC in HBV carriers. This effect was described to result in their ability to interfere with liver cell metabolism and inflammatory processes engaged in cell carcinogenesis. In a cohort of more than 7000 patients with CHB, statins were associated with a cumulative dose-response reduction of HCC risk of 74% over an observation period of 7 years, after adjustment for important confounders like age, sex, cirrhosis, antiviral therapy, and correlates of metabolic syndrome[71]. These observations confirm previous observations and aligned with other studies of statins showing potential anticancer activity in other cancer types (i.e., breast, colon, and prostate cancer) through inhibition of the downstream products of the mevalonate pathway, which are crucial for malignant cell proliferation while inhibiting hepatic fibrogenesis, another significant risk factor of HCC[72-74]. Last but not least, statins may also counteract HBV by slowing down cholesterol synthesis and HBV replication[75]. In Taiwan, a nationwide cohort study of more than 10,000 patients with CHB showed a statistically significant risk reduction of HCC in patients who received daily aspirin compared with 1:4 matched controls[76]. Prevention of HCC by aspirin is biologically plausible, considering that this drug may prevent the progression of liver disease and liver carcinogenesis through different mechanisms involving blockade of platelets, modulation of bioactive lipids, and inhibition of the proinflammatory cyclooxygenase-2 enzyme[77-79].
Hepatitis C
While HBV is the most common underlying HCC etiology worldwide, HCV is responsible for most cases in Western countries[1]. In patients with a chronic HCV infection, the risk of HCC gradually increases as liver fibrosis progresses. Once cirrhosis is established, the annual incidence of HCC is high, at 1% to 7% per year[17]. Overall, it has been established that HCV infected patients have a 15-20 fold increased risk of developing HCC compared with HCV negative patients[14,80,81].
Risk factors for HCC in HCV patients
The rate of HCC progression varies greatly among patients with chronic HCV infection, and this is due to the existence of a complex interplay between host, viral, and environmental factors. Similar to what was described for HBV, the most important risk factor for the development of HCC in patients with a chronic HCV infection is the underlying liver disease[82]. Apart from that, several other concurrent risk factors that impact the HCC risk in patients with HCV have been identified. To begin, male sex and older age have universally been described as independent risk factors for the development of HCC in patients with a chronic HCV infection[82-84]. Also, a coinfection with HBV or HIV seems to influence the course of an HCV infection. Several studies have demonstrated that coinfection with HIV promotes the progression of fibrosis and cirrhosis in patients with HCV, resulting in a significantly increased risk for severe liver disease[85-88]. As a result, it is widely accepted that an HIV coinfection in HCV patients also increases the risk of HCC compared to HCV mono-infected patients[89]. However, recent data from two prospective French cohorts demonstrate that this is no longer the case in the current context of more effective combination antiretroviral therapies and increased access to HCV therapy. In this analysis, the 5-year cumulative incidence of HCC and liver decompensation did not differ significantly between HIV/HCV coinfected and HCV mono-infected patients (8.5% vs. 13.2%, P = 0.12 and 12.8% vs. 15.6%, P = 0.40, respectively)[90]. Also, patients with a dual HBV/HCV infection have a higher risk of progression to cirrhosis and decompensated liver disease compared to patients with an HCV mono-infection[91,92]. Already in 1998, a meta-analysis of more than 30 case-control studies demonstrated a synergistic effect of HCV and HBV on the incidence of HCC[14]. This observation was later confirmed by a second, Chinese meta-analysis indicating that a dual infection by HBV and HCV was associated with a higher risk of HCC than each infection alone [odds ratio (OR) for the development of HCC for coinfected patients: 35.7][13]. Similarly, an Italian study reported a yearly HCC incidence of 6.4% in HBV/HCV coinfected patients as compared to 2.0% and 3.7% in HBV and HCV mono-infected patients, respectively. At 10-years, this translated into a cumulative HCC rate of 45%, 16%, and 28%, respectively[93]. For HBV/HCV dual infection, the research data suggest that the HBV replication status is the crucial factor affecting the risk for HCC. HCV patients with active HBV replication have twice the risk of HCC compared to those with latent HBV and HCV, while the risk in coinfected patients with undetectable HBV DNA levels is similar to that of mono-infected HCV patients[83]. As discussed in the section on HBV, more research is needed to shed light on the potential effect of OBI on the risk for HCC in HCV-infected patients[40]. Intriguingly, certain HCV genotypes seem to be associated with a higher risk of HCC, particularly genotype 3 which is associated with an 80% higher risk of HCC compared to genotype 1[94], contradicting a previous meta-analysis that associated genotype 1 with a 78% increased risk of HCC relative to all other genotypes and a 60% increased risk among patients with cirrhosis[95].
Similar to what was described for HBV, there are significant associations between lifestyle factors and the HCC risk in HCV patients. A meta-analysis has shown a significant increase in the relative risk of HCC in smokers (relative risk 23) compared to non-smokers (relative risk 7.9) with HCV[24]. Also, alcohol consumption was shown to accelerate liver fibrosis in HCV-infected patients, resulting in an increased risk for progression to cirrhosis and HCC. In a study evaluating the natural history of liver fibrosis progression in 2,235 HCV patients, daily alcohol consumption of at least 50 g resulted in a 34% increase in the rate of fibrosis progression[96]. In line with this, a meta-analysis involving more than 15,000 HCV patients demonstrated that heavy alcohol intake (210-560 g per week) was associated with a 3.54 relative risk for the development of decompensated cirrhosis[97]. A study that specifically looked into the effect of alcohol on the development of HCC in HCV patients revealed a 2-fold increase in HCC risk for drinkers of more than 60 g per day[98]. Similarly, a case-control study revealed an OR for HCC development of 26.1 in HCV-carriers with an alcohol intake of 0-40 g/day, rising to 62.6 and 126 among patients drinking a daily dose of 41-80 g and more than 80 g per day, respectively[99]. Interestingly, studies have indicated that the synergistic effect of alcohol on the HCC risk in HCV patients is not restricted to heavy drinkers and that even light to modest alcohol use can promote the development of cirrhosis (and subsequent HCC) in HCV patients[100]. In line with this, a study including 192 HCV patients with compensated cirrhosis, reported a 5-year cumulative HCC rate of 10.6% among abstainers as compared to 23.8% among HCV patients with a light to moderate alcohol consumption (median intake: 15 g/day)[101].
HCV also comes with an increased risk of HCC in patients with DM. A meta-analysis evaluating the association between DM and HCC in chronic HCV patients indicated a 2- to 3-fold increased risk[102]. Similarly, a population-based cohort study from Taiwan indicated that DM increases the risk of HCC in HCV-infected patients (HR = 1.36, 95%CI: 1.16-1.68;)[103]. Finally, NAFLD is a prominent characteristic of a chronic HCV infection. In patients with genotype 3 HCV, the presence of NAFLD is directly linked to the viral load, and in this setting, the NAFLD is considered to be of viral origin[104]. In contrast, in patients with other HCV genotypes, the NAFLD is linked to host factors such as obesity. Studies have consistently identified (NAFLD-related) steatosis as an independent factor associated with fibrosis progression in HCV patients[105-108]. As such, it is not surprising to see that several studies (both retrospective and prospective) have demonstrated that steatosis in HCV patients is also strictly associated with the development of HCC[109-113]. An interesting study in this respect demonstrated that American chronic HCV patients were found to progress more rapidly to HCC than their counterparts in China, with underlying fatty liver disease as the prominent factor fueling this difference[114].
Several prediction models for HCC have been developed for patients with an HCV infection (e.g., HALT-C, REVEAL-HCV, and SCORE
Carcinogenic mechanisms
In contrast to HBV, HCV is a single-stranded RNA virus with little potential to integrate its genetic material into the host. Therefore, HCV must exhibit its tumorigenic potential less directly. The mechanisms involved in this process mainly involve two parts: induction of chronic inflammation and the expression of viral proteins. HCV-induced HCC development is a multistep process that involves the establishment of chronic HCV infection, persistent chronic hepatic inflammation, progressive liver fibrogenesis, initiation of neoplastic clones accompanied by irreversible somatic genetic/epigenetic alterations, and progression of the malignant clones in a carcinogenic tissue microenvironment[118].
Several proinflammatory cytokines, including TNF-α, IL-1, IL-23, IL-6, and lymphotoxins α and β have been implicated in chronic liver inflammation and the development of HCC[119]. Specifically, for HCV-mediated hepatocarcinogenesis, a high ratio of TNF-α/IL-10 levels has been observed in the sera of patients with severe liver damage and HCC[120]. Apart from immunological disturbances, epigenetic processes were found to be involved in the development of HCV-related HCC. Like HBV-related HCC, HCV also seems to profoundly impact the expression of certain miRNAs[121]. Similar to HBV, miRNA-155 also plays a role in the development of HCC in HCV-infected patients. miRNA-155 levels are markedly increased in patients infected with HCV, and this overexpression was found to stimulate hepatocyte proliferation and tumorigenesis by activating Wnt signaling[122]. Other epigenetic studies have suggested a prominent role for noncoding RNAs in the development of HCV-related HCC[123,124].
With respect to the role of HCV-related proteins in the development of liver cancer, the HCV core protein and NS5A seem to be of interest[62]. In 1996, the HCV core protein was found to induce a carcinogenic phenotype in primary rat embryo through a RAS-mediated mechanism[125]. The HCV core protein was also shown to activate the MAPK signaling pathway, upregulate Wnt/β-catenin signaling, suppress apoptosis pathways, and activate TGF-β, PI3K/Akt/mTOR, NF-κB, p53, IL-6/Stat3, and the androgen receptor pathways. Through these pathways, the HCV core protein could regulate cell growth, differentiation, apoptosis, transcription, and angiogenesis[62,126]. Similarly, NS5A interacts with multiple pro-oncogenic pathways, including β-catenin, PI3K/Akt/mTOR, NF-κB, and p53[62,126].
Finally, insulin resistance, commonly observed in patients with HCV, plays a crucial role in the development of HCC in HCV patients. Intensive research identified that cross-talk between the HCV core protein and molecules regulating insulin signaling might affect HCV-related hepatocarcinogenesis[62].
Prevention of HCV-related HCC
Every year, de novo HCV affects 1.75 million persons, and more than 350,000 people die of HCV-related cirrhosis or liver cancer[66,127]. Since new HCV infections outnumber the sum of people who die of end-stage hepatitis C and those pharmacologically cured, HCV elimination is also a priority of the WHO[63]. This goal is achievable because the virus lacks a non-human reservoir, cannot amplify in the environment and can be identified with simple and accurate diagnostic tests. At the same time, practical interventions can be delivered to interrupt transmission and cure both acute and chronic infections[128]. With an articulated package of interventions similar to the one in place for HBV elimination, except for vaccine prophylaxis, which is not available, the strategy designed by WHO is expected to provide treatment to 90% of all infected individuals by 2030 worldwide[63]. The clinical benefits of HCV elimination are undisputed and are already well-established since the ages of interferon therapy for HCV. The treatment strategy is also well acknowledged by all international liver societies, which strongly recommend antiviral treatment of all HCV infected patients, independent of the severity of the underlying liver disease[129,130]. A sustained virologic response (SVR) heralds improvement of portal hypertension and fibrosis progression in patients with chronic hepatitis and may reduce HCC incidence and all-cause mortality[130]. Significant HCC chemoprevention already surfaced in the interferon studies, where an SVR was found to be associated with a relative risk reduction of 74% compared to non-responders[131]. This HCC chemoprevention by SVR was further amplified by the advent of safe and effective DAA against HCV. These agents extended the range and clinical benefits of antiviral therapy. Also, DAAs can be indicated in patients with decompensated liver disease who were either ineligible for treatment with interferon or did not respond to the therapy. HCC chemoprevention by DAA was firmly established through the retrospective scrutiny of large cohorts of patients in the US, Europe, and Asia. Moreover, this finding was further confirmed by prospective population studies. In a Veterans Health Administration cohort, which included more than 20,000 patients with advanced liver disease and multiple comorbidities, DAAs reduced the risk of developing HCC by 72% compared to non-responders[132]. More recently, an analysis of the Veterans Affair cohorts including more than 15,000 HCV patients demonstrated that patients who achieved an SVR after DAA treatment had a significantly lower all-cause mortality (78.9% reduction) and a lower HCC incidence (83.5% reduction) than those who did not achieve an SVR [Figure 1][133,134].
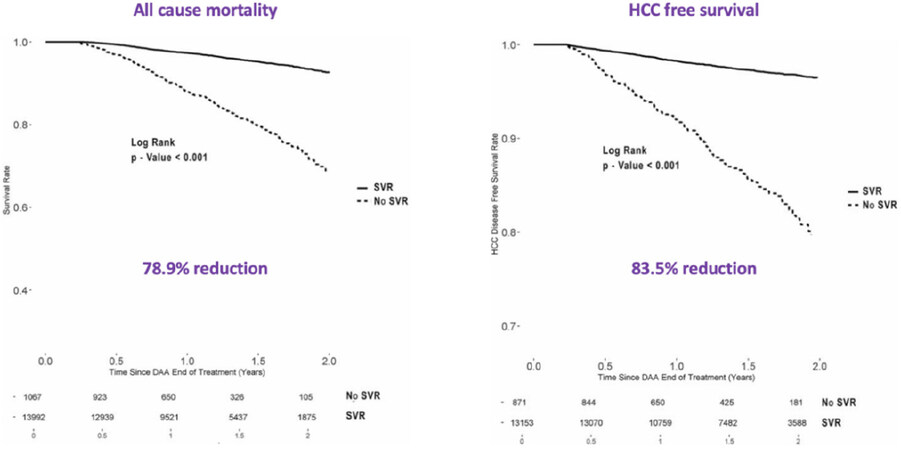
Figure 1. Reduction in HCC incidence and all-cause mortality in patients with advanced hepatitis C who achieve an SVR to DAA. Data from the Veteran Health Administration cohort[133]. HCC: Hepatocellular carcinoma; SVR: sustained virologic response; DAA: direct-acting antiviral
In a prospective population study in Sicily, Italy, the incidence rate of HCC at one year was 2.6% in SVR patients compared to 8% in non-responders with a clear cut association between HCC risk and liver disease severity[134]. Importantly, these studies had the additional merit of wiping out any doubt about the safety of DAA therapy in patients with advanced hepatitis C as they counteracted the initial observations of high rates of clinically aggressive de novo HCC after an SVR to DAA that were identified in small groups of cirrhotic patients in Spain and Italy[135-137]. Nowadays, it is clear that early occurrence of de novo HCC is confined to patients who harbor magnetic resonance imaging (MRI)-undefined liver nodules at the onset of DAA therapy and is promoted by the imbalance of field immunity caused by the swift eradication of HCV[136]. Early, aggressive recurrence was also a complication of HCV eradication with DAAs in patients with a history of HCC[137,138], where antiviral therapy aims to halt the progression of hepatitis C towards liver failure and prevent the onset of second primary tumors which result from both direct and indirect carcinogenic damage of hepatocellular DNA fueled by unrested HCV replication[139]. The latter usually takes place 1 to 2 years after the cure of a primary HCC (late recurrence), at variance with earlier recurrence that is caused by the proliferation of pre-existing cancer cells surviving the removal of the primary tumor, for which no effective adjuvant therapy exists. The early recurrence depends on tumor size and cell grading, reflecting the cancer cells that invade the tumor vessels and/or to tumor satellites emerging far from parental HCC[140], the evidence is mounting that early recurrence of HCC in DAA-treated patients is often bound to pre-existing liver nodules with undefined vascular patterns at MRI[138].
In a final note on anti-HCV therapy, it is important to underscore that achieving an SVR to antiviral therapy does not tell the whole story. In fact, data from Lens et al.[141] reported in 2017 indicate that obtaining an SVR to all-oral anti-HCV therapy in patients with HCV-associated cirrhosis indeed leads to decrease in a hepatic venous pressure gradient, but that clinically significant hypertension did persist in 78% of patients. These patients have a continued risk for liver decompensation, and subsequently, maintain a higher risk of HCC[141].
A recent study based on Swedish nationwide registries has demonstrated a significant reduction of HCC risk in a population including more than 50,000 HCV infected and 10,000 HBV infected adults. The latter had been chronically exposed to low doses of aspirin (HR = 0.69, 95%CI: 0.62-0.76). After adjusting for relevant confounding morbidities, chemoprevention of liver cancer was confirmed and found to be associated with a similar risk reduction of liver-related mortality[142]. These findings align with studies done in HBV patients showing chemoprevention of HBV-related liver cancer following long-term exposure to aspirin.
Hepatitis D
HDV is a small replication-defective RNA virus that relies on HBV to replicate and propagate. Due to a lack of dedicated studies assessing the prevalence of HDV, the global disease burden of HDV is likely underestimated[143]. In a recently published systematic review and meta-analysis, 650-700 million people have a chronic HBV infection globally, of whom 60-70 million have an HDV coinfection, which is almost twice as much as the previous estimate[144]. HDV does not integrate into the genome of hepatocytes, making a direct oncogenic mechanism unlikely. However, preliminary data have indicated the potential indirect oncogenic effects of this virus. HDV can modify several key signaling pathways with a known role in cirrhosis and hepatocarcinogenesis, including the activation of the TGF-β, NF-κB, and JAK-STAT signaling pathways[145-147].
Studies comparing HCC incidence between HBV/HDV coinfected and HBV mono-infected patients provide better insights on the oncogenic impact of HDV. A critical Eurohep study demonstrated that HBV/HDV-positive cirrhotic patients followed for a median of 6.6 years had a twofold increase in mortality risk compared to patients with HBV-related cirrhosis[148]. Moreover, the estimated risk for HCC was 13% among cirrhotic HBV/HDV patients compared to 2%-4% for cirrhotic patients with an HBV mono-infection, corresponding to a threefold increase in HCC risk for coinfected patients[148]. Also, a large Swedish retrospective cohort study demonstrated a significantly higher risk of HCC for patients with acute (RR = 6.1, 95%CI: 2.8-11.7) or chronic (RR = 3.9, 95%CI: 1.6-7.2) HDV[149]. Similarly, an American study, including 2,175 HBV patients, found a 2.9-fold increase in the incidence of HCC in individuals with an HBV/HDV coinfection (OR = 2.1, 95%CI: 1.1-3.9)[150].
Cohort studies evaluating the HCC incidence among patients with HDV yielded variable results. Among 299 HDV infected patients who were included in a single-center Italian study (diagnosed between 1987 and 2006), 46 HCC cases were reported, accounting for an annual rate of 2.8%[151]. Interestingly, the authors of this analysis later identified high serum levels of HDV RNA as a predictor of cirrhosis and liver cancer in HDV patients[152]. In a large cohort study which included 1,576 HDV patients, the annual HCC incidence was slightly lower at 1.9%[153]. The results obtained in other cohort studies with a fewer patients were inconsistent and reported an annual HCC incidence among HDV patients of 3%-13%[154-156]. Finally, a recent study addressed the HDV-associated mortality in HIV/HBV coinfected patients. HDV infection appeared to be strongly associated with overall death (HR = 2.33, 95%CI: 1.41-3.84), liver-related death (HR = 7.71, 95%CI: 3.13-18.97), and HCC occurrence (HR = 9.3, 95%CI: 3.03-28.61)[157].
Very recently, a meta-analysis pooling data from 93 studies compared the HCC risk in patients with HBV/HDV and HBV alone (68 case-control studies with 22,862 patients and 25 cohort studies with 75,427 patients) was published[5]. Patients with HBV/HDV had a significantly higher HCC risk than patients with an HBV mono-infection (pooled OR = 1.28, 95%CI: 1.05-1.57). Of note, this association was particularly pronounced in studies with HIV-infected patients (pooled OR = 7.13, 95%CI: 2.83-17.92)[5].
In summary, though most studies evaluating the incidence of HCC in HDV-infected patients have provided low-level evidence, data support an association between HDV and the development of HCC. Similar to what was described for HBV and HCV, the objective of antiviral treatment in patients with a chronic HDV infection is to eliminate HDV and HBV and, as such, prevent the long-term detrimental effects of hepatitis D on the liver. Unfortunately, the treatment options for patients with HDV are limited, and HDV elimination is not commonly achieved. In fact, 1-year of interferon-α (IFN-α) only induces a sustained HDV clearance in 10%-20% of patients[158]. More recently, several studies have evaluated the use of pegylated IFN-α in HDV patients. While response rates in these trials were higher than what was seen with classical IFN-α, sustained clearance of HDV RNA proved feasible in only about a quarter of patients[159-161]. Data on the impact of IFN-α treatment on the natural history of hepatitis D are scarce. In an extended follow-up of 36 chronic HDV patients treated with one year of IFN-α found that some patients experienced regression of their advanced stage fibrosis, indicating a positive effect on the natural HDV disease course[162]. Less favorable data come from the HIDIT trial in which pegylated IFN-α was used to treat chronic HDV. In this trial, 58% of patients who were shown to be HDV RNA negative 6 months after therapy experienced a late HDV relapse[163]. Interestingly, these late relapses were not associated with clinical complications. This might indicate that a prolonged virologic response to pegylated IFN-α, even if not sustained, can be clinically relevant in patients with chronic hepatitis D[163].
Conclusion
Blood-borne viral hepatitis is a dominant cause of HCC worldwide, and the positive effect of their eradication on the risk of developing HCC has been extensively demonstrated even in patients carrying significant metabolic comorbidities that predispose them to the neoplastic transformation of the liver. Along this line, intriguing data have emerged, suggesting a significant chemoprophylactic activity of liver metabolism modifiers such as statins and aspirin. All in all, the implementation of articulated interventions of sanitation make reaching the WHO goal of viral elimination more realistic in some small countries like Georgia or Iceland, where elimination programs can easily be run, than in large countries where it is difficult even to identify hepatitis carriers. Further mitigating our belief of being on the right track for global elimination of viral hepatitis was the increasing hesitance against hepatitis B vaccination. In recent years, it has amounted to the level of reaching an alarming proportion of 30% in many high-income countries. While this could be counteracted by the recent pandemic of 2019 coronavirus disease (shortly, COVID-19) that has restored confidence in public health policies based on vaccination, another critical point in the fight against viral HCC is the existence of awareness campaigns and screening programs, since the first essential step in the viral hepatitis cure pathway is to be aware of the infection. Identification of defined risk cohorts, including baby-boomers, people who inject drugs, prisoners, and men who have sex with men, improves the cost-effectiveness of screening programs aimed to target infected populations; however, determining the infectious status is irrelevant if effective linkage-to-care programs are not in place. The availability of generic antivirals can partially overcome the bottleneck represented by the lack of resources in low and middle-income countries where HBV and HCV prevail as risk factors for HCC.
Declarations
Authors’ contributionsEqually made substantial contributions to the conception and design of the study and performed data analysis and interpretation: Alqahtani SA, Colombo M
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestBoth authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Akinyemiju T, Abera S, Ahmed M, Alam N, Alemayohu MA, et al; Global Burden of Disease Liver Cancer Collaboration. The Burden of Primary Liver Cancer and Underlying Etiologies From 1990 to 2015 at the Global, Regional, and National Level: Results From the Global Burden of Disease Study 2015. JAMA Oncol 2017;3:1683-91.
2. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394-424.
3. Caviglia GP, Rizzetto M. Treatment of hepatitis D: an unmet medical need. Clin Microbiol Infect 2020;26:824-7.
4. Liu KSH, Seto WK, Lau EHY, Wong DK, Lam YF, et al. A territorywide prevalence study on blood-borne and enteric viral hepatitis in Hong Kong. J Infect Dis 2019;219:1924-33.
5. Alfaiate D, Clément S, Gomes D, Goossens N, Negro F. Chronic hepatitis D and hepatocellular carcinoma: a systematic review and meta-analysis of observational studies. J Hepatol 2020; doi: 10.1016/j.jhep.2020.02.030.
6. Liu Z, Jiang Y, Yuan H, Fang Q, Cai N, et al. The trends in incidence of primary liver cancer caused by specific etiologies: results from the Global Burden of Disease Study 2016 and implications for liver cancer prevention. J Hepatol 2019;70:674-83.
7. Maticic M, Mondelli MU. Elimination of viral hepatitis: where do we stand in the year 2020? Clin Microbiol Infect 2020;26:816-7.
8. Lucifora J, Protzer U. Attacking hepatitis B virus cccDNA--The holy grail to hepatitis B cure. J Hepatol 2016;64:S41-8.
10. Raffetti E, Fattovich G, Donato F. Incidence of hepatocellular carcinoma in untreated subjects with chronic hepatitis B: a systematic review and meta-analysis. Liver Int 2016;36:1239-51.
12. Baecker A, Liu X, La Vecchia C, Zhang ZF. Worldwide incidence of hepatocellular carcinoma cases attributable to major risk factors. Eur J Cancer Prev 2018;27:205-12.
13. Shi J, Zhu L, Liu S, Xie WF. A meta-analysis of case-control studies on the combined effect of hepatitis B and C virus infections in causing hepatocellular carcinoma in China. Br J Cancer 2005;92:607-12.
14. Donato F, Boffetta P, Puoti M. A meta-analysis of epidemiological studies on the combined effect of hepatitis B and C virus infections in causing hepatocellular carcinoma. Int J Cancer 1998;75:347-54.
15. Tawada A, Kanda T, Imazeki F, Yokosuka O. Prevention of hepatitis B virus-associated liver diseases by antiviral therapy. Hepatol Int 2016;10:574-93.
16. Yang H, Yuen M, Chan HL, Han K, Chen P, et al. Risk estimation for hepatocellular carcinoma in chronic hepatitis B (REACH-B): development and validation of a predictive score. Lancet Oncol 2011;12:568-74.
17. Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, et al. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol 2019;16:589-604.
18. Wong VW, Chan SL, Mo F, Chan TC, Loong HH, et al. Clinical scoring system to predict hepatocellular carcinoma in chronic hepatitis B carriers. J Clin Oncol 2010;28:1660-5.
19. Wong VW, Janssen HL. Can we use HCC risk scores to individualize surveillance in chronic hepatitis B infection? J Hepatol 2015;63:722-32.
20. Yang HI, Lu SN, Liaw YF, You SL, Sun CA, et al; Taiwan Community-Based Cancer Screening Project Group. Hepatitis B e antigen and the risk of hepatocellular carcinoma. N Engl J Med 2002;347:168-74.
21. Zhang BH, Yang BH, Tang ZY. Randomized controlled trial of screening for hepatocellular carcinoma. J Cancer Res Clin Oncol 2004;130:417-22.
22. Zhang H, Zhai Y, Hu Z, Wu C, Qian J, et al. Genome-wide association study identifies 1p36.22 as a new susceptibility locus for hepatocellular carcinoma in chronic hepatitis B virus carriers. Nat Genet 2010;42:755-8.
23. Iida-Ueno A, Enomoto M, Tamori A, Kawada N. Hepatitis B virus infection and alcohol consumption. World J Gastroenterol 2017;23:2651-9.
24. Chuang SC, Lee YC, Hashibe M, Dai M, Zheng T, et al. Interaction between cigarette smoking and hepatitis B and C virus infection on the risk of liver cancer: a meta-analysis. Cancer Epidemiol Biomarkers Prev 2010;19:1261-8.
25. Liu X, Baecker A, Wu M, Zhou JY, Yang J, et al. Interaction between tobacco smoking and hepatitis B virus infection on the risk of liver cancer in a Chinese population. Int J Cancer 2018;142:1560-7.
26. Kim K, Choi S, Park SM. Association of high body mass index and hepatocellular carcinoma in patients with chronic hepatitis B virus infection: a Korean population-based cohort study. JAMA Oncol 2018;4:737-9.
27. Tan Y, Wei S, Zhang W, Yang J, Yang J, et al. Type 2 diabetes mellitus increases the risk of hepatocellular carcinoma in subjects with chronic hepatitis B virus infection: a meta-analysis and systematic review. Cancer Manag Res 2019;11:705-13.
28. Joo EJ, Chang Y, Yeom JS, Ryu S. Hepatitis B virus infection and decreased risk of nonalcoholic fatty liver disease: a cohort study. Hepatology 2017;65:828-35.
29. Hui RWH, Seto WK, Cheung KS, Mak LY, Liu KSH, et al. Inverse relationship between hepatic steatosis and hepatitis B viremia: results of a large case-control study. J Viral Hepat 2018;25:97-104.
30. Seto WK, Hui RWH, Mak LY, Fung J, Cheung KS, et al. Association between hepatic steatosis, measured by controlled attenuation parameter, and fibrosis burden in chronic hepatitis B. Clin Gastroenterol Hepatol 2018;16:575-83.e2.
31. Wong GL, Chan HL, Yu Z, Chan AW, Choi PC, et al. Coincidental metabolic syndrome increases the risk of liver fibrosis progression in patients with chronic hepatitis B--a prospective cohort study with paired transient elastography examinations. Aliment Pharmacol Ther 2014;39:883-93.
32. Chen CJ, Yang HI, Su J, Jen CL, You SL, et al; REVEAL-HBV Study Group. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA level. JAMA 2006;295:65-73.
33. Wong VW, Sung JJ. Diagnosis and personalized management of hepatitis B including significance of genotypes. Curr Opin Infect Dis 2012;25:570-7.
34. Kao JH, Chen PJ, Lai MY, Chen DS. Basal core promoter mutations of hepatitis B virus increase the risk of hepatocellular carcinoma in hepatitis B carriers. Gastroenterology 2003;124:327-34.
35. Wong GL, Chan HL, Yiu KK, Lai JW, Chan VK, et al. Meta-analysis: the association of hepatitis B virus genotypes and hepatocellular carcinoma. Aliment Pharmacol Ther 2013;37:517-26.
36. Lee HW, Yoo EJ, Kim BK, Kim SU, Park JY, et al. Prediction of development of liver-related events by transient elastography in hepatitis B patients with complete virological response on antiviral therapy. Am J Gastroenterol 2014;109:1241-9.
37. Sumi H, Yokosuka O, Seki N, Arai M, Imazeki F, et al. Influence of hepatitis B virus genotypes on the progression of chronic type B liver disease. Hepatology 2003;37:19-26.
38. Raimondo G, Allain JP, Brunetto MR, Buendia MA, Chen DS, et al. Statements from the Taormina expert meeting on occult hepatitis B virus infection. J Hepatol 2008;49:652-7.
39. Pollicino T, Saitta C. Occult hepatitis B virus and hepatocellular carcinoma. World J Gastroenterol 2014;20:5951-61.
40. Raimondo G, Locarnini S, Pollicino T, Levrero M, Zoulim F, et al; Taormina Workshop on Occult HBV Infection Faculty Members. Update of the statements on biology and clinical impact of occult hepatitis B virus infection. J Hepatol 2019;71:397-408.
41. Wang H, Swann R, Thomas E, Innes HA, Valerio H, et al. Impact of previous hepatitis B infection on the clinical outcomes from chronic hepatitis C? A population-level analysis. J Viral Hepat 2018;25:930-8.
42. Ikeda K, Marusawa H, Osaki Y, Nakamura T, Kitajima N, et al. Antibody to hepatitis B core antigen and risk for hepatitis C-related hepatocellular carcinoma: a prospective study. Ann Intern Med 2007;146:649-56.
43. Coppola N, Onorato L, Sagnelli C, Sagnelli E, Angelillo IF. Association between anti-HBc positivity and hepatocellular carcinoma in HBsAg-negative subjects with chronic liver disease: a meta-analysis. Medicine (Baltimore) 2016;95:e4311.
44. Stroffolini T, Almasio PL, Persico M, Bollani S, Benvegnù L, et al; Italian Association of the Study of the Liver Disease (AISF). Lack of correlation between serum anti-HBcore detectability and hepatocellular carcinoma in patients with HCV-related cirrhosis. Am J Gastroenterol 2008;103:1966-72.
45. Kitab B, Ezzikouri S, Alaoui R, Nadir S, Badre W, et al. Occult HBV infection in Morocco: from chronic hepatitis to hepatocellular carcinoma. Liver Int 2014;34:e144-50.
46. Kim JH, Kim MN, Han KH, Kim SU. Clinical application of transient elastography in patients with chronic viral hepatitis receiving antiviral treatment. Liver Int 2015;35:1103-15.
47. Jung KS, Kim SU, Ahn SH, Park YN, Kim DY, et al. Risk assessment of hepatitis B virus-related hepatocellular carcinoma development using liver stiffness measurement (FibroScan). Hepatology 2011;53:885-94.
48. Huang R, Jiang N, Yang R, Geng X, Lin J, et al. Fibroscan improves the diagnosis sensitivity of liver fibrosis in patients with chronic hepatitis B. Exp Ther Med 2016;11:1673-7.
49. Yuen MF, Tanaka Y, Fong DY, Fung J, Wong DK, et al. Independent risk factors and predictive score for the development of hepatocellular carcinoma in chronic hepatitis B. J Hepatol 2009;50:80-8.
50. Wong GL, Chan HL, Wong CK, Leung C, Chan CY, et al. Liver stiffness-based optimization of hepatocellular carcinoma risk score in patients with chronic hepatitis B. J Hepatol 2014;60:339-45.
51. Papatheodoridis G, Dalekos G, Sypsa V, Yurdaydin C, Buti M, et al. PAGE-B predicts the risk of developing hepatocellular carcinoma in Caucasians with chronic hepatitis B on 5-year antiviral therapy. J Hepatol 2016;64:800-6.
52. Jung KS, Kim SU, Song K, Park JY, Kim DY, et al. Validation of hepatitis B virus-related hepatocellular carcinoma prediction models in the era of antiviral therapy. Hepatology 2015;62:1757-66.
53. Sung WK, Zheng H, Li S, Chen R, Liu X, et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet 2012;44:765-9.
54. Wang M, Xi D, Ning Q. Virus-induced hepatocellular carcinoma with special emphasis on HBV. Hepatol Int 2017;11:171-80.
55. Wu S, Kanda T, Nakamoto S, Jiang X, Nakamura M, et al. Cooperative effects of hepatitis B virus and TNF may play important roles in the activation of metabolic pathways through the activation of NF-κB. Int J Mol Med 2016;38:475-81.
56. Xie Y. Hepatitis B virus-associated hepatocellular carcinoma. In: Cai Q, Yuan Z, Lan K, editors. Infectious Agents Associated Cancers: Epidemiology and Molecular Biology. Singapore: Springer; 2017. pp. 11-21.
57. Wang W, Bian H, Li F, Li X, Zhang D, et al. HBeAg induces the expression of macrophage miR-155 to accelerate liver injury via promoting production of inflammatory cytokines. Cell Mol Life Sci 2018;75:2627-41.
58. Gao P, Wong CC, Tung EK, Lee JM, Wong CM, et al. Deregulation of microRNA expression occurs early and accumulates in early stages of HBV-associated multistep hepatocarcinogenesis. J Hepatol 2011;54:1177-84.
59. Tian Y, Ou JH. Genetic and epigenetic alterations in hepatitis B virus-associated hepatocellular carcinoma. Virol Sin 2015;30:85-91.
60. Kanda T, Wu S, Sasaki R, Nakamura M, Haga Y, et al. HBV core protein enhances cytokine production. Diseases 2015;3:213-20.
61. Kim CM, Koike K, Saito I, Miyamura T, Jay G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 1991;351:317-20.
62. Kanda T, Goto T, Hirotsu Y, Moriyama M, Omata M. Molecular mechanisms driving progression of liver cirrhosis towards hepatocellular carcinoma in chronic hepatitis B and C infections: a review. Int J Mol Sci 2019;20:1358.
63. WHO. Global health sector strategy on viral hepatitis 2016-2021. Geneva, Switzerland: WHO; 2016.
64. Ward JW, Hinman AR. What is needed to eliminate hepatitis b virus and hepatitis C virus as global health threats. Gastroenterology 2019;156:297-310.
65. Poovorawan Y, Chongsrisawat V, Theamboonlers A, Bock HL, Leyssen M, et al. Persistence of antibodies and immune memory to hepatitis B vaccine 20 years after infant vaccination in Thailand. Vaccine 2010;28:730-6.
66. WHO. Global hepatitis report, 2017. Geneva, Switzerland: WHO; 2017.
67. European Association for the Study of the Liver. Electronic address: easloffice@easloffice.eu., European Association for the Study of the Liver. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J Hepatol 2017;67:370-98.
68. Chiang CJ, Yang YW, Chen JD, You SL, Yang HI, et al. Significant reduction in end-stage liver diseases burden through the national viral hepatitis therapy program in Taiwan. Hepatology 2015;61:1154-62.
69. Papatheodoridis GV, Idilman R, Dalekos GN, Buti M, Chi H, et al. The risk of hepatocellular carcinoma decreases after the first 5 years of entecavir or tenofovir in Caucasians with chronic hepatitis B. Hepatology 2017;66:1444-53.
70. Li M, Lv T, Wu S, Wei W, Wu X, et al. Tenofovir versus entecavir in lowering the risk of hepatocellular carcinoma development in patients with chronic hepatitis B: a critical systematic review and meta-analysis. Hepatol Int 2020;14:105-14.
71. Goh MJ, Sinn DH, Kim S, Woo SY, Cho H, et al. Statin use and the risk of hepatocellular carcinoma in patients with chronic hepatitis B. Hepatology 2020;71:2023-32.
72. Singh S, Singh PP, Singh AG, Murad MH, Sanchez W. Statins are associated with a reduced risk of hepatocellular cancer: a systematic review and meta-analysis. Gastroenterology 2013;144:323-32.
73. Jiang P, Mukthavaram R, Chao Y, Nomura N, Bharati IS, et al. In vitro and in vivo anticancer effects of mevalonate pathway modulation on human cancer cells. Br J Cancer 2014;111:1562-71.
74. Eberlein M, Heusinger-Ribeiro J, Goppelt-Struebe M. Rho-dependent inhibition of the induction of connective tissue growth factor (CTGF) by HMG CoA reductase inhibitors (statins). Br J Pharmacol 2001;133:1172-80.
75. Tsan YT, Lee CH, Wang JD, Chen PC. Statins and the risk of hepatocellular carcinoma in patients with hepatitis B virus infection. J Clin Oncol 2012;30:623-30.
76. Lee TY, Hsu YC, Tseng HC, Yu SH, Lin JT, et al. Association of daily aspirin therapy with risk of hepatocellular carcinoma in patients with chronic hepatitis B. JAMA Intern Med 2019;179:633-40.
77. Kern MA, Schubert D, Sahi D, Schöneweiss MM, Moll I, et al. Proapoptotic and antiproliferative potential of selective cyclooxygenase-2 inhibitors in human liver tumor cells. Hepatology 2002;36:885-94.
78. Sitia G, Aiolfi R, Di Lucia P, Mainetti M, Fiocchi A, et al. Antiplatelet therapy prevents hepatocellular carcinoma and improves survival in a mouse model of chronic hepatitis B. Proc Natl Acad Sci U S A 2012;109:E2165-72.
79. Calvisi DF. Inhibition of hepatitis B virus-associated liver cancer by antiplatelet therapy: a revolution in hepatocellular carcinoma prevention? Hepatology 2013;57:848-50.
80. Mancebo A, González-Diéguez ML, Cadahía V, Varela M, Pérez R, et al. Annual incidence of hepatocellular carcinoma among patients with alcoholic cirrhosis and identification of risk groups. Clin Gastroenterol Hepatol 2013;11:95-101.
81. Mahale P, Torres HA, Kramer JR, Hwang LY, Li R, et al. Hepatitis C virus infection and the risk of cancer among elderly US adults: a registry-based case-control study. Cancer 2017;123:1202-11.
82. Chang KC, Wu YY, Hung CH, Lu SN, Lee CM, et al. Clinical-guide risk prediction of hepatocellular carcinoma development in chronic hepatitis C patients after interferon-based therapy. Br J Cancer 2013;109:2481-8.
83. Kruse RL, Kramer JR, Tyson GL, Duan Z, Chen L, et al. Clinical outcomes of hepatitis B virus coinfection in a United States cohort of hepatitis C virus-infected patients. Hepatology 2014;60:1871-8.
84. Huang CF, Yeh ML, Huang CI, Lin YJ, Tsai PC, et al. Risk of hepatitis C virus related hepatocellular carcinoma between subjects with spontaneous and treatment-induced viral clearance. Oncotarget 2017;8:43925-33.
85. Graham CS, Baden LR, Yu E, Mrus JM, Carnie J, et al. Influence of human immunodeficiency virus infection on the course of hepatitis C virus infection: a meta-analysis. Clin Infect Dis 2001;33:562-9.
86. Giordano TP, Kramer JR, Souchek J, Richardson P, El-Serag HB. Cirrhosis and hepatocellular carcinoma in HIV-infected veterans with and without the hepatitis C virus: a cohort study, 1992-2001. Arch Intern Med 2004;164:2349-54.
87. López-Diéguez M, Montes ML, Pascual-Pareja JF, Quereda C, Von Wichmann MA, et al; GESIDA 37/03-FIPSE 36465/03-NEAT IG5 Study Group. The natural history of liver cirrhosis in HIV-hepatitis C virus-coinfected patients. AIDS 2011;25:899-904.
88. Tovo CV, Becker SC, Almeida PR, Galperim B, Chaves S. Progression of liver fibrosis in monoinfected patients by hepatitis C virus and coinfected by HCV and human immunodeficiency virus. Arq Gastroenterol 2013;50:19-22.
89. Puoti M, Prestini K, Putzolu V, Zanini B, Baiguera C, et al. HIV/HCV co-infection: natural history. J Biol Regul Homeost Agents 2003;17:144-6.
90. Salmon-Ceron D, Nahon P, Layese R, Bourcier V, Sogni P, et al; ANRS CO12 CirVir and ANRS CO13 HEPAVIH study groups. Human Immunodeficiency Virus/Hepatitis C Virus (HCV) Co-infected Patients With Cirrhosis Are No Longer at Higher Risk for Hepatocellular Carcinoma or End-Stage Liver Disease as Compared to HCV Mono-infected Patients. Hepatology 2019;70:939-54.
91. Konstantinou D, Deutsch M. The spectrum of HBV/HCV coinfection: epidemiology, clinical characteristics, viralinteractions and management. Ann Gastroenterol 2015;28:221-8.
92. Lee LP, Dai CY, Chuang WL, Chang WY, Hou NJ, et al. Comparison of liver histopathology between chronic hepatitis C patients and chronic hepatitis B and C-coinfected patients. J Gastroenterol Hepatol 2007;22:515-7.
93. Chiaramonte M, Stroffolini T, Vian A, Stazi MA, Floreani A, et al. Rate of incidence of hepatocellular carcinoma in patients with compensated viral cirrhosis. Cancer 1999;85:2132-7.
94. Kanwal F, Kramer JR, Ilyas J, Duan Z, El-Serag HB. HCV genotype 3 is associated with an increased risk of cirrhosis and hepatocellular cancer in a national sample of U.S. Veterans with HCV. Hepatology 2014;60:98-105.
95. Raimondi S, Bruno S, Mondelli MU, Maisonneuve P. Hepatitis C virus genotype 1b as a risk factor for hepatocellular carcinoma development: a meta-analysis. J Hepatol 2009;50:1142-54.
96. Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. Lancet 1997;349:825-32.
97. Hutchinson SJ, Bird SM, Goldberg DJ. Influence of alcohol on the progression of hepatitis C virus infection: a meta-analysis. Clin Gastroenterol Hepatol 2005;3:1150-9.
98. Donato F, Tagger A, Gelatti U, Parrinello G, Boffetta P, et al. Alcohol and hepatocellular carcinoma: the effect of lifetime intake and hepatitis virus infections in men and women. Am J Epidemiol 2002;155:323-31.
99. Tagger A, Donato F, Ribero ML, Chiesa R, Portera G, et al; Brescia HCC Study. Case-control study on hepatitis C virus (HCV) as a risk factor for hepatocellular carcinoma: the role of HCV genotypes and the synergism with hepatitis B virus and alcohol. Int J Cancer 1999;81:695-9.
100. Monto A, Patel K, Bostrom A, Pianko S, Pockros P, et al. Risks of a range of alcohol intake on hepatitis C-related fibrosis. Hepatology 2004;39:826-34.
101. Vandenbulcke H, Moreno C, Colle I, Knebel JF, Francque S, et al. Alcohol intake increases the risk of HCC in hepatitis C virus-related compensated cirrhosis: a prospective study. J Hepatol 2016;65:543-51.
102. Dyal HK, Aguilar M, Bartos G, Holt EW, Bhuket T, et al. Diabetes mellitus increases risk of hepatocellular carcinoma in chronic hepatitis C virus patients: a systematic review. Dig Dis Sci 2016;61:636-45.
103. Huang TS, Lin CL, Lu MJ, Yeh CT, Liang KH, et al. Diabetes, hepatocellular carcinoma, and mortality in hepatitis C-infected patients: a population-based cohort study. J Gastroenterol Hepatol 2017;32:1355-62.
104. Adinolfi LE, Rinaldi L, Guerrera B, Restivo L, Marrone A, et al. NAFLD and NASH in HCV infection: prevalence and significance in hepatic and extrahepatic manifestations. Int J Mol Sci 2016;17:803.
105. Castéra L, Hézode C, Roudot-Thoraval F, Bastie A, Zafrani ES, et al. Worsening of steatosis is an independent factor of fibrosis progression in untreated patients with chronic hepatitis C and paired liver biopsies. Gut 2003;52:288-92.
106. Monto A, Alonzo J, Watson JJ, Grunfeld C, Wright TL. Steatosis in chronic hepatitis C: relative contributions of obesity, diabetes mellitus, and alcohol. Hepatology 2002;36:729-36.
107. Cross TJ, Quaglia A, Hughes S, Joshi D, Harrison PM. The impact of hepatic steatosis on the natural history of chronic hepatitis C infection. J Viral Hepat 2009;16:492-9.
108. Lonardo A, Adinolfi LE, Restivo L, Ballestri S, Romagnoli D, et al. Pathogenesis and significance of hepatitis C virus steatosis: an update on survival strategy of a successful pathogen. World J Gastroenterol 2014;20:7089-103.
109. Salomao M, Yu WM, Brown RS Jr, Emond JC, Lefkowitch JH. Steatohepatitic hepatocellular carcinoma (SH-HCC): a distinctive histological variant of HCC in hepatitis C virus-related cirrhosis with associated NAFLD/NASH. Am J Surg Pathol 2010;34:1630-6.
110. Tanaka A, Uegaki S, Kurihara H, Aida K, Mikami M, et al. Hepatic steatosis as a possible risk factor for the development of hepatocellular carcinoma after eradication of hepatitis C virus with antiviral therapy in patients with chronic hepatitis C. World J Gastroenterol 2007;13:5180-7.
111. Pekow JR, Bhan AK, Zheng H, Chung RT. Hepatic steatosis is associated with increased frequency of hepatocellular carcinoma in patients with hepatitis C-related cirrhosis. Cancer 2007;109:2490-6.
112. Kumar D, Farrell GC, Kench J, George J. Hepatic steatosis and the risk of hepatocellular carcinoma in chronic hepatitis C. J Gastroenterol Hepatol 2005;20:1395-400.
113. Ohata K, Hamasaki K, Toriyama K, Matsumoto K, Saeki A, et al. Hepatic steatosis is a risk factor for hepatocellular carcinoma in patients with chronic hepatitis C virus infection. Cancer 2003;97:3036-43.
114. Rao H, Wu E, Fu S, Yang M, Feng B, et al. The higher prevalence of truncal obesity and diabetes in American than Chinese patients with chronic hepatitis C might contribute to more rapid progression to advanced liver disease. Aliment Pharmacol Ther 2017;46:731-40.
115. Singal AG, Mukherjee A, Elmunzer BJ, Higgins PD, Lok AS, et al. Machine learning algorithms outperform conventional regression models in predicting development of hepatocellular carcinoma. Am J Gastroenterol 2013;108:1723-30.
116. El-Serag HB, Kanwal F, Davila JA, Kramer J, Richardson P. A new laboratory-based algorithm to predict development of hepatocellular carcinoma in patients with hepatitis C and cirrhosis. Gastroenterology 2014;146:1249-55.e1.
117. Ikeda M, Fujiyama S, Tanaka M, Sata M, Ide T, et al. Risk factors for development of hepatocellular carcinoma in patients with chronic hepatitis C after sustained response to interferon. J Gastroenterol 2005;40:148-56.
118. Hoshida Y, Fuchs BC, Bardeesy N, Baumert TF, Chung RT. Pathogenesis and prevention of hepatitis C virus-induced hepatocellular carcinoma. J Hepatol 2014;61:S79-90.
120. Aroucha DC, do Carmo RF, Moura P, Silva JL, Vasconcelos LR, et al. High tumor necrosis factor-α/interleukin-10 ratio is associated with hepatocellular carcinoma in patients with chronic hepatitis C. Cytokine 2013;62:421-5.
121. Braconi C, Valeri N, Gasparini P, Huang N, Taccioli C, et al. Hepatitis C virus proteins modulate microRNA expression and chemosensitivity in malignant hepatocytes. Clin Cancer Res 2010;16:957-66.
122. Zhang Y, Wei W, Cheng N, Wang K, Li B, et al. Hepatitis C virus-induced up-regulation of microRNA-155 promotes hepatocarcinogenesis by activating Wnt signaling. Hepatology 2012;56:1631-40.
123. Hou W, Bonkovsky HL. Non-coding RNAs in hepatitis C-induced hepatocellular carcinoma: dysregulation and implications for early detection, diagnosis and therapy. World J Gastroenterol 2013;19:7836-45.
124. Plissonnier ML, Herzog K, Levrero M, Zeisel MB. Non-coding RNAs and hepatitis C virus-induced hepatocellular carcinoma. Viruses 2018;10:591.
125. Ray RB, Lagging LM, Meyer K, Ray R. Hepatitis C virus core protein cooperates with ras and transforms primary rat embryo fibroblasts to tumorigenic phenotype. J Virol 1996;70:4438-43.
126. Kanda T, Yokosuka O, Omata M. Hepatitis C virus and hepatocellular carcinoma. Biology (Basel) 2013;2:304-16.
127. WHO. Global Health Estimates 2016: Deaths by Cause, Age, Sex, by Country and by Region, 2000-2016.2018; Available from: http://origin.who.int/healthinfo/global_burden_disease/estimates/en/. [Last accessed on 22 Jul 2017].
128. Edlin BR, Winkelstein ER. Can hepatitis C be eradicated in the United States? Antiviral Res 2014;110:79-93.
129. European Association for the Study of the Liver, European Association for the Study of the Liver. EASL Recommendations on Treatment of Hepatitis C 2018. J Hepatol 2018;69:461-511.
130. Cooke GS, Andrieux-meyer I, Applegate TL, Atun R, Burry JR, et al. Accelerating the elimination of viral hepatitis: a lancet gastroenterology & hepatology commission. Lancet Gastroenterol Hepatol 2019;4:135-84.
131. Morgan RL, Baack B, Smith BD, Yartel A, Pitasi M, et al. Eradication of hepatitis C virus infection and the development of hepatocellular carcinoma: a meta-analysis of observational studies. Ann Intern Med 2013;158:329-37.
132. Kanwal F, Kramer J, Asch SM, Chayanupatkul M, Cao Y, et al. Risk of hepatocellular cancer in HCV patients treated with direct-acting antiviral agents. Gastroenterology 2017;153:996-1005.e1.
133. Backus LI, Belperio PS, Shahoumian TA, Mole LA. Impact of sustained virologic response with direct-acting antiviral treatment on mortality in patients with advanced liver disease. Hepatology 2019;69:487-97.
134. Calvaruso V, Cabibbo G, Cacciola I, Petta S, Madonia S, et al; Rete Sicilia Selezione Terapia-HCV (RESIST-HCV). Incidence of hepatocellular carcinoma in patients with HCV-associated cirrhosis treated with direct-acting antiviral agents. Gastroenterology 2018;155:411-21.e4.
135. Conti F, Buonfiglioli F, Scuteri A, Crespi C, Bolondi L, et al. Early occurrence and recurrence of hepatocellular carcinoma in HCV-related cirrhosis treated with direct-acting antivirals. J Hepatol 2016;65:727-33.
136. Colombo M, Boccaccio V. HCV therapy and risk of liver cancer recurrence: who to treat? Nat Rev Gastroenterol Hepatol 2018;15:392-3.
137. Reig M, Mariño Z, Perelló C, Iñarrairaegui M, Ribeiro A, et al. Unexpected high rate of early tumor recurrence in patients with HCV-related HCC undergoing interferon-free therapy. J Hepatol 2016;65:719-26.
138. Sangiovanni A, Alimenti E, Gattai R, Filomia R, Parente E, et al. Undefined/non-malignant hepatic nodules are associated with early occurrence of HCC in DAA-treated patients with HCV-related cirrhosis. J Hepatol 2020; doi: 10.1016/j.jhep.2020.03.030.
140. Sasaki A, Kai S, Iwashita Y, Hirano S, Ohta M, et al. Microsatellite distribution and indication for locoregional therapy in small hepatocellular carcinoma. Cancer 2005;103:299-306.
141. Lens S, Alvarado-Tapias E, Mariño Z, Londoño MC, LLop E, et al. Effects of all-oral anti-viral therapy on HVPG and systemic hemodynamics in patients with hepatitis C Virus-associated cirrhosis. Gastroenterology 2017;153:1273-83.e1.
142. Simon TG, Ma Y, Ludvigsson JF, Chong DQ, Giovannucci EL, et al. Association between aspirin use and risk of hepatocellular carcinoma. JAMA Oncol 2018;4:1683-90.
144. Chen HY, Shen DT, Ji DZ, Han PC, Zhang WM, et al. Prevalence and burden of hepatitis D virus infection in the global population: a systematic review and meta-analysis. Gut 2019;68:512-21.
145. Choi SH, Jeong SH, Hwang SB. Large hepatitis delta antigen modulates transforming growth factor-beta signaling cascades: implication of hepatitis delta virus-induced liver fibrosis. Gastroenterology 2007;132:343-57.
146. Park CY, Oh SH, Kang SM, Lim YS, Hwang SB. Hepatitis delta virus large antigen sensitizes to TNF-alpha-induced NF-kappaB signaling. Mol Cells 2009;28:49-55.
147. Williams V, Brichler S, Khan E, Chami M, Dény P, et al. Large hepatitis delta antigen activates STAT-3 and NF-κB via oxidative stress. J Viral Hepat 2012;19:744-53.
148. Fattovich G, Giustina G, Christensen E, Pantalena M, Zagni I, et al. Influence of hepatitis delta virus infection on morbidity and mortality in compensated cirrhosis type B. The European Concerted Action on Viral Hepatitis (Eurohep). Gut 2000;46:420-6.
149. Ji J, Sundquist K, Sundquist J. A population-based study of hepatitis D virus as potential risk factor for hepatocellular carcinoma. J Natl Cancer Inst 2012;104:790-2.
150. Kushner T, Serper M, Kaplan DE. Delta hepatitis within the Veterans Affairs medical system in the United States: prevalence, risk factors, and outcomes. J Hepatol 2015;63:586-92.
151. Romeo R, Del Ninno E, Rumi M, Russo A, Sangiovanni A, et al. A 28-year study of the course of hepatitis Delta infection: a risk factor for cirrhosis and hepatocellular carcinoma. Gastroenterology 2009;136:1629-38.
152. Romeo R, Foglieni B, Casazza G, Spreafico M, Colombo M, et al. High serum levels of HDV RNA are predictors of cirrhosis and liver cancer in patients with chronic hepatitis delta. PLoS One 2014;9:e92062.
153. Wranke A, Pinheiro Borzacov LM, Parana R, Lobato C, Hamid S, et al; Hepatitis Delta International Network. Clinical and virological heterogeneity of hepatitis delta in different regions world-wide: The Hepatitis Delta International Network (HDIN). Liver Int 2018;38:842-50.
154. Niro GA, Smedile A, Ippolito AM, Ciancio A, Fontana R, et al. Outcome of chronic delta hepatitis in Italy: a long-term cohort study. J Hepatol 2010;53:834-40.
155. Gheorghe L, Iacob S, Simionov I, Vadan R, Gheorghe C, et al. Natural history of compensated viral B and D cirrhosis. Rom J Gastroenterol 2005;14:329-35.
156. Buti M, Homs M, Rodriguez-Frias F, Funalleras G, Jardí R, et al. Clinical outcome of acute and chronic hepatitis delta over time: a long-term follow-up study. J Viral Hepat 2011;18:434-42.
157. Béguelin C, Moradpour D, Sahli R, Suter-Riniker F, Lüthi A, et al; Swiss HIV Cohort Study. Hepatitis delta-associated mortality in HIV/HBV-coinfected patients. J Hepatol 2017;66:297-303.
159. Wedemeyer H, Yurdaydìn C, Dalekos GN, Erhardt A, Çakaloğlu Y, et al; HIDIT Study Group. Peginterferon plus adefovir versus either drug alone for hepatitis delta. N Engl J Med 2011;364:322-31.
160. Kabaçam G, Dalekos GN, Çakaloğlu Y, Zachou K, Bock T, et al. Pegylated interferon-based treatment in patients with advanced liver disease due to chronic delta hepatitis. Turk J Gastroenterol 2012;23:560-8.
161. Heller T, Rotman Y, Koh C, Clark S, Haynes-Williams V, et al. Long-term therapy of chronic delta hepatitis with peginterferon alfa. Aliment Pharmacol Ther 2014;40:93-104.
162. Farci P, Roskams T, Chessa L, Peddis G, Mazzoleni AP, et al. Long-term benefit of interferon alpha therapy of chronic hepatitis D: regression of advanced hepatic fibrosis. Gastroenterology 2004;126:1740-9.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Alqahtani SA, Colombo M. Viral hepatitis as a risk factor for the development of hepatocellular carcinoma. Hepatoma Res 2020;6:58. http://dx.doi.org/10.20517/2394-5079.2020.49
AMA Style
Alqahtani SA, Colombo M. Viral hepatitis as a risk factor for the development of hepatocellular carcinoma. Hepatoma Research. 2020; 6: 58. http://dx.doi.org/10.20517/2394-5079.2020.49
Chicago/Turabian Style
Alqahtani, Saleh A., Massimo Colombo. 2020. "Viral hepatitis as a risk factor for the development of hepatocellular carcinoma" Hepatoma Research. 6: 58. http://dx.doi.org/10.20517/2394-5079.2020.49
ACS Style
Alqahtani, SA.; Colombo M. Viral hepatitis as a risk factor for the development of hepatocellular carcinoma. Hepatoma. Res. 2020, 6, 58. http://dx.doi.org/10.20517/2394-5079.2020.49
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 51 clicks
Cite This Article 51 clicks

 Like This Article 19
likes
Like This Article 19
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.