
Mechanisms and immunotherapies of HBV- and NAFLD-related hepatocellular carcinoma
 0
0Abstract
Hepatitis B virus (HBV) infection remains the most important risk factor for hepatocellular carcinoma (HCC) worldwide and nonalcoholic fatty liver disease (NAFLD) has developed as major etiology of chronic liver diseases, cirrhosis and eventually HCC in the last decades. Although nucleos(t)ide analogs are recommended as the first-line drug for patients with chronic hepatitis B, incomplete eradication of HBV serves as an obstacle for effective cure of chronic hepatitis B and even HCC. NAFLD refers to a spectrum of hepatic metabolic disorders, compromised with multi-system diseases. Considering the specificity of hepatocytes and enrichment of immune cells in liver, this review aims to summarize the mechanisms of direct pro-tumorigenesis to hepatocytes induced by HBV infection and abnormal lipid metabolism, and indirect oncogenic processes mediated by immune cells. We also discuss similarities and differences of immune cells between HBV- and NAFLD-HCC and finally focus on the novel immunotherapies concerning preclinical and clinical studies for liver cancer.
Keywords
Introduction
Liver cancer is one of the most common causes of cancer-related deaths worldwide, with an estimated of 1.1 million new cases to be diagnosed in 10 years (GLOBOCAN, 2018). Hepatocellular carcinoma (HCC) accounts for nearly 80% of primary liver cancer and the occurrence is globally gender and geography biased. The estimated age-standardized incidence rates in men are mostly 3 times than that of women (13.9/1,000,000 vs. 4.9/1,000,000) in 2018. The high rates in Pacific Asia, West and Central Africa have persisted for decades, although declining trends have been observed in China and Japan in the past two decades due to therapy advancement and control of etiology. In contrast, incidence and mortality rates in North America have been increasing since 1978, for which the main reasons are cumulative emergence of new risk factors and lack of efficient therapy strategies.
HCC is prominently induced by chronic hepatitis B virus (HBV)/hepatitis C virus (HCV) infection, alcoholic or nonalcoholic fatty liver disease, aflatoxin intake and parasitic infection, the incidence of which are related with geographic variance. About 80% of HCC arises from persistent HBV infection in Asia-Pacific and Africa countries, while non-alcoholic fatty liver disease (NAFLD) is the major contributor in North America, followed by HCV infection. The direct-acting antiviral therapy with high rates of safety and sustained virologic response (> 95%), is considered as the contributing factor for eliminating HCV[1,2], which helps decrease the pathogenesis of HCC. However, HBV-associated HCC remains to be a global problem, because current nucleos(t)ide analogue therapy functionally reduces HBsAg level and suppresses HBV DNA but fails to control remission[3,4]. Since obesity has emerged as a new global burden, NAFLD, which is one of the most common metabolic syndrome manifesting in liver with estimated prevalence of 20%~30% in many countries, will rank as the leading cause of HCC in future decades[5-9].
Although patients diagnosed with early stage of HCC undergo surgical resection or radiofrequency ablation for curative intent, up to 70% of them are susceptible to recurrence within 5 years[10]. During the last decades, conventional chemotherapy and combination of transarterial chemoembolization and sorafenib have been developed for patients at intermediate and advanced stages, however, therapy efficacy and adverse effects should be further assessed. Although sorafenib has been approved for treatment of HCC, the tyrosine kinase inhibitors-induced adverse events and drug resistance still remain an issue during clinical management[11]. Therefore, more novel target agents and immunotherapies are being investigated and incorporated to achieve better outcomes[12,13]. Here, this review mainly discusses the current progress in molecular and cellular mechanisms of HBV- and NAFLD-related HCC respectively, specifically the immune responses in liver, along with current immunotherapy options.
Direct pro-oncogenic mechanisms
HBV
Genomic features are significantly different in the group of HBV-HCC compared with non-infected patients. These results suggest that HBV-related HCC use alternative mechanisms for tumorigenesis to some extent. A high frequency of p53 inactivation and stem cell genes overexpression provide a potential pathogenic link between impaired cell reprogramming and HBV infection. Notably, these observations have clinical implication since TP53 mutations were associated with poor prognosis only in HBV-related tumors.
Virally oncogenic proteins
HBV genome contains four overlapping open reading frames, including preS1/preS2/S, preCore/Core, X and Pol, encoding viral proteins. Among them, hepatitis B X protein (HBx) is the most important oncogenic protein. Mutated or deleted HBx is frequently detected in HCC and plays critical role in liver carcinogenesis[14-16]. Although it is still debated, a growing body of evidence suggests that both wild type HBx and truncated HBx promote tumorigenesis by abrogating cell-cycle arrest and apoptosis inhibition[17-19]. HBx promotes HCC progression mainly through interaction with host factors including proteins and non-coding RNAs. HBx mutants interact with Bcl-2 and farnesoid X receptor leading to enhanced carcinogenesis[20-22]. HBx-cortactin (CTTN) interaction promotes HCC progression by up-regulating expression of CREB1 and its downstream targets, cyclin D1 and MMP9[23]. As a major conserved cellular pathway that controls critical cell processes, the ubiquitin proteasome system is often hijacked by HBx, leading to dysregulated ubiquitination. HBx stabilizes critical transcriptional oncoproteins Myc and PAX8 via blocking Skp2-mediated ubiquitination[24,25]. Silencing lncRNA-MALAT1/miR-124 axis or miR-5188-FOXO1/β-catenin-c-Jun feedback loop significantly block HBx-mediated upregulating stemness markers and reprogramming proteins in HCC[26,27]. Besides, HBx targets genes at transcriptional level. HBx complexed with HDAC1 binds to glycogen synthase 2 (GYS2) and transcriptionally inhibit the expression[28][Figure 1].
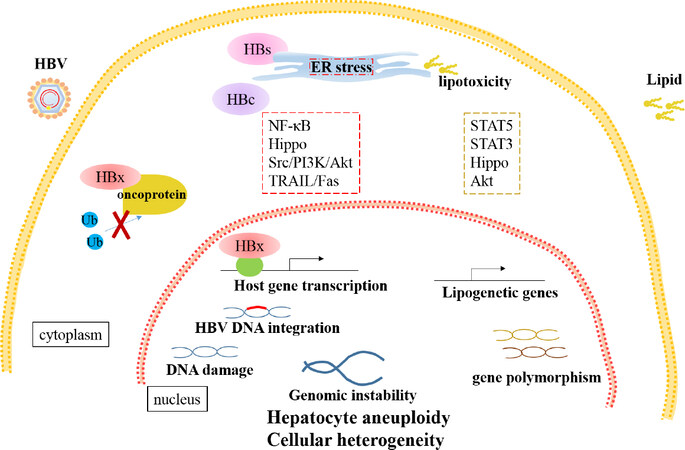
Figure 1. Direct oncogenic mechanisms of HBV- and NAFLD- HCC. HBV-encoded proteins lead to cellular heterogeneity via multiple manners. HBx stabilizes oncoproteins by suppressing ubiquitination or regulates transcription of host genes through transactivation. Cytoplasmic accumulation of HBs induces ER stress and thereby initiates cellular hippo pathways. Besides ER response, NF-κB, Src/PI3K/Akt and TRAIL/Fas pathways are involved in HBc-induced abnormal proliferation and metabolism of hepatocytes. Furthermore, HBV DNA integration and viral protein result in DNA damage and genomic instability, which drive cellular heterogeneity. Excessive lipid accumulation stimulates ER stress and dysregulates apoptosis and fibrogenesis via STAT3 and hippo pathways. The STAT5 and Akt pathways are involved in upregulation of the lipogenetic genes and downstream targets. Moreover, gene polymorphism is significantly related with NAFLD progression. HBV: hepatitis B virus; NAFLD: nonalcoholic fatty liver disease; HCC: hepatocellular carcinoma; ER: endoplasmic reticulum
As the most abundant proteins, HBV surface (HBs) proteins are composed of three proteins, the large (L), middle (M), and small (S) HBs, encoded by the preS1/preS2/S gene respectively[29-31]. Particular deletion mutants in preS2 have been reported to be associated with progression of chronic liver diseases. Mutated HBs accumulated in hepatocytes induce endoplasmic reticulum (ER) stress and initiate multiple cellular signal pathways, leading to cell growth advantages[32,33]. Intrahepatic LHBs induce DNA damage and up-regulation of Plk1 to provoke hyperploidy, which disrupts the genomic stability of host cells[34]. Pre-S2 deletion mutant also promote carcinogenesis by up-regulating expression of various oncogenes, including hTERT, TAZ in hippo pathways, and GPC3[35-38][Figure 1].
As the major capsid protein and important viral replication mediator, HB core-related antigen (HBcrAg) is reported to be an independent risk factor for HCC development[39]. Mechanistically, hepatitis B core protein (HBc) function as an immunogen as well as an important mediator of hepatocarcinogenesis via several mechanisms. HBc mutations P5T/H/L stimulate ER response, further increasing production of radical oxidative species (ROS) and activating NF-κB signaling pathway, which directly promote infected hepatocytes to malignant transformation. Furthermore, HBc promotes proliferation, glycolysis and amino acid metabolism or suppress apoptosis via regulating Src/PI3K/Akt pathway, blocking TRAIL/Fas pathway or expression of p53 in transcriptional/post-transcriptional manner[40-44][Figure 1].
HBV DNA integration
HBV DNA integration occurs early once HBV entering into hepatocytes and remains stable throughout tumor progression, which explains the existence of monoclonal or polyclonal origins of HCC from the view of virus infection[45,46]. HBV DNA susceptibly integrates into rare fragile sites or functional genomic regions, which are proximate to protein coding or non-coding genes. Functionally, the protein-coding genes are deeply associated with tumorigenesis and the non-coding genes are involved in telomere maintenance, protein modification processes, and chromosome localization[47-49]. For example, integration drives mutation in tumor suppressor gene ZNF717 and over-expression of critical oncogene c-MYC. Meanwhile, integration into intron of cyclin A2 generates a pseudo-exon forming a chimeric fusion with CCNA2, which encodes protein promoting cell cycle progression[46,50,51]. Moreover, HBV DNA integration is closely correlated with gender-bias and recurrence of HCC. Androgen receptor, but not estrogen receptor, enhances transcription responsive of TERT promoter with HBV integration to sex hormones via hepatocyte nuclear factor 4 alpha (HNF4α)- dependent manner[52]. Multiple studies have identified fragile integration sites more in non-cancerous tissue than those in neoplastic tissues, which implies tumor recurrence[48,53].
Genomic instability
In infected hepatocytes, both HBV DNA integration and viral proteins lead to genomic instability. Sequencing analysis of 373 liver cancer samples demonstrated ultra-high structural instability and preserved un-methylation in HBV integrated regions[54]. HBx directly induces genomic instability via inhibiting mono-ubiquitylation of an evolutionarily conserved E3 ligase and impairing homologous recombination, which contribute to tumorigenesis[55]. LHBs are the major viral protein leading to genomic instability. By inducing DNA damage and G2/M checkpoint failure, LHBs promote formation of hepatocyte aneuploidy and further self-propagating cycles of chromosomal instability, which drives cellular heterogeneity and clonal cancer evolution[56,57].
NAFLD
NAFLD and its complication nonalcoholic steatohepatitis (NASH) are becoming the leading cause of HCC. Multiple pathways, such as abnormal metabolism, dysbiosis of gut microbiota and dysregulated immune responses, are involved in NAFLD initiated hepatocarcinogenesis and have been well summarized recently[58]. Here, we mainly focus on dysregulated pathway mediated by lipid accumulation which is fundamental in progression of NAFLD related HCC. The abnormal intrahepatic lipid metabolism invokes insulin resistance, alteration of signaling pathways and oncogenes, followed by inflammation, fibrogenesis and hepatocarcinogenesis. In clinical samples, level of p-STAT5 is positively correlated with expression of sterol regulatory element binding protein-1 (SREBP1) and further in vivo and in vitro essays demonstrate that mTORC1 interacts and phosphorylates STAT5 to upregulate expression of lipogenetic genes, including SREBP1, fatty acid synthase (FASN) and acetyl-CoA carboxylase (ACC)[59]. Both oxidative and ER stress caused by chronic lipotoxicity play critical roles in NAFLD-HCC. Oxidized LDL (oxLDL) uptake triggers CEBPβ expression to directly upregulate Nogo-B, an ER-residential protein, and promote lipophagy leading to lysophosphatidic acid-enhanced YAP oncogenic activity[60]. In addition to hippo signaling pathway, STAT3-mediated pathways are frequently involving in pathogenesis of NAFLD-HCC, which mediated lipid accumulation, apoptosis and fibrogenesis[61-66].
Other than metabolic disorder, gene polymorphism is significantly responsible for NAFLD. Recent genome-wide association studies (GWASs) in European ancestry suggest a robust relationship of PNPLA3 gene cluster with NAFLD activity, progression to HCC and liver-associated death[67-69]. This study also demonstrated an association of novel loci near IL17RA and ZFP90-CDH1 with NAFLD disease severity and fibrosis[67]. rs368234815 variant in IFNL4 and FNDC5 rs3480 polymorphism are respectively identified to be associated with liver damage and fibrosis in patients with NAFLD[70,71]. In addition, gender-bias is as well observed in NAFLD-HCC. As the confirmed risk factor for male HCC, androgen receptor (AR) transcription activity is enhanced by key enzymes and specific unsaturated fatty acid produced in lipogenesis via activation of Akt kinase[72][Figure 1].
Alteration of immune microenvironment
Multiple resident or migratory immune cells, including innate and adaptive immune cells, are responsible for maintaining immune homeostasis in the liver. In addition, the dysregulation of hepatic immune cells play critical roles in liver cancer pathogenesis. Liver is rich of innate immune cells including myeloid cells and innate immune cells. The myeloid cells contain macrophages, dendritic cells and myeloid-derived suppressor cells (MDSCs). Liver innate lymphocytes are represented by natural killer (NK) cells, NKT cells, mucosal associated invariant T cells and γδT cells. CD4+ T, CD8+ T and B cells comprise major adaptive cells in liver[73]. Both virus infection and metabolic disorder invoke the immune environment from tolerogenic status to active inflammation, leading to further cellular injury, fibrosis and eventually hepatocarinogenesis. Moreover, various subtype immune cells and related markers in tumor microenvironment are significantly predictors for clinical outcome of patients[74]. Here, this part will discuss the similarities and differences of immune cells responses between HBV and NAFLD-associated HCC [Figure 2].
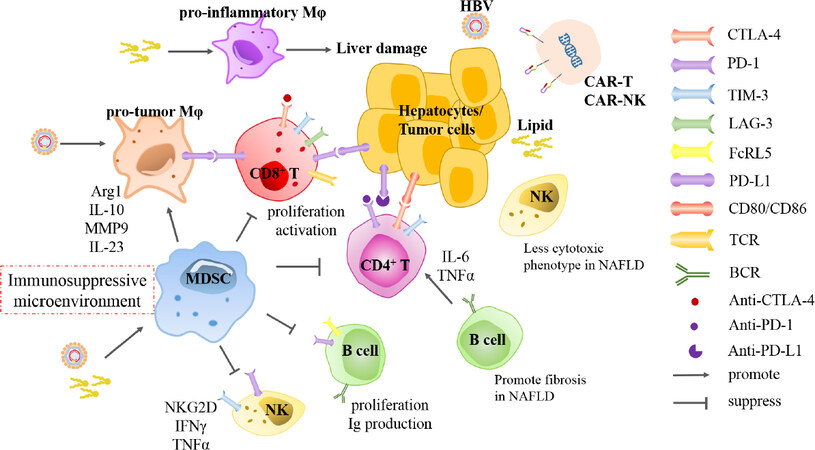
Figure 2. Alteration of immune cells in HBV- and NAFLD- HCC and corresponding immunotherapies. In HBV-infected liver microenvironment, MDSCs are recruited and macrophages are educated to polarization of pro-tumor phenotype, which constitute immunosuppressive microenvironment. However, excessive lipid accumulation and lipotoxic hepatocytes induce macrophages to pro-inflammatory phenotype which promotes liver damage. In both etiologies- associated HCC, cytotoxic T cells and NK cells inhibit progression of tumor formation through cytokines secretion or phenotype switch. B cells control progression of HBV infection by antibodies production and cytokines secretion, whereas promote NAFLD development towards fibrosis via inducing CD4+ T cells. Overall, activation and function of effector immune cells are suppressed by MDSCs and macrophages through interaction of checkpoint molecules, such as PD-1, PD-L1, CTLA-4, TIM-3 and LAG-3. Furthermore, several drugs blocking inhibitory checkpoint receptors have been developed and approved for treatment of cancer diseases. Collaboratively, CAR-T and CAR-NK cell therapy are investigated to be promising in treatment of liver cancer. HBV: hepatitis B virus; NAFLD: nonalcoholic fatty liver disease; HCC: hepatocellular carcinoma; MDSCs: myeloid-derived suppressor cells; CAR: chimeric antigen receptor; NK: natural killer; Mφ: macrophage; Arg1: arginase 1; MMP9: matrix metallopeptidase 9; IL: interleukin; IFN: interferon; TNF-α: tumor necrosis factor-alpha; TCR: T-cell receptor; BCR: B-cell receptor
Macrophages
Macrophages compromise almost 20% of overall immune cells in liver, containing residential Kupffer cells (KCs) and migratory monocyte-derived macrophages. Hepatic macrophages are extremely plastic and accommodative to signals from the microenvironment. Both inflammatory stimuli and viral proteins reprogram macrophages towards M2-like tumor macrophages, which in turn promote HCC progression[75-77]. In chronic HBV-infected tissue, macrophages are manipulated by HBV for persistent maintenance in liver. HBV suppresses pattern recognition receptors (PRRs) sensing or downstream interferon (IFN) response in hepatocytes but activate macrophages to secrete cytokines at high titer[78]. Furthermore, HBc protein is detected within macrophages in liver tissues from HBV-infected patients. Macrophages exposed to HBV or HBV-producing cells are prone to secrete more IL-10, which subsequently impairs lymphocyte activation, but less pro-inflammatory cytokines such as IL-6 and IL-1β with property of inhibition on HBV infection[76]. Accordingly, HBV drives macrophages to be suppressive immune cells. In offspring with horizontal transmission of HBV, HBeAg induces up-regulation of checkpoint molecular PD-L1 on macrophage and polarization to M2 protumor subtype, which impairs responses of CD8+ T cell to HBV and lead to virus persistence[79]. Moreover, macrophages produce matrix metalloproteinase 9 (MMP9) and IL-23 under the stimulation of HBV, which blocks binding of IFN-α to IFNAR1 and facilitate tumor angiogenesis and progression[80,81].
Contrarily, pro-inflammatory macrophages are essential for progression of NASH-associated HCC. In a high-fat diet (HFD) zebrafish model, hepatic macrophage morphology is changed and number of TNF-α positive positive macrophages is increased, and liver size is subsequently reduced in macrophage-ablation NAFLD/NASH associated HCC larvae but not in HCC or NAFLD alone[82]. The process from NAFLD to HCC is proximately accompanied by inflammation and fibrosis, in which, innate and adaptive immune cells are enriched and trafficking in liver. A recent study demonstrates that KCs induce recruitment of platelet in early stage of NAFLD, which is associated with hepatic injury. Furthermore, antiplatelet treatment abrogates infiltration of CD8+ T cells, Ly6G+ granulocytes, especially CD11b+ F4/80+ monocyte-derived macrophages and KCs, which alleviates the progression of carcinogenesis[83].Mechanistically, excessive toxic lipids and cholesterol crystals accumulate in macrophages, forcing cells polarization to pro-inflammatory phenotype, and depletion of KCs dampens release of pro-inflammatory cytokines and liver damage[84-87]. Meanwhile, lipotoxic hepatocytes activate macrophages polarization via releasing exosomes containing miR-192-5p[88]. Generally, NF-κB and JNK signaling pathways mainly mediate the activation of macrophages towards the pro-inflammatory phenotype. Stimulator of interferon genes (STING) invokes the polarization of macrophage to produce TNF-α and IL-1β via JNK/NF-κB pathways, which further modulate increase lipid deposition in hepatocytes and exacerbate progression of NAFLD[89].
MDSCs
MDSCs, generated as immature myeloid cells from bone marrow, are the major immunosuppressive cells which inhibit T cells proliferation and function and enhance induction of Treg cells and tumor-associated macrophages in chronic inflammation and cancers. Multiple studies have reported that MDSCs are closely related with HBV persistence and HCC. In patients with chronic HBV infection (CHB), HBeAg induces expansion of monocytic MDSCs (mMDSCs) via indoleamine-2,3-dioxygenase (IDO), to suppress autologous T cell proliferation and IFN-γ production, favoring the establishment of tolerant immune environment[90]. Similarly, HBsAg promotes differentiation of mMDSCs through activation of ERK/IL-6/STAT3 signaling feedback[91]. Mechanistically, cell cycle-related kinase (CCRK), as a direct AR-regulated oncogene, mediates virus-host signaling to promote progression of HBV-associated HCC[92], and further induces polymorphonuclear (PMN) MDSCs in HCC[93]. Moreover, AR-CCRK pathway is consistently shared by tumorigenesis in NASH-related HCC. Induction of CCRK activates mTORC1/4E-BP1/S6K/SREBP1 cascades and recruits PMN MDSCs to initiate metabolic reprogramming and immunosuppressive microenvironment to facilitate the progression of HCC[94]. Concordantly, hepatic lipid deposit promotes accumulation of MDSCs and further enhances the cellular production of ROS in NASH-model mice[95,96].
NK cells
Lymphocytes represent another crucial population in intrahepatic innate immune cells, including NKT cells, ILCs (NK cells and other ILC subpopulations) and γδT cells. NKT and NK cells, which mainly contain conventional circulating NK and liver-resident NK cells, comprise about 50% of lymphocytes in liver. Generally, hepatic NK cells recognize and restrain virus-infected cells via direct cytotoxicity or secreting immunoregulatory cytokines, such as IFN-γ and TNF-α, to activate T cells[97,98]. Recent studies demonstrate that liver-resident CD56 (bright) NK cells, which show increased expression of NKG2D and TRAIL and low production of IFN-γ, represent a cell population adapted for tolerogenic liver microenvironment and inducible anti-viral immunity[99,100]. Accumulating evidence illustrates that function of NK cells is impaired in CHB and HBV progressed diseases. NK cells in blood from CHB patients express higher level of suppressive cell surface molecules and cytokines, such as Tim-3, PD-1 and IL-10, which is induced by HBsAg-mediated increase of monocytes[101,102]. Besides, HBV nucleic acids-contained exosomes entering into NK cells inhibit expression of pattern-recognition receptors and thus NF-κB and p38 MAPK pathways, which results in dysfunction of NK cells[103]. Furthermore, intrahepatic NK cells-mediated apoptosis of hepatic stellate cells (HSCs) is reversed via blockade of TGF-β in HBV-infected liver cirrhosis patients, which leads to impairment of anti-fibrosis capacity in NK cells[104]. In HBV-associated HCC, LINC01149 variant upregulates MICA expression through serving as miR-128-3p sponge to recruit NK cells to lyse infected cells, which process releases highly soluble MICA and thus induces exhaustion of NK cells[105].
IL-15 is crucial for homeostasis of NK cells. Deficiency of IL-15 or IL-15Rα inhibits high fat diet-induced accumulation of lipids and inflammation in liver[106], which probably suggests dysregulation of NK cells involving in NAFLD progression. Although NK cells is a cell population with cytotoxicity in infected tissues, these cells convert to less cytotoxic ILC1-like phenotype in NAFLD, which probably protect obese liver from severe NAFLD but dampened capability to kill cancer cells[107]. In advanced stages of fibrosis in NAFLD accompanied by insulin resistance, the ability of NK cells to restrain HSCs is impaired, mediated by low expression of insulin receptors, which leads to deterioration of the liver and fibrosis[108]. Furthermore, NKp46+ NK cells, as immunoregulatory cells, induce polarization of hepatic macrophages towards M1-like phenotype via IFN-γ, which prevents NASH progression to fibrosis[109].
T cells
T cells represent the major adaptive lymphocytes in transformation from inflammation to cancer. CD8+ T cells frequently show restricted proliferation and exhausted function with high expression of inhibitory checkpoint molecules such as CTLA-4, PD-1 and TIM-3 in CHB and HBV-HCC. High expression of PD-1 on HBV-specific T cells or B cells induces exhaustion of T cells or reduced antibodies production, which is partially reversed by PD-1 blockade[110-112]. In HCC tissue, there exist CD8+ T cells expressing different levels of PD-1. The population with high-level PD-1 expresses TIM-3 and/or LAG-3 and produce limited IFNγ and TNFα when exposed to anti-CD3[113]. Beyond, thymocyte selection associated high mobility group box (TOX) in CD8+ T cells promotes PD-1 translocation to cell surface by regulating endocytic recycling of PD-1[114].
Abundant exhausted CD8+ T cells and Tregs exist and potentially clonally expand in cancerous tissues from HCC patients[115], contributing to evasion of tumor cells from immune surveillance. However, several studies illustrate that CD8+ T cells potentiate to promote persistent hepatic inflammation susceptible to HCC development when HBV-specific CD8+ T cells stay in activation but lack of capability to constrain virus replication[116]. Consistently, CD8+ T cells possess dual functions in progression of NAFLD-related HCC. In diet-induced obese mice, CD8+ T cells are recruited to liver to promote insulin resistance and glucose metabolism, leading to steatohepatitis[117]. Meanwhile, another study demonstrates that activation of CD8+ T cells is suppressed by liver-resident immunoglobulin-A-producing (IgA+) cells, which express high levels of PD-L1 and IL-10[118].
Compared to CD8+ T cells, HBV-specific CD4+ cytotoxic cells in PBMCs from HBV-associated HCC present at the similar level, but display weaker cytolysis capability and suppress cytotoxicity of CD8+ T cells in the absence of Tregs[119]. The antigen-experienced T cells, CD4+ follicular helper T (Tfh) cells diminish HBV via response to HBsAg, which is impaired by CTLA-4-mediated Treg suppression[120]. Contrary with CTLA-4, increased expression of co-stimulatory molecule OX40 on peripheral CD4+ T cells is associated with HBV clearance[121]. Furthermore, combined OX40 stimulation and PD-L1 blockade effectively activate IFN-γ producing Th1 cells and IL-21 producing Tfh cells, which potentiates the application of immunotherapeutic approaches[122]. Consistent with anti-tumor function in HBV-related HCC, surveillance from CD4+ T cells are crucial for NAFLD[123,124]. However, selective loss of CD4+ T cells has been recently detected in NAFLD, which promotes hepatocarcinogenesis[125]. Mechanistically, linoleic acid (C18:2) is released from dead hepatocytes caused by lipotoxicity into CD4+ T cells. C18:2 then co-localizes within mitochondria, upregulating expression of carnitine palmitoyltransferase, which further promotes production of ROS and cell apoptosis[124].
B cells
HBV viral load as well as HBsAg and HBeAg levels differed with progression of distinct phases of chronic HBV infection, accompanied by alteration of immune cells. Immunoglobulin-encoding genes and B cell function-related genes are more enriched in immune active patients than those in immune tolerant and inactive carrier patients[126]. B-cell response and humoral immunity are essential for controlling progression of chronic HBV infection[127,128]. However, differentiated HBsAg-specific B cells from patients with CHB are defective in antibody production and express high levels of inhibitory receptors, such as PD-1, FcRL4 and FcRL5[112,129]. Blockade of PD-1 on B cells effectively reverses cellular dysfunction to maturation and cytokines secretion[111]. Mechanistically, HBsAg inhibit toll-like receptor (TLR) promoter activity by suppressing phosphorylation of CAMP responsive element binding protein (CREB), thereby comprehensively inhibits expression of TLR9 in B cells, leading to reduced proliferation of B cells and decreased proinflammatory cytokines secretion[130].
Intrahepatic B cells are highly associated with NAFLD. CD20+ B cells are increased in liver tissues from NAFLD patients with activity score higher than 5, compared with those of 0 or 1~4[131]. Consistently, hepatic B cells account for more in HFD-induced NAFLD mice than in control mice. Moreover, the B cells in NAFLD liver tissues potentiate to produce higher levels of IL-6 and TNF-α, and promote differentiation of CD4+ T cells to Th1 cells[132]. Functionally, the maturation of hepatic B2-cells and the increase of IgG in circulation targeting oxidative stress-derived epitopes predict the onset of steatohepatitis, and B cells-mediated activation of Th1 cells contributes to NAFLD progression toward fibrosis[133].
Immunotherapies
As discussed above, a long duration is taken for both HBV- and NAFLD-related HCC, therefore, treatments aiming at eliminating HBV infection or blocking NASH progression and fibrogenesis can prevent the development of tumors. Abundant alterations of hepatic immune cells during liver carcinogenesis provide wide-range targets. Several immunotherapies have been approved for clinical application, nonetheless, the induced resistance and adverse events remain to be further investigated. The following sections will discuss immunotherapies in HCC, such as immune checkpoint inhibitors, virus or tumor vaccines, and adoptive cells transfer.
Immune checkpoint blockade
Inhibitory checkpoint receptors (ICRs) including CTLA-4, PD-1 and PD-L1, are recognized as master regulators for anti-tumor immunity. Immune checkpoint inhibition (ICI) therapy using antibody against PD-1, PD-L1 and CTLA-4 have been since been approved by the Food and Drug Administration[134]. Moreover, function of exhausted CD8+ T cells is restored through implementation of anti-PD-1 therapy and even combined blockade of ICRs[113,114]. In a cohort study with 262 patients enrolled between 2012 and 2016, nivolumab, a monoclonal antibody against PD-1, showed a decent safety profile, including acceptable tolerability[135]. Consistently, safety and efficacy of nivolumab was comparable in Asian patients, compared to intent-to-treat overall population[136]. However, a phase III trial showed that pembrolizumab, another PD-1 inhibitor, did not display significant increase in overall survival (OS) and progression-free survival (PFS), compared with control groups[137]. Since medicines based on anti-PD-L1 monoclonal antibodies have been validated to be effective for several cancer diseases, a phase Ib clinical trial of atezolizumab combined with bevacizumab, the anti-PD-L1 antibody and VEGF blocking antibody respectively (ClinicalTrials.gov NCT02715531), has announced an overall response rate of 32% among 73 patients with advanced HCC[138]. Moreover, this combined therapy-based IMbrave150 phase III trial (ClinicalTrials.gov NCT03434379) is currently implemented and significant improvements in OS and PFS compared with sorafenib have been reported by the sponsor[139,140][Figure 2].
CTLA-4 is another inhibitory molecule that attenuates activation and function of T cells. Upregulation of CTLA-4 promote the apoptosis, secretion of anti-inflammatory cytokines and exhaustion of CD4+ Th and CD8+ T cells in liver tissues from CHB and HCC patients[141-144]. Tremelimumab, which is the monoclonal antibody blocking CTLA-4, was administered in HCV-related HCC patients and assessed to possess antitumor and antiviral activity, along with good safety profile. The disease control rate was 76.4% and time to progression was 6.48 months (95%CI: 3.95-9.14)[145]. The similar efficacy displayed in another cohort with 32 HCC patients enrolled. Combinational administration of Tremelimumab and tumor ablation results in accumulation of intratumoral CD8+ T cells and median OS of treated patients was 12.3 months (95%CI: 9.3 to 15.4 months), which is a potential new therapy for patients with advanced HCC[146]. Furthermore, a preclinical study of durvalumab combined with tremelimumab was analyzed in patients with unresectable HCC (11 HBV+, 9 HCV+, 20 uninfected). Among 40 patients, 60% (24/40) had discontinued treatment. The uninfected patients after treatment showed predominantly clinical activity and no unexpected safety signals were observed. The phase II portion of this study is still ongoing[147][Figure 2].
Adoptive cells transfer
Chimeric antigen receptor (CAR) therapy is a promising strategy for cancer immunotherapy. Accumulating evidence has demonstrated CAR-T cell therapy as effective strategy against lymphoid leukemia and multiple myeloma[148-150], whereas, efficacy and safety of CAR-T therapy for treatment of solid tumors remain elusive. Glypican-3 (GPC3) is broadly expressed on liver cancer cells, but minority on normal hepatocytes. GPC3-targeted CAR-T cells efficiently kill GPC3-positive hepatoma cells and the T cells expressing third-generation CAR potentiate to suppress growth of xenografts with even low GPC3 expression level in mice[151]. Moreover, GPC3-specific CAR-T therapy combination with sorafenib could effectively promote apoptosis of liver cancer cells[152][Figure 2].
However, side effects to CAR-T therapy contain high manufacturing and fatal toxicities, such as cytokine release syndrome[153]. CAR-NK therapy is under investigation and probably provide better options. Similar to CAR-T, FLT3- and CS1-targeted CAR-NK cells respectively suppress proliferation of cancer cells in myeloid leukemia and multiple myeloma[154,155]. Furthermore, DAP12-targeted CAR-NK cells efficiently suppressed tumor cells in ascites and proliferation of liver-metastatic tumors in colorectal cancer patients[156]. In vivo studies have shown that injection of ErbB2-specific NK-92/5.28.z cells possess the ability to prolong survival of glioblastoma-xenograft mice[157][Figure 2].
Conclusion
Chronic HBV infection-induced progression of HCC mainly depends on direct intrusion of viral components in hepatocytes, which disturbs its normal cellular metabolism and function. This subsequently stimulates immune imbalance in liver. Frequently accompanied by systemic metabolic syndrome, advancement of NALFD- associated HCC encompasses excessive lipid accumulation in hepatocytes and immune cells, insulin resistance and inflammation induced malignant transformation. The hepatic complexity of immune cells mediates tumorigenesis in situation of anergy of cytotoxic cells and enhancement of immunosuppressive cells. Cytotoxic T cells are the major factor which eradicates HBV-infected cells or malignant cells. The exhaustion mediated by MDSCs or Tregs leads to persistence of HBV and evasion of cancer cells. Flexibility of macrophages facilitates themselves to polarize according to different signals from microenvironment. Macrophages mostly function as suppressive cells to inhibit proliferation and activation of CTLs in HBV-related HCC, while promotes liver damage and fibrosis via releasing pro-inflammatory cytokines.
Since there still remains lack of effective drug specifically targeting eradication of HBV or controlling development of NAFLD, illustrating molecular mechanisms of immune cells and establishing novel immunotherapies will provide promising options for treatment of HCC. Unbridling cytotoxic cells (ICI) and stimulating the cells specifically engineered with tumor-associated antigens (CAR-T and CAR-NK) mutually contribute to effectively restrain progression of HBV- and NAFLD-induced HCC. Taken together, the drug resistance and adverse effect induced by immunotherapies should be further recorded and investigated in future studies.
Declarations
Authors’ contributionsConceived of the presented idea: Ma CH, Song XJ
Performed the basic writing: Song XJ
Developed the further revision: Ma CH
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by grants from the National Science Foundation of China (Key project 81830017 and 81902443), Taishan Scholarship (No.tspd20181201), National Key Research and Development Program (2018YFE0126500), Shandong Provincial Key Innovation project (No.2018FYJH0503), Collaborative Innovation Center of Technology and Equipment for Biological Diagnosis and Therapy in Universities of Shandong.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2020.
REFERENCES
1. Ghany MG, Marks KM, Morgan TR, Wyles DL, Aronsohn AI, et al. Hepatitis C guidance 2019 update: AASLD-IDSA recommendations for testing, managing, and treating hepatitis C virus infection. Hepatology 2020;71:686-721.
2. Falade-Nwulia O, Suarez-Cuervo C, Nelson DR, Fried MW, Segal JB, et al. Oral direct-acting agent therapy for hepatitis C virus infection: a systematic review. Ann Intern Med 2017;166:637-48.
3. Cornberg M, Lok AS, Terrault NA, Zoulim F; 2019 EASL-AASLD HBV Treatment Endpoints Conference Faculty. Guidance for design and endpoints of clinical trials in chronic hepatitis B - Report from the 2019 EASL-AASLD HBV Treatment Endpoints Conference. J Hepatol 2019. Epub ahead of print. doi: 10.1002/hep.31030
4. Yip TC, Wong VW, Chan HL, Tse YK, Lui GC, et al. Tenofovir is associated with lower risk of hepatocellular carcinoma than entecavir in patients with chronic HBV infection in China. Gastroenterology 2020;158:215-25.e6.
5. Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol 2019;16:411-28.
6. Kanwal F, Kramer J, Li L, Dai J, Natarajan Y, et al. Effect of metabolic traits on the risk of cirrhosis and hepatocellular cancer in non-alcoholic fatty liver disease. Hepatology 2020;71:808-19.
7. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 2018;15:11-20.
8. Younossi Z, Stepanova M, Ong JP, Jacobson IM, Bugianesi E, et al. Nonalcoholic steatohepatitis is the fastest growing cause of hepatocellular carcinoma in liver transplant candidates. Clin Gastroenterol Hepatol 2019;17:748-55.e3.
9. Estes C, Anstee QM, Arias-Loste MT, Bantel H, Bellentani S, et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016-2030. J Hepatol 2018;69:896-904.
10. Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, et al. Hepatocellular carcinoma. Nat Rev Dis Primers 2016;2:16018.
11. Rimassa L, Danesi R, Pressiani T, Merle P. Management of adverse events associated with tyrosine kinase inhibitors: Improving outcomes for patients with hepatocellular carcinoma. Cancer Treat Rev 2019;77:20-8.
12. Bruix J, da Fonseca LG, Reig M. Insights into the success and failure of systemic therapy for hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 2019;16:617-30.
13. Brown ZJ, Greten TF, Heinrich B. Adjuvant treatment of hepatocellular carcinoma: prospect of immunotherapy. Hepatology 2019;70:1437-42.
14. Amaddeo G, Cao Q, Ladeiro Y, Imbeaud S, Nault JC, et al. Integration of tumour and viral genomic characterizations in HBV-related hepatocellular carcinomas. Gut 2015;64:820-9.
15. Hsia CC, Yuwen H, Tabor E. Hot-spot mutations in hepatitis B virus X gene in hepatocellular carcinoma. Lancet 1996;348:625-6.
16. Iavarone M, Trabut JB, Delpuech O, Carnot F, Colombo M, et al. Characterisation of hepatitis B virus X protein mutants in tumour and non-tumour liver cells using laser capture microdissection. J Hepatol 2003;39:253-61.
17. Ali A, Abdel-Hafiz H, Suhail M, Al-Mars A, Zakaria MK, et al. Hepatitis B virus, HBx mutants and their role in hepatocellular carcinoma. World J Gastroenterol 2014;20:10238-48.
18. Kim CM, Koike K, Saito I, Miyamura T, Jay G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 1991;351:317-20.
19. Kew MC. Hepatitis B virus x protein in the pathogenesis of hepatitis B virus-induced hepatocellular carcinoma. J Gastroenterol Hepatol 2011;26:144-52.
20. Jiang T, Liu M, Wu J, Shi Y. Structural and biochemical analysis of Bcl-2 interaction with the hepatitis B virus protein HBx. Proc Natl Acad Sci U S A 2016;113:2074-9.
21. Niu Y, Xu M, Slagle BL, Huang H, Li S, et al. Farnesoid X receptor ablation sensitizes mice to hepatitis b virus X protein-induced hepatocarcinogenesis. Hepatology 2017;65:893-906.
22. Jegal ME, Jung SY, Han YS, Kim YJ. C-terminal truncated HBx reduces doxorubicin cytotoxicity via ABCB1 upregulation in Huh-7 hepatocellular carcinoma cells. BMB Rep 2019;52:330-5.
23. Li Y, Fu Y, Hu X, Sun L, Tang D, et al. The HBx-CTTN interaction promotes cell proliferation and migration of hepatocellular carcinoma via CREB1. Cell Death Dis 2019;10:405.
24. Lee S, Kim W, Ko C, Ryu WS. Hepatitis B virus X protein enhances Myc stability by inhibiting SCF(Skp2) ubiquitin E3 ligase-mediated Myc ubiquitination and contributes to oncogenesis. Oncogene 2016;35:1857-67.
25. Wang J, Li N, Huang ZB, Fu S, Yu SM, et al. HBx regulates transcription factor PAX8 stabilization to promote the progression of hepatocellular carcinoma. Oncogene 2019;38:6696-710.
26. He B, Peng F, Li W, Jiang Y. Interaction of lncRNA-MALAT1 and miR-124 regulates HBx-induced cancer stem cell properties in HepG2 through PI3K/Akt signaling. J Cell Biochem 2019;120:2908-18.
27. Lin X, Zuo S, Luo R, Li Y, Yu G, et al. HBX-induced miR-5188 impairs FOXO1 to stimulate beta-catenin nuclear translocation and promotes tumor stemness in hepatocellular carcinoma. Theranostics 2019;9:7583-98.
28. Chen SL, Zhang CZ, Liu LL, Lu SX, Pan YH, et al. A GYS2/p53 negative feedback loop restricts tumor growth in HBV-related hepatocellular carcinoma. Cancer Res 2019;79:534-45.
29. Liu WC, Wu IC, Lee YC, Lin CP, Cheng JH, et al. Hepatocellular carcinoma-associated single-nucleotide variants and deletions identified by the use of genome-wide high-throughput analysis of hepatitis B virus. J Pathol 2017;243:176-92.
30. Bai X, Jia JA, Fang M, Chen S, Liang X, et al. Deep sequencing of HBV pre-S region reveals high heterogeneity of HBV genotypes and associations of word pattern frequencies with HCC. PLoS Genet 2018;14:e1007206.
31. Huang JT, Liu YJ, Wang J, Xu ZG, Yang Y, et al. Next generation digital PCR measurement of hepatitis B virus copy number in formalin-fixed paraffin-embedded hepatocellular carcinoma tissue. Clin Chem 2015;61:290-6.
32. Hsieh YH, Su IJ, Wang HC, Chang WW, Lei HY, et al. Pre-S mutant surface antigens in chronic hepatitis B virus infection induce oxidative stress and DNA damage. Carcinogenesis 2004;25:2023-32.
33. Pollicino T, Cacciola I, Saffioti F, Raimondo G. Hepatitis B virus PreS/S gene variants: pathobiology and clinical implications. J Hepatol 2014;61:408-17.
34. Li TN, Wu YJ, Tsai HW, Sun CP, Wu YH, et al. Intrahepatic hepatitis B virus large surface antigen induces hepatocyte hyperploidy via failure of cytokinesis. J Pathol 2018;245:502-13.
35. Liu P, Zhang H, Liang X, Ma H, Luan F, et al. HBV preS2 promotes the expression of TAZ via miRNA-338-3p to enhance the tumorigenesis of hepatocellular carcinoma. Oncotarget 2015;6:29048-59.
36. Luan F, Liu B, Zhang J, Cheng S, Zhang B, et al. Correlation between HBV protein preS2 and tumor markers of hepatocellular carcinoma. Pathol Res Pract 2017;213:1037-42.
37. Zhang R, Real CI, Liu C, Baba HA, Gerken G, et al. Hepatic expression of oncogenes Bmi1 and Dkk1 is up-regulated in hepatitis B virus surface antigen-transgenic mice and can be induced by treatment with HBV particles or lipopolysaccharides in vitro. Int J Cancer 2017;141:354-63.
38. Luan F, Liu H, Gao L, Liu J, Sun Z, et al. Hepatitis B virus protein preS2 potentially promotes HCC development via its transcriptional activation of hTERT. Gut 2009;58:1528-37.
39. Tseng TC, Liu CJ, Hsu CY, Hong CM, Su TH, et al. High level of hepatitis B core-related antigen associated with increased risk of hepatocellular carcinoma in patients with chronic HBV infection of intermediate viral load. Gastroenterology 2019;157:1518-29.e3.
40. Liu W, Guo TF, Jing ZT, Yang Z, Liu L, et al. Hepatitis B virus core protein promotes hepatocarcinogenesis by enhancing Src expression and activating the Src/PI3K/Akt pathway. FASEB J 2018;32:3033-46.
41. Liu D, Cui L, Wang Y, Yang G, He J, et al. Hepatitis B e antigen and its precursors promote the progress of hepatocellular carcinoma by interacting with NUMB and decreasing p53 activity. Hepatology 2016;64:390-404.
42. Xie Q, Fan F, Wei W, Liu Y, Xu Z, et al. Multi-omics analyses reveal metabolic alterations regulated by hepatitis B virus core protein in hepatocellular carcinoma cells. Sci Rep 2017;7:41089.
43. Du J, Liang X, Liu Y, Qu Z, Gao L, et al. Hepatitis B virus core protein inhibits TRAIL-induced apoptosis of hepatocytes by blocking DR5 expression. Cell Death Differ 2009;16:219-29.
44. Liu W, Lin YT, Yan XL, Ding YL, Wu YL, et al. Hepatitis B virus core protein inhibits Fas-mediated apoptosis of hepatoma cells via regulation of mFas/FasL and sFas expression. FASEB J 2015;29:1113-23.
45. Tu T, Budzinska MA, Vondran FWR, Shackel NA, Urban S. Hepatitis B virus DNA integration occurs early in the viral life cycle in an in vitro infection model via sodium taurocholate cotransporting polypeptide-dependent uptake of enveloped virus particles. J Virol 2018;92.
46. Duan M, Hao J, Cui S, Worthley DL, Zhang S, et al. Diverse modes of clonal evolution in HBV-related hepatocellular carcinoma revealed by single-cell genome sequencing. Cell Res 2018;28:359-73.
47. Zhao LH, Liu X, Yan HX, Li WY, Zeng X, et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat Commun 2016;7:12992.
48. Yang X, Wu L, Lin J, Wang A, Wan X, et al. Distinct hepatitis B virus integration patterns in hepatocellular carcinoma and adjacent normal liver tissue. Int J Cancer 2017;140:1324-30.
49. Wong DK, Cheng SCY, Mak LL, To EW, Lo RC, et al. Among patients with undetectable hepatitis B surface antigen and hepatocellular carcinoma, a high proportion has integration of HBV DNA into hepatocyte DNA and no cirrhosis. Clin Gastroenterol Hepatol 2020;18:449-56.
50. Yan H, Yang Y, Zhang L, Tang G, Wang Y, et al. Characterization of the genotype and integration patterns of hepatitis B virus in early- and late-onset hepatocellular carcinoma. Hepatology 2015;61:1821-31.
51. Chiu YT, Wong JK, Choi SW, Sze KM, Ho DW, et al. Novel pre-mRNA splicing of intronically integrated HBV generates oncogenic chimera in hepatocellular carcinoma. J Hepatol 2016;64:1256-64.
52. Li CL, Li CY, Lin YY, Ho MC, Chen DS, et al. Androgen receptor enhances hepatic telomerase reverse transcriptase gene transcription after hepatitis B virus integration or point mutation in promoter region. Hepatology 2019;69:498-512.
53. Yoo S, Wang W, Wang Q, Fiel MI, Lee E, et al. A pilot systematic genomic comparison of recurrence risks of hepatitis B virus-associated hepatocellular carcinoma with low- and high-degree liver fibrosis. BMC Med 2017;15:214.
54. Hama N, Totoki Y, Miura F, Tatsuno K, Saito-Adachi M, et al. Epigenetic landscape influences the liver cancer genome architecture. Nat Commun 2018;9:1643.
55. Ren L, Zeng M, Tang Z, Li M, Wang X, et al. The antiresection activity of the X protein encoded by hepatitis virus B. Hepatology 2019;69:2546-61.
56. Musa J, Li J, Grunewald TG. Hepatitis B virus large surface protein is priming for hepatocellular carcinoma development via induction of cytokinesis failure. J Pathol 2019;247:6-8.
57. Hsieh YH, Chang YY, Su IJ, Yen CJ, Liu YR, et al. Hepatitis B virus pre-S2 mutant large surface protein inhibits DNA double-strand break repair and leads to genome instability in hepatocarcinogenesis. J Pathol 2015;236:337-47.
58. Takakura K, Oikawa T, Nakano M, Saeki C, Torisu Y, et al. Recent insights into the multiple pathways driving non-alcoholic steatohepatitis-derived hepatocellular carcinoma. Front Oncol 2019;9:762.
59. Li T, Weng J, Zhang Y, Liang K, Fu G, et al. mTOR direct crosstalk with STAT5 promotes de novo lipid synthesis and induces hepatocellular carcinoma. Cell Death Dis 2019;10:619.
60. Tian Y, Yang B, Qiu W, Hao Y, Zhang Z, et al. ER-residential Nogo-B accelerates NAFLD-associated HCC mediated by metabolic reprogramming of oxLDL lipophagy. Nat Commun 2019;10:3391.
61. Kodama T, Yi J, Newberg JY, Tien JC, Wu H, et al. Molecular profiling of nonalcoholic fatty liver disease-associated hepatocellular carcinoma using SB transposon mutagenesis. Proc Natl Acad Sci U S A 2018;115:e10417-26.
62. Ye J, Li TS, Xu G, Zhao YM, Zhang NP, et al. JCAD promotes progression of nonalcoholic steatohepatitis to liver cancer by inhibiting LATS2 kinase activity. Cancer Res 2017;77:5287-300.
63. Jeong SH, Kim HB, Kim MC, Lee JM, Lee JH, et al. Hippo-mediated suppression of IRS2/AKT signaling prevents hepatic steatosis and liver cancer. J Clin Invest 2018;128:1010-25.
64. Inoue-Yamauchi A, Itagaki H, Oda H. Eicosapentaenoic acid attenuates obesity-related hepatocellular carcinogenesis. Carcinogenesis 2018;39:28-35.
65. Kumar DP, Santhekadur PK, Seneshaw M, Mirshahi F, Uram-Tuculescu C, et al. A regulatory role of apoptosis antagonizing transcription factor in the pathogenesis of nonalcoholic fatty liver disease and hepatocellular carcinoma. Hepatology 2019;69:1520-34.
66. He Y, Hwang S, Cai Y, Kim SJ, Xu M, et al. MicroRNA-223 ameliorates nonalcoholic steatohepatitis and cancer by targeting multiple inflammatory and oncogenic genes in hepatocytes. Hepatology 2019;70:1150-67.
67. Namjou B, Lingren T, Huang Y, Parameswaran S, Cobb BL, et al. GWAS and enrichment analyses of non-alcoholic fatty liver disease identify new trait-associated genes and pathways across eMERGE Network. BMC Med 2019;17:135.
68. Grimaudo S, Pipitone RM, Pennisi G, Celsa C, Camma C, et al. Association between PNPLA3 rs738409 C>G variant and liver-related outcomes in patients with non-alcoholic fatty liver disease. Clin Gastroenterol Hepatol 2020;18:935-44.e3.
69. Tepper CG, Dang JHT, Stewart SL, Fang DM, Wong KA, et al. High frequency of the PNPLA3 rs738409 [G] single-nucleotide polymorphism in Hmong individuals as a potential basis for a predisposition to chronic liver disease. Cancer 2018;124:1583-9.
70. Petta S, Valenti L, Tuttolomondo A, Dongiovanni P, Pipitone RM, et al. Interferon lambda 4 rs368234815 TT>deltaG variant is associated with liver damage in patients with nonalcoholic fatty liver disease. Hepatology 2017;66:1885-93.
71. Petta S, Valenti L, Svegliati-Baroni G, Ruscica M, Pipitone RM, et al. Fibronectin type III domain-containing protein 5 rs3480 A>G polymorphism, irisin, and liver fibrosis in patients with nonalcoholic fatty liver disease. J Clin Endocrinol Metab 2017;102:2660-9.
72. Cheng YW, Chen KW, Kuo HC, Kuo CH, Lin WH, et al. Specific diacylglycerols generated by hepatic lipogenesis stimulate the oncogenic androgen receptor activity in male hepatocytes. Int J Obes (Lond) 2019;43:2469-79.
73. Campisano S, La Colla A, Echarte SM, Chisari AN. Interplay between early-life malnutrition, epigenetic modulation of the immune function and liver diseases. Nutr Res Rev 2019;32:128-45.
74. Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer 2012;12:298-306.
75. Yan W, Liu X, Ma H, Zhang H, Song X, et al. Tim-3 fosters HCC development by enhancing TGF-beta-mediated alternative activation of macrophages. Gut 2015;64:1593-604.
76. Faure-Dupuy S, Delphin M, Aillot L, Dimier L, Lebosse F, et al. Hepatitis B virus-induced modulation of liver macrophage function promotes hepatocyte infection. J Hepatol 2019;71:1086-98.
77. Bility MT, Cheng L, Zhang Z, Luan Y, Li F, et al. Hepatitis B virus infection and immunopathogenesis in a humanized mouse model: induction of human-specific liver fibrosis and M2-like macrophages. PLoS Pathog 2014;10:e1004032.
78. Cheng X, Xia Y, Serti E, Block PD, Chung M, et al. Hepatitis B virus evades innate immunity of hepatocytes but activates cytokine production by macrophages. Hepatology 2017;66:1779-93.
79. Tian Y, Kuo CF, Akbari O, Ou JH. Maternal-derived hepatitis B virus e antigen alters macrophage function in offspring to drive viral persistence after vertical transmission. Immunity 2016;44:1204-14.
80. Chen J, Xu W, Chen Y, Xie X, Zhang Y, et al. Matrix metalloproteinase 9 facilitates hepatitis B virus replication through binding with type i interferon (IFN) receptor 1 to repress IFN/JAK/STAT signaling. J Virol 2017;91.
81. Zang M, Li Y, He H, Ding H, Chen K, et al. IL-23 production of liver inflammatory macrophages to damaged hepatocytes promotes hepatocellular carcinoma development after chronic hepatitis B virus infection. Biochim Biophys Acta Mol Basis Dis 2018;1864:3759-70.
82. de Oliveira S, Houseright RA, Graves AL, Golenberg N, Korte BG, et al. Metformin modulates innate immune-mediated inflammation and early progression of NAFLD-associated hepatocellular carcinoma in zebrafish. J Hepatol 2019;70:710-21.
83. Malehmir M, Pfister D, Gallage S, Szydlowska M, Inverso D, et al. Platelet GPIbalpha is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat Med 2019;25:641-55.
84. Eckert C, Klein N, Kornek M, Lukacs-Kornek V. The complex myeloid network of the liver with diverse functional capacity at steady state and in inflammation. Front Immunol 2015;6:179.
85. Walenbergh SM, Houben T, Hendrikx T, Jeurissen ML, van Gorp PJ, et al. Weekly treatment of 2-hydroxypropyl-beta-cyclodextrin improves intracellular cholesterol levels in LDL receptor knockout mice. Int J Mol Sci 2015;16:21056-69.
86. Leroux A, Ferrere G, Godie V, Cailleux F, Renoud ML, et al. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J Hepatol 2012;57:141-9.
87. Alisi A, Carpino G, Oliveira FL, Panera N, Nobili V, et al. The role of tissue macrophage-mediated inflammation on NAFLD pathogenesis and its clinical implications. Mediators Inflamm 2017;2017:8162421.
88. Liu XL, Pan Q, Cao HX, Xin FZ, Zhao ZH, et al. Lipotoxic hepatocyte-derived exosomal mir-192-5p activates macrophages via Rictor/Akt/FoxO1 signaling in NAFLD. Hepatology 2019. Epub ahead of print. doi: 10.1002/hep.31050
89. Luo X, Li H, Ma L, Zhou J, Guo X, et al. Expression of STING is increased in liver tissues from patients with NAFLD and promotes macrophage-mediated hepatic inflammation and fibrosis in mice. Gastroenterology 2018;155:1971-84.e4.
90. Yang F, Yu X, Zhou C, Mao R, Zhu M, et al. Hepatitis B e antigen induces the expansion of monocytic myeloid-derived suppressor cells to dampen T-cell function in chronic hepatitis B virus infection. PLoS Pathog 2019;15:e1007690.
91. Fang Z, Li J, Yu X, Zhang D, Ren G, et al. Polarization of monocytic myeloid-derived suppressor cells by hepatitis b surface antigen is mediated via ERK/IL-6/STAT3 signaling feedback and restrains the activation of T cells in chronic hepatitis B virus infection. J Immunol 2015;195:4873-83.
92. Yu Z, Gao YQ, Feng H, Lee YY, Li MS, et al. Cell cycle-related kinase mediates viral-host signalling to promote hepatitis B virus-associated hepatocarcinogenesis. Gut 2014;63:1793-804.
93. Zhou J, Liu M, Sun H, Feng Y, Xu L, et al. Hepatoma-intrinsic CCRK inhibition diminishes myeloid-derived suppressor cell immunosuppression and enhances immune-checkpoint blockade efficacy. Gut 2018;67:931-44.
94. Sun H, Yang W, Tian Y, Zeng X, Zhou J, et al. An inflammatory-CCRK circuitry drives mTORC1-dependent metabolic and immunosuppressive reprogramming in obesity-associated hepatocellular carcinoma. Nat Commun 2018;9:5214.
95. Tsunashima H, Tsuneyama K, Moritoki Y, Hara M, Kikuchi K. Accumulated myeloid-derived suppressor cells demonstrate distinct phenotypes and functions in two non-alcoholic steatohepatitis mouse models. Hepatobiliary Surg Nutr 2015;4:313-9.
96. Yao L, Abe M, Kawasaki K, Akbar SM, Matsuura B, et al. Characterization of liver monocytic myeloid-derived suppressor cells and their role in a murine model of non-alcoholic fatty liver disease. PLoS One 2016;11:e0149948.
97. Pallmer K, Oxenius A. Recognition and regulation of T cells by NK cells. Front Immunol 2016;7:251.
98. Biron CA, Altfeld M. Is there natural killer cell memory and can it be harnessed by vaccination? Can natural killer and CD8 T cells switch jobs? Cold Spring Harb Perspect Biol 2018;10.
99. Harmon C, Robinson MW, Fahey R, Whelan S, Houlihan DD, et al. Tissue-resident Eomes(hi) T-bet(lo) CD56(bright) NK cells with reduced proinflammatory potential are enriched in the adult human liver. Eur J Immunol 2016;46:2111-20.
100. Stegmann KA, Robertson F, Hansi N, Gill U, Pallant C, et al. CXCR6 marks a novel subset of T-bet(lo)Eomes(hi) natural killer cells residing in human liver. Sci Rep 2016;6:26157.
101. Ju Y, Hou N, Meng J, Wang X, Zhang X, et al. T cell immunoglobulin- and mucin-domain-containing molecule-3 (Tim-3) mediates natural killer cell suppression in chronic hepatitis B. J Hepatol 2010;52:322-9.
102. Li H, Zhai N, Wang Z, Song H, Yang Y, et al. Regulatory NK cells mediated between immunosuppressive monocytes and dysfunctional T cells in chronic HBV infection. Gut 2018;67:2035-44.
103. Yang Y, Han Q, Hou Z, Zhang C, Tian Z, et al. Exosomes mediate hepatitis B virus (HBV) transmission and NK-cell dysfunction. Cell Mol Immunol 2017;14:465-75.
104. Shi J, Zhao J, Zhang X, Cheng Y, Hu J, et al. Activated hepatic stellate cells impair NK cell anti-fibrosis capacity through a TGF-beta-dependent emperipolesis in HBV cirrhotic patients. Sci Rep 2017;7:44544.
105. Zhong R, Tian J, Fu M, Ma S, Liu L, et al. LINC01149 variant modulates MICA expression that facilitates hepatitis B virus spontaneous recovery but increases hepatocellular carcinoma risk. Oncogene 2020;39:1944-56.
106. Cepero-Donates Y, Lacraz G, Ghobadi F, Rakotoarivelo V, Orkhis S, et al. Interleukin-15-mediated inflammation promotes non-alcoholic fatty liver disease. Cytokine 2016;82:102-11.
107. Cuff AO, Sillito F, Dertschnig S, Hall A, Luong TV, et al. The obese liver environment mediates conversion of NK cells to a less cytotoxic ILC1-like phenotype. Front Immunol 2019;10:2180.
108. Amer J, Salhab A, Noureddin M, Doron S, Abu-Tair L, et al. Insulin signaling as a potential natural killer cell checkpoint in fatty liver disease. Hepatol Commun 2018;2:285-98.
109. Tosello-Trampont AC, Krueger P, Narayanan S, Landes SG, Leitinger N, et al. NKp46(+) natural killer cells attenuate metabolism-induced hepatic fibrosis by regulating macrophage activation in mice. Hepatology 2016;63:799-812.
110. Li Y, Tang L, Guo L, Chen C, Gu S, et al. CXCL13-mediated recruitment of intrahepatic CXCR5(+)CD8(+) T cells favors viral control in chronic HBV infection. J Hepatol 2020;72:420-30.
111. Salimzadeh L, Le Bert N, Dutertre CA, Gill US, Newell EW, et al. PD-1 blockade partially recovers dysfunctional virus-specific B cells in chronic hepatitis B infection. J Clin Invest 2018;128:4573-87.
112. Burton AR, Pallett LJ, McCoy LE, Suveizdyte K, Amin OE, et al. Circulating and intrahepatic antiviral B cells are defective in hepatitis B. J Clin Invest 2018;128:4588-603.
113. Kim HD, Song GW, Park S, Jung MK, Kim MH, et al. Association between expression level of PD1 by tumor-infiltrating CD8(+) T cells and features of hepatocellular carcinoma. Gastroenterology 2018;155:1936-50.e17.
114. Wang X, He Q, Shen H, Xia A, Tian W, et al. TOX promotes the exhaustion of antitumor CD8(+) T cells by preventing PD1 degradation in hepatocellular carcinoma. J Hepatol 2019;71:731-41.
115. Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 2017;169:1342-56.e16.
116. Kim GA, Lim YS, Han S, Choi J, Shim JH, et al. High risk of hepatocellular carcinoma and death in patients with immune-tolerant-phase chronic hepatitis B. Gut 2018;67:945-52.
117. Ghazarian M, Revelo XS, Nohr MK, Luck H, Zeng K, et al. Type I interferon responses drive intrahepatic T cells to promote metabolic syndrome. Sci Immunol 2017;2:pii: eaai7616.
118. Shalapour S, Lin XJ, Bastian IN, Brain J, Burt AD, et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 2017;551:340-5.
119. Meng F, Zhen S, Song B. HBV-specific CD4+ cytotoxic T cells in hepatocellular carcinoma are less cytolytic toward tumor cells and suppress CD8+ T cell-mediated antitumor immunity. APMIS 2017;125:743-51.
120. Wang X, Dong Q, Li Q, Li Y, Zhao D, et al. Dysregulated response of follicular helper T cells to hepatitis B surface antigen promotes HBV persistence in mice and associates with outcomes of patients. Gastroenterology 2018;154:2222-36.
121. Publicover J, Gaggar A, Jespersen JM, Halac U, Johnson AJ, et al. An OX40/OX40L interaction directs successful immunity to hepatitis B virus. Sci Transl Med 2018;10.
122. Jacobi FJ, Wild K, Smits M, Zoldan K, Csernalabics B, et al. OX40 stimulation and PD-L1 blockade synergistically augment HBV-specific CD4 T cells in patients with HBeAg-negative infection. J Hepatol 2019;70:1103-13.
123. Walker CM, Lemon SM. Getting the skinny on CD4(+) T cell survival in fatty livers. Immunity 2016;44:725-7.
124. Brown ZJ, Fu Q, Ma C, Kruhlak M, Zhang H, et al. Carnitine palmitoyltransferase gene upregulation by linoleic acid induces CD4(+) T cell apoptosis promoting HCC development. Cell Death Dis 2018;9:620.
125. Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016;531:253-7.
126. Vanwolleghem T, Hou J, van Oord G, Andeweg AC, Osterhaus AD, et al. Re-evaluation of hepatitis B virus clinical phases by systems biology identifies unappreciated roles for the innate immune response and B cells. Hepatology 2015;62:87-100.
128. Zhang S, Zhao J, Zhang Z. Humoral immunity, the underestimated player in hepatitis B. Cell Mol Immunol 2018;15:645-8.
129. Poonia B, Ayithan N, Nandi M, Masur H, Kottilil S. HBV induces inhibitory FcRL receptor on B cells and dysregulates B cell-T follicular helper cell axis. Sci Rep 2018;8:15296.
130. Tout I, Gomes M, Ainouze M, Marotel M, Pecoul T, et al. Hepatitis B virus Blocks the CRE/CREB Complex and Prevents TLR9 Transcription and Function in Human B Cells. J Immunol 2018;201:2331-44.
131. Schwenger KJP, Chen L, Chelliah A, Da Silva HE, Teterina A, et al. Markers of activated inflammatory cells are associated with disease severity and intestinal microbiota in adults with nonalcoholic fatty liver disease. Int J Mol Med 2018;42:2229-37.
132. Zhang F, Jiang WW, Li X, Qiu XY, Wu Z, et al. Role of intrahepatic B cells in non-alcoholic fatty liver disease by secreting pro-inflammatory cytokines and regulating intrahepatic T cells. J Dig Dis 2016;17:464-74.
133. Bruzzi S, Sutti S, Giudici G, Burlone ME, Ramavath NN, et al. B2-Lymphocyte responses to oxidative stress-derived antigens contribute to the evolution of nonalcoholic fatty liver disease (NAFLD). Free Radic Biol Med 2018;124:249-59.
134. Zhang JC, Chen WD, Alvarez JB, Jia K, Shi L, et al. Cancer immune checkpoint blockade therapy and its associated autoimmune cardiotoxicity. Acta Pharmacol Sin 2018;39:1693-8.
135. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017;389:2492-502.
136. Yau T, Hsu C, Kim TY, Choo SP, Kang YK, et al. Nivolumab in advanced hepatocellular carcinoma: Sorafenib-experienced Asian cohort analysis. J Hepatol 2019;71:543-52.
137. Finn RS, Ryoo BY, Merle P, Kudo M, Bouattour M, et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: a randomized, double-blind, phase III trial. J Clin Oncol 2020;38:193-202.
138. Pishvaian MJ, Lee MS, Ryoo BY, Stein S, Lee KH, et al. Updated safety and clinical activity results from a phase Ib study of atezolizumab + bevacizumab in hepatocellular carcinoma (HCC). Ann Oncol 2018;29:viii718-9.
139. Roche’s Tecentriq in combination with Avastin increased overall survival and progression-free survival in people with unresectable hepatocellular carcinoma. Available from: https://www.roche.com/media/releases/med-cor-2019-10-21.htm [Last accessed on 14 Apr 2020].
140. Faivre S, Rimassa L, Finn RS. Molecular therapies for HCC: looking outside the box. J Hepatol 2020;72:342-52.
141. Tang ZS, Hao YH, Zhang EJ, Xu CL, Zhou Y, et al. CD28 family of receptors on T cells in chronic HBV infection: Expression characteristics, clinical significance and correlations with PD-1 blockade. Mol Med Rep 2016;14:1107-16.
142. Yin Y, Wu C, Song J, Wang J, Zhang E, et al. DNA immunization with fusion of CTLA-4 to hepatitis B virus (HBV) core protein enhanced Th2 type responses and cleared HBV with an accelerated kinetic. PLoS One 2011;6:e22524.
143. Schurich A, Khanna P, Lopes AR, Han KJ, Peppa D, et al. Role of the coinhibitory receptor cytotoxic T lymphocyte antigen-4 on apoptosis-Prone CD8 T cells in persistent hepatitis B virus infection. Hepatology 2011;53:1494-503.
144. Wang X, He Q, Shen H, Lu XJ, Sun B. Genetic and phenotypic difference in CD8(+) T cell exhaustion between chronic hepatitis B infection and hepatocellular carcinoma. J Med Genet 2019;56:18-21.
145. Sangro B, Gomez-Martin C, de la Mata M, Inarrairaegui M, Garralda E, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol 2013;59:81-8.
146. Duffy AG, Ulahannan SV, Makorova-Rusher O, Rahma O, Wedemeyer H, et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J Hepatol 2017;66:545-51.
147. Kelley RK, Abou-Alfa GK, Bendel JC, Kim TY, Borad MJ, et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. J Clin Oncol 2017;35.
148. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. New Engl J Med 2013;368:1509-18.
149. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. New Engl J Med 2014;371:1507-17.
150. Brudno JN, Maric I, Hartman SD, Rose JJ, Wang M, et al. T cells genetically modified to express an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J Clin Oncol 2018;36:2267.
151. Gao H, Li K, Tu H, Pan X, Jiang H, et al. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin Cancer Res 2014;20:6418-28.
152. Wu X, Luo H, Shi B, Di S, Sun R, et al. Combined antitumor effects of sorafenib and GPC3-CAR T cells in mouse models of hepatocellular carcinoma. Mol Ther 2019;27:1483-94.
153. Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J 2014;20:119-22.
154. Oelsner S, Waldmann A, Billmeier A, Roder J, Lindner A, et al. Genetically engineered CAR NK cells display selective cytotoxicity against FLT3-positive B-ALL and inhibit in vivo leukemia growth. Int J Cancer 2019;145:1935-45.
155. Chu J, Deng Y, Benson DM, He S, Hughes T, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia 2014;28:917-27.
156. Xiao L, Cen D, Gan H, Sun Y, Huang N, et al. Adoptive transfer of NKG2D CAR mRNA-engineered natural killer cells in colorectal cancer patients. Mol Ther 2019;27:1114-25.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Song XJ, Ma CH. Mechanisms and immunotherapies of HBV- and NAFLD-related hepatocellular carcinoma. Hepatoma Res 2020;6:27. http://dx.doi.org/10.20517/2394-5079.2020.05
AMA Style
Song XJ, Ma CH. Mechanisms and immunotherapies of HBV- and NAFLD-related hepatocellular carcinoma. Hepatoma Research. 2020; 6: 27. http://dx.doi.org/10.20517/2394-5079.2020.05
Chicago/Turabian Style
Song, Xiao-Jia, Chun-Hong Ma. 2020. "Mechanisms and immunotherapies of HBV- and NAFLD-related hepatocellular carcinoma" Hepatoma Research. 6: 27. http://dx.doi.org/10.20517/2394-5079.2020.05
ACS Style
Song, X.J.; Ma C.H. Mechanisms and immunotherapies of HBV- and NAFLD-related hepatocellular carcinoma. Hepatoma. Res. 2020, 6, 27. http://dx.doi.org/10.20517/2394-5079.2020.05
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 8 clicks
Cite This Article 8 clicks

 Like This Article 32
likes
Like This Article 32
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.