
Cancer immunotherapy for hepatocellular carcinoma


Abstract
Most hepatocellular carcinomas (HCCs) arise on a background of chronically inflamed liver, and thus are considered typical immunogenic cancers. Although there have been advances in treatment options for HCC, many patients still struggle with a limited chance of survival requiring further innovative approach. Especially for the advanced HCC, many other molecular targeted therapies had been evaluated without success. Based on the immunological mechanisms thought to be acting during HCC development, the effects of diverse immunomodulatory regimens such as therapeutic vaccination, immune checkpoint inhibitors, and adoptive cellular immunotherapy have been investigated. Notably, many strategies have been developed in adoptive cellular immunotherapy, including dendritic cells, cytotoxic T cells, natural killer cells, cytokine-induced killer (CIK) cells, and genetically engineered T cells. In recent clinical trials, adjuvant CIK cell immunotherapy increased progression free survival after curative treatment of HCC. Most recently, new immunomodulatory agents were introduced for oncological treatment, eventually leading to the clinical breakthrough of checkpoint inhibitors targeting cytotoxic T lymphocyte antigen-4 (CTLA-4) and programmed cell death-1 (PD-1). To date, very promising published evidence with checkpoint inhibitors in HCC has been reported in the clinical trials with anti-CTLA-4 agent tremelimumab and a large phase II trial with anti-PD-1 agent nivolumab. Further investigations of immuno-oncology potentially popularized the applications of immunotherapy in the various stages of HCCs, and thus immune-based therapies are the promising innovative approach for patients with HCC. Hopefully, the immuno-oncology will bring about a paradigm shift of anti-cancer treatment for HCC.
Keywords
Introduction
Hepatocellular carcinoma (HCC) ranks fifth as the most common cancer in the world and the second most common cause of cancer-related death, accounting for 70%-85% of primary liver cancers[1-3]. The current standard treatments for HCC offer a fair chance of survival but there are still many patients who struggle with only a limited chance of survival[4]. A majority of patients present with disease too advanced to be treated with curative modalities such as surgical resection, transplantation, or radiofrequency ablation (RFA)[2]. Although the Barcelona Clinic Liver Cancer (BCLC) guideline recommends sorafenib in advanced HCC, which proved a survival benefit of 2.8 months compared to the placebo group[5], many liver cancer centers still select multimodality approaches including transarterial chemoembolization (TACE), radiotherapy (RT), and hepatic arterial infusion chemotherapy (HAIC)[6,7].
Most HCCs arise on a background of chronically inflamed liver, and thus are considered typical immunogenic cancers[8]. Based on the immunological mechanisms thought to be acting during HCC development, the effects of diverse immunomodulatory regimens such as therapeutic vaccination, immune checkpoint inhibitors, and transfer of adoptive cellular immunotherapy, have been investigated[8,9]. In the 21st century, cell-based therapies developed to bolster human anti-tumor immunity represent a growing component of cancer therapeutics[10,11]. Of note, adoptive cellular immunotherapies have employed several types of immune cells, including dendritic cells (DCs), cytotoxic T lymphocytes (CTLs), lymphokine-activated killer (LAK) cells, cytokine-induced killer (CIK) cells, and natural killer (NK) cells[12]. In addition, therapeutic cancer vaccines utilizing tumor antigens with or without DCs have been investigated.
Immune suppressor cells comprising tumor-associated macrophages (TAMs), regulatory T cells (Tregs), or myeloid-derived suppressive cells (MDSCs) in the HCC tumor microenvironment, could disturb the immune surveillance resulting in cancer immune evasion or immune escape[13]. It is well known that the interactions of HCC cells with the immune cells and their factors of immune system play a major role in its progression[14,15]. Inadequate co-stimulation, failure of tumor-associated antigens (TAAs) processing and presentation by antigen-presenting cells (APCs), along with suppression of effector cells are proposed mechanisms that result in weakened immune response in HCC patients[15,16]. To complement these immunosuppressive tumor microenvironment of HCC, previous cancer immunotherapy has aimed so far to enhance immune cell activity to kill the HCC tumor cells.
In this regard, cancer vaccines help the immune system recognize and attack cancer cells[17]. Unlike preventive vaccine, which prevents a development of a certain disease in advance, therapeutic cancer vaccine aims to treat the existing cancer. DCs are professional APCs that serve as a key player for inducing and activating the effector anti-tumor CTLs. There is ample evidence to justify therapeutic DC vaccines in HCC[18]. Decreased function of peripheral blood DCs in patients with HCC is well established[19]. Up to date, although DC vaccines are used in various stages of clinical trials of HCC, unfortunately, no therapeutic cancer vaccine has been approved for HCC[19]. Meanwhile, the failure of these approaches for boosting immune responses by cancer vaccine using peptides or DCs, could be associated with the brake function in immunity (i.e., immune checkpoints)[20]. It is now clear that tumors modulate immune checkpoints as one of the mechanisms to escape anti-cancer immune surveillance.
These immune checkpoints are known to regulate different stages and signaling processes of the immune response[21]. At the initial stage of "priming" of naïve T cell activation, cytotoxic T lymphocyte associated antigen-4 (CTLA-4):B7 binding blocks stimulatory signals, and stops the development of potentially autoreactive T cells[22]. Compared to CTLA-4, the major role of programmed cell death 1 protein (PD-1) and its ligand, PD-L1, is related to regulate previously activated CTLs at the later "effector" stage of immune response[23]. In the tumor microenvironment, antigen-specific T cells induce PD-1 expression on reactive CTLs and upregulate PD-L1 in cancer cells[8,23].
The above immune checkpoint molecules are highly expressed in HCCs that are recognized as immunogenic tumors[24]. Also, the hepatitis B (HBV) and hepatitis C virus (HCV) infections, two major pathogens of HCC, have been shown to interfere with antiviral immunity via the immune checkpoint pathways[25-27]. Blocking these immune checkpoint molecules restores T cell function, which release the brakes on the anti-tumor immune surveillance, allowing the immune system to more effectively detect and kill the HCC tumor cells[2,20,28].
As "cancer immunotherapy comes of age"[29] in this era, the topic of "immuno-oncology in HCC" could be a timely one. In this review, we focus on the human clinical immunotherapy trials in HCC, according to the four major categories: (1) adoptive immunotherapies using CIK, NK and engineered T cells; (2) therapeutic cancer vaccine; (3) immune checkpoint blockades; and (4) combination of immunotherapies with other cancer treatments.
Adoptive cellular immunotherapy
Adoptive cellular immunotherapy is a form of passive immunization in which autologous effector cells are ex vivo sensitized and or expanded and then given back to the cancer patients[30]. To date, adoptive immunotherapy is one stone in the pillar of cancer immunotherapy, which relies on the various lymphocytes including tumor-infiltrating lymphocytes (TILs), CD8+ CTLs, CD56+ NK cells, LAK cells, CIK cells, and engineering T cells. As one of main immunotherapeutic strategies, adoptive immunotherapy is widely used in the current cancer clinical trials. A sizable portion of immunotherapy clinical trials for HCCs are adoptive cellular immunotherapies [Table 1][30].
Selected clinical trials with adoptive cellular immunotherapy for HCC
| Registered No. | Recruitment status | Start year | Phase | Immunotherapy | Included patients of HCC |
|---|---|---|---|---|---|
| NCT00161187 | Completed | 2001 | I | Therapeutic allogeneic lymphocytes: irradiated lymphocytes from a donor | Unresectable or metastatic disease |
| NCT01828762 | Completed | 2005 | I | Autologous immune killer cell | Locally advanced or metastatic HCC |
| NCT00699816 | Completed | 2008 | III | Immuncell-LC | Stage I/II, after curative treatment |
| NCT01749865 | Completed | 2008 | III | CIK | After radical resection |
| NCT00769106 | Completed | 2008 | III | CIK | After radical resection |
| NCT01024530 | Unknown | 2009 | II/III | Autologous immune killer cells with TACE | BCLC stage B/C |
| NCT01212341 | Completed | 2010 | I | MG4101: allogeneic NK cells | Solid tumors |
| NCT01147380 | Completed | 2010 | I | Liver NK cell inoculation with liver transplantation | Liver transplant recipient |
| NCT01174121 | Recruiting | 2010 | II | Autologous TILs and IL-2 with cyclophosphamide, fludarabine and pembrolizumab | Metastatic HCC who has received sorafenib |
| NCT01218867 | Completed | 2010 | I/II | Anti-VEGFR2 CAR CD8 and PBL with cyclophosphamide, IL-2 and fludarabine | Metastatic cancer |
| NCT01462903 | Unknown | 2011 | I | Autologous TILs and IL-2 | Metastatic HCC after primary operation, radiotherapy and chemotherapy |
| NCT01758679 | Recruiting | 2012 | IV | CIK and Licartin | Postoperative HCC |
| NCT01801852 | Recruiting | 2013 | I | Autologous NKT cell infusion | Refractory to conventional treatment |
| NCT01897610 | Recruiting | 2013 | II | Immuncell-LC with sorafenib | Stage III/IV |
| NCT02008929 | Recruiting | 2014 | II | MG4101: allogeneic NK cell | After curative resection |
| NCT01914263 | Recruiting | 2014 | I | Cord blood-derived CIKs | After radical resection |
| NCT02587689 | Recruiting | 2015 | I/II | Anti-MUC1 CAR T cells | MUC1+ malignancies |
| NCT02959151 | Recruiting | 2015 | I/II | GPC3-CAR T cell | HCC with GPC3 high expression |
| NCT02725996 | Not yet recruiting | 2016 | II | Autologous NK cells | Stage I/II, after curative treatment |
| NCT02856815 | Not yet recruiting | 2016 | II | Immuncell-LC | BCLC stage B, tumor removal has been confirmed after TACE |
| NCT02715362 | Recruiting | 2016 | I/II | GPC3-CAR T cells with transcatheter arterial infusion (TAI) | Persistent cancer after at least one prior standard of care chemotherapy |
| NCT02839954 | Recruiting | 2016 | I/II | Anti-MUC1 CAR-pNK cells | MUC1+ malignancies |
| NCT02959151 | Recruiting | 2016 | I/II | GPC3-CAR T cell | HCC with GPC3 expression |
| NCT02854839 | Recruiting | 2016 | IIA | MG4101: allogeneic NK cells | Complete remission after TACE |
| NCT03175679 | Recruiting | 2017 | I | iNKT cells and IL-2 with 5-fluorouracil | Relapsed/advanced HCC, BCLC stage C |
| NCT03199807 | Not yet recruiting | 2017 | IB/II | Personalized new antigen reactive immune cells (NRT), radiotherapy | Advanced HCC, unresectable and no chemotherapy before |
| NCT03130712 | Recruiting | 2017 | I/II | GPC3-CAR T cells intratumor injection | Advanced HCC, persistent cancer after at least one prior standard of chemotherapy or surgery |
| NCT03132792 | Recruiting | 2017 | I | Autologous genetically modified AFPᶜ³³²T cells: genetically changed T cells that target alpha-fetoprotein | Positive for HLA-A*02:01 or HLA-A*02:642 allele |
| NCT03302403 | Not yet recruiting | 2017 | N/A | Autologous T cells transduced with CAR recognizing CD19, BCMA, GPC3 and Claudin18.2 | Advanced HCC with previous ablation or resection in the last 4 to 12 weeks |
| NCT02905188 | Not yet recruiting | 2018 | I | GPC3-CAR T cells with fludarabine and cytoxan | BCLC stage A/B/C |
| NCT03441100 | Not yet recruiting | 2018 | I | IMA202 Product (CAR T cell) with fludarabine and cyclophosphamide | HCC not amenable to treatments with curative intent |
In 1989, regression of tumor size in ten HCC patients was shown after treatment with LAK cells combined with interleukin-2 (IL-2)[31]. Later, two separate, but similar, clinical trials combining adriamycin chemotherapy with LAK cells after hepatoma resection were performed in 1991 and 1995[32,33]. The former study showed a decrease in postoperative recurrence rate of HCC[32]. However, in the latter study in 1995, there was no statistically significant difference between the two groups in the survival rate[33].
Another source of adjuvant immunotherapy is TILs[34]. TILs acquired from patients with hepatic malignancies, activated by IL-2 and anti-CD3 antibody and labeled with indium-111 were found to move to the tumor sites preferentially[34]. This might augment the antitumor effects of adoptive immunotherapy. In 1997, TILs isolated from resected tumors of 12 patients with HCC were activated and expanded in vitro by IL-2, and then infused to the patients[35]. In this study, TIL infusion as an adjuvant immunotherapy for HCC patients significantly decreased recurrence rate at 6 and 12 months compared to the control group.
Another promising cellular immunotherapy as the adjuvant setting for HCC involves CIK cell immunotherapies. Also, the recent clinical trials from many Asian-pacific countries reported that adjuvant CIK cell immunotherapy increased progression free survival (PFS) after curative treatment for HCC[30,36,37].
Adoptive immunotherapy using CIK cells
CIK cells are heterogeneous cell population consisting of CD8+ CTLs, CD56+ NK cells and both CD3+CD56+ NK like T (NKT) cells that were first discovered in the 1990s[11,37]. CIK cells display both anti-tumor ability of antigen specific CD8+ CTLs and non-major histocompatibility complex (MHC) restricted cancer cell killing capacity of NK cells [Figure 1][38]. Earlier clinical studies have shown a potent antitumor activity of CIK cells against various types of tumors[36].
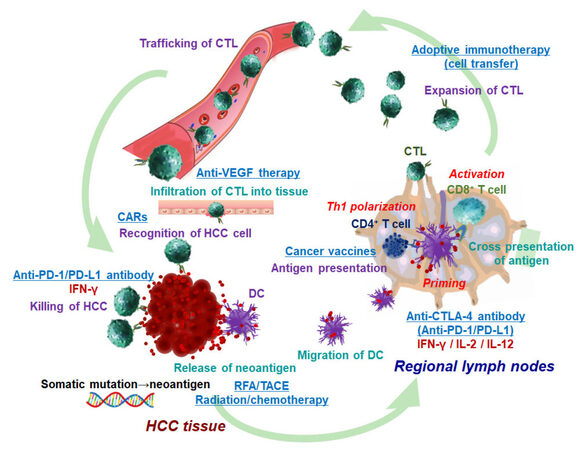
Figure 1. Cancer-immunity cycle and targets of immune therapies. Hepatocellular carcinoma (HCC) cells produce various tumor-associated antigens (TAAs) and neoantigens; the latter derive cancer-specific somatic mutations. The initial steps of anti-tumor immune response include uptake of TAAs and neoantigens by dendric cells (DCs). After that, the DCs migrate into regional lymph nodes and present processed antigen to CD4+ T cells. Antigen recognition leads to proliferation of CD4+ T cells and induction of interferon (IFN)-γ in the presence of IL-12 and type I IFN (Th1 polarization). The cross-presentation of antigenic peptide to CD8+ T cells by DCs facilitates the development of antigen-specific CD8+ cytotoxic T lymphocytes (CTLs). After the trafficking of CTLs to HCC tissues, the antigen-specific CTLs exert anti-tumor effecter function through release of humoral factors, such as granzyme B and perforin, and interaction with death receptors on tumor cells. Locoregional therapies and systemic chemotherapies should enhance the release of neoantigens and TAAs through HCC cell death. Cancer vaccines can promote the antigen presentation; anti-CTLA-4 antibody mainly acts in priming phase and facilitates the Th1 polarization and activation of CD8+ T cells. Adoptive immune therapies (immune cell transfer) increase the peripheral anti-tumor immune cells; chimeric antigen receptor (CAR) T cells can more directly targets cancer cells compared to conventional adoptive immunotherapies. Anti-vascular endothelial growth factor (VEGF) potentially induce infiltration of T cell into tumor tissues. Anti-PD-1/PD-L1 antibodies block the co-inhibitory signal of CD8+ T cells and induce cancer cell killing. CTLA-4: cytotoxic T lymphocyte associated antigen-4; PD-1: programmed cell death 1 protein; PD-L1: PD-1 ligand
In 2000, CIK cell immunotherapy is demonstrated to be a safe and feasible treatment that can lower recurrence rate and improve PFS after curative resection of HCC[37]. In this randomized trial, CIK cells were infused 5 times during the first 6 postoperative months. During the median follow-up of 4.4 years, recurrence rate reduced remarkably by 18% in the CIK cell treatment group (59%, 45/76) compared with that in the control group (77%, 57/74). Moreover, the PFS was significantly improved in the CIK treatment group (P = 0.01). All of the adverse events (AEs) were grade I or II and self-limiting. AEs associated with treatment were fever (47%), headache (4%), nausea (4%), dizziness (1%), itching (1%) and tachycardia (1%).
In 2004, the influence of autologous CIK cells was investigated in terms of phenotypes of CIK effector cells, peripheral T lymphocyte subsets and DC subsets in 13 HCC patients who had liver cirrhosis and chronic HBV infection[39]. Peripheral blood mononuclear cells (PBMCs) were collected by a blood cell separator, and then expanded by priming them with interferon-gamma (IFN-γ), monoclonal antibody against CD3 and IL-2. After two weeks of in vitro incubation, the percentages of CD8+ CTL and CD3+CD56+ NKT cells increased significantly from 33.5% and 7.7% to 36.6% and 18.9%, respectively. CIK cell therapy increased the proportions of type I DC and type II DC from 0.59% and 0.26% to 0.85% and 0.43%, respectively (all P < 0.01). These results indicated that autologous CIK cells could efficiently improve the immunological status in HCC patients.
In 2009, a randomized trial was conducted to investigate the impact of postoperative adjuvant CIK immunotherapy on the prognosis[40]. In 127 HCC patients who underwent radical hepatic resection, CIK cell therapy significantly increased the disease-free survival rate compared with the control group. However, the overall survival (OS) was not significantly different[40].
In 2010, the impact of adjuvant CIK therapy after TACE combined with sequential RFA on tumor recurrence was demonstrated in relation to serum AFP level[41]. After curative TACE plus RFA therapy, 83 patients with AFP level less than 37.5 ng/mL (1.5 times the normal range) were randomly assigned for CIK immunotherapy or for best supportive treatments. CIK cell infusions were given either intravenously or via common hepatic arteries every week for at least 4 times. During the follow-up of 12 months, AFP levels in the CIK group but not in the control group gradually decreased from the baseline levels, and those reduced levels were maintained. Furthermore, the reduced AFP levels of the CIK group were lower than the AFP levels of the control group with statistical significance both in 1 month (P < 0.05) and in 3 months (P < 0.05) after treatment. The 1-year recurrence rate was 7.1% for the CIK study group and 23.1% for the control group (P = 0.04). In addition, the authors showed that HBV DNA titer decreased after CIK cell therapy. They concluded that the adjuvant CIK cell therapy can reduce the serum AFP and HBV DNA levels and decrease the 1-year recurrence rate of patients with HCC after curative TACE plus RFA[41].
The most recent clinical trial, reported in 2015, demonstrated that adjuvant CIK cell immunotherapy after curative treatment for HCC increased not only the PFS but also the OS[36]. In this study, 230 patients with HCC who were treated by surgical resection, RFA, or percutaneous ethanol injection were included. Patients were assigned randomly to receive adjuvant CIK cell immunotherapy 16 times during 60 weeks or no adjuvant therapy. The median time of PFS was 44.0 months in the CIK cell therapy group and 30.0 months in the control group (P = 0.01). Hazard ratios (HR) of all-cause death (0.21; 95% CI, 0.06-0.75; P = 0.008) and HR of cancer-related death (0.19; 95% CI, 0.04-0.87; P = 0.02) were significantly lower in the CIK cell immunotherapy group compared with the control group. This study proved that adjuvant immunotherapy with activated CIK cells increase PFS as well as OS of HCC patients after the curative treatments including surgery and RFA[36]. However, the efficacy of CIK immunotherapy for HCC needs to be further validated, by extending the sample size and follow up duration of the HCC research cohort.
NK cell based immunotherapy
Human NK cell, recognized as a CD3-CD56+ lymphocyte, is a very important part of innate immune system. It provides surveillance toward tumor cells eliminating those when detected. Thus, NK cell was suggested to be used for cancer therapy[12]. NK cells are characterized by an inborn receptor diversity which allows NK cells to recognize and to respond to different pathogens including virus-infected cells and neoplastic cells without prior sensitization or acquired receptor rearrangement[17]. It is well known that NK cells can be long-lived, remember past exposures, and interact with MHC class I molecules to acquire full function. NK cell function is tightly regulated by signals from natural cytotoxicity receptors, CD16 receptor for antibody-dependent cellular cytotoxicity (ADCC), C-type lectins, and killer cell immunoglobulin-like receptors (KIR).
Recently, there have been advances in ex vivo techniques of NK cell activation and expansion[17]. Autologous cytokine-stimulated NK cell therapy has been tried with multiple tumors such as renal cell carcinoma, glioblastoma and myeloma[42]. On the other hand, allogeneic NK cell therapy is particularly beneficial because it can enhance the anti-cancer efficacy of NK cells via donor-recipient incompatibility in terms of KIRs on donor NK cells and MHC class I on recipient tissues[43]. Thus, the use of allogeneic NK cell therapy is being actively investigated in hematologic malignancies with or without hematopoietic stem cell transplantation[12]. In these settings, HLA-haploidentical NK cells have been used mostly.
In HCC patients, impaired functions of DC and NK cell were observed in relation to elevated level of serum MHC class I-related chain A (MICA), an inhibitory ligand for NKG2D[44]. Increase of Tregs and MDSCs were also known to contribute to the functional impairment of NK cells and in turn the reduced anti-tumor immune response[30,45]. In contrast, increased number of NK cells in peripheral blood and tumor tissues accompanied by an upregulation of related chemokines was an immune-gene signature which determines a long-term survival in resectable HCC[46].
A group in the University of Miami suggested that NK cells extracted from donor liver graft perfusate could be used as a source of a treatment to reduce recurrence rate after liver transplantation (LT)[47]. When the NK cells acquired from donor graft was activated with IL-2, activation markers and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), which is critical for NK cell mediated cancer cell death, were greatly upregulated. The authors concluded that the adoptive transfer of IL-2 stimulated NK cells from deceased donor liver graft could be a promising treatment for LT patients with HCC[47]. Moreover, the cytokines and chemokines released by activated NK cells may stimulate both innate and adaptive immune responses toward cancer.
In this regard, a phase I clinical trial was conducted to evaluate the feasibility and safety of the adoptive transfer of activated NK cells extracted from cadaveric donor liver graft perfusate after LT (NCT01147380). According to preliminary results posted on the website clinicaltrials.gov, there seemed to be no side effects or serious adverse events. There are also ongoing clinical trials on adoptive NK cell therapy. A phase II clinical trial (NCT02008929) initiated in August 2014 aims to evaluate the safety and efficacy of ex vivo expanded allogeneic NK cells (MG4101) as a secondary treatment after curative liver resection on advanced HCC patients with a high risk of recurrence. Adoptive cell transfer of allogeneic NK cells that came from a totally unrelated donor had been demonstrated to be safe without any significant side effects [Figure 1][30]. Notably, another multi-center, open label, phase IIA clinical trial (NCT02854839) with a purpose to evaluate the safety and efficacy of allogeneic NK cell (MG4101) therapy for intermediate-stage HCC patients after TACE started in September, 2016.
Adoptive immunotherapy using genetically engineered T cell receptor or chimeric antigen receptors
The emergence of immuno-oncology as the first broadly successful strategy for metastatic cancer will require clinicians to integrate this new pillar of medicine with the pillars of already established therapeutic methods such as chemotherapy, RT and targeted small molecule compounds[48]. Chimeric antigen receptor (CAR) T cell therapy combines adoptive cellular immunotherapy with targeted molecular therapy, and it has proven that engineered immune cells can serve as a powerful new class of cancer therapeutics [Figure 1]. Adoptive immunotherapy retargeting T cells to CD19 via CAR is an investigational treatment capable of inducing complete tumor regression of B-cell malignancies[49]. The major hurdle in developing CAR T cell therapy is the on-target off-tumor toxicity as was shown in a metastatic colon cancer patient who died 5 days after infusion of ErbB2 targeting CAR T cell[50]. Expression of ErbB2 on lung epithelium even with a low level brought a detrimental result. Therefore, finding a target antigen which is effective enough for cancer-killing and at the same time safe enough for the normal tissue is a key requirement in the development of CAR T cell therapy for HCC[51].
It has been demonstrated that HBV antigens can serve as a tumor specific antigen in HBV related HCC and can be targeted by adoptively transferred HBV-specific T cell receptor (TCR) redirected T cells in preclinical models[52,53]. Recently, Qasim et al.[54] have reported the clinical results of immunotherapy for HCC metastases with autologous TCR redirected T cells, targeting HBV surface antigen (HBsAg) in a liver transplant patient. Autologous T cells genetically modified to express an HBsAg specific TCR were infused with no immediate infusion-related toxicities despite the patient’s frail condition. The authors confirmed that HBV antigens were expressed in metastatic lesions of HCC and demonstrated that tumor cells were recognized in vivo by the engineered T lymphocytes. Furthermore, the engineered T cells successfully survived, expanded, and mediated a reduction in HBsAg levels without exacerbation of liver inflammation or other toxicity. Although the clinical efficacy in this patient was not established with end-stage metastatic HCC, these results confirm the feasibility of autologous CAR T cell immunotherapy targeting HBsAg in HBV associated HCC[54].
In 2010, Food and Drug Administration (FDA) approved a phase I/II study of CAR T cell immunotherapy targeting vascular endothelial growth factor receptor (VEGFR)-2 (NCT01218867), where HCC patients without hepatitis B and C were included[55]. The result of this study is still awaited. Recently, phase I/II clinical trials with CAR T cells targeting glypian-3 (GPC3), alpha-fetoprotein (AFP), and mucin 1 (MUC1) are being conducted [Table 1][56,57]. Moreover, a CAR NK cell immunotherapy targeting MUC1 is being conducted (NCT02839954)[58].
Taken together, adoptive cellular immunotherapy in HCC is a safe and feasible treatment. However, its efficacy in preventing recurrence and prolonging survival in advanced HCC patients remains controversial[59]. Indeed, cellular immunotherapy seems to be more effective in patients with low burden of micrometastases[36]. The current situation lacking sufficiently effective cellular immunotherapy for advanced stages of HCC calls for further improvement in immunotherapeutic strategies and additional approaches with immune checkpoints modulators.
Therapeutic cancer vaccines
Therapeutic cancer vaccine is an important part of cancer immunotherapy. Vaccination with cancer antigens or peptides is believed to help the immune system to recognize cancer cells and attack them more easily [Tables 2 and 3]. In therapeutic cancer vaccine, DC is an important component. As professional APCs, DCs serve as an essential link between innate and adaptive immune systems[17]. Two functional states of DC are described, as immature or mature DCs. Several factors can induce maturation of DCs. Mature DCs are specialized APCs, which express high levels of surface MHC I and MHC II class, as well as the appropriate costimulatory molecules required for T-cell activation. One of the most important functions of mature DCs is the rapid production of high amounts of type I IFN, especially in response to virus-derived nucleic acids through activation of Toll-like receptors (TLRs), both TLRs 7 and 9.
Selected clinical trials with therapeutic cancer vaccine immunotherapy for HCC
| Registered No. | Recruitment status | Start year | Phase | Immunotherapy | Included patients of HCC |
|---|---|---|---|---|---|
| NCT00004604 | Completed | 1997 | I | CEA RNA-pulsed DC cancer vaccine | Metastatic adenocarcinoma expressing CEA that has failed conventional therapy |
| NCT00019331 | Completed | 1997 | II | Ras peptides and IL-2 or GM-CSF | Solid tumors potentially expressing mutant Ras |
| NCT00005629 | Completed | 1999 | I/II | AFP gene HCC vaccine | HLA-A*0201 positive, serum AFP levels > 2 times above the upper limit of normality |
| NCT00022334 | Completed | 2001 | I/II | AFP peptide-pulsed autologous DC | HLA-A*0201 positive, HCC with a serum AFP determination > 30 ng/mL |
| NCT00028496 | Completed | 2001 | I | Recombinant fowlpox-CEA(6D)/TRICOM vaccine, sargramostim and recombinant fowlpox GM-CSF vaccine adjuvant | Failed standard curative options and no standard palliative options required within the next 8weeks |
| NCT00027534 | Completed | 2002 | I | TRICOM-CEA(6D) | Histologically confirmed advanced or metastatic malignancy expressing CEA |
| NCT00629759 | Completed | 2006 | I | JX-594: recombinant vaccinia virus (TK-deletion plus GM-CSF) | Progressing HCC |
| NCT00610389 | Unknown | 2008 | II | DC with PEG-IFN alfa and GM-CSF | HCC not amenable of curative treatment with Child´s stage A or B |
| NCT01266707 | Unknown | 2010 | I | VEGFR1 and VEGFR2 specific epitope vaccine | Unresectable or treatment-resistant HCC |
| NCT01828762 | Completed | 2012 | N/A | DC incubated with irradiated autologous tumor stem cells in GM-CSF | BCLC stage A/B, after resection and TACE |
| NCT01974661 | Completed | 2013 | I | COMBIG-DC (ilixadencel): allogenic dendrite-cell based therapeutic vaccine | BCLC stage B/C, not eligible for curative treatment or TACE |
| NCT02232490 | Recruiting | 2014 | III | Hepcortespenlisimut-L (V5) | HCC with AFP serum test higher or equal to 30 IU/mL |
| NCT02409524 | Recruiting | 2016 | II | AlloVax, AlloStim and CRCL | Unresectable HCC with minimum 90 days of sorafenib treatment |
| NCT03203005 | Recruiting | 2017 | I/II | A new cancer vaccine called IMA970A combined with CV8102 with cyclophosphamide | BLCL stage 0/A/B following any standard treatment |
Current trials on combinational immunotherapy strategies in HCC
| Registered No. | Recruitment status | Start year | Phase | Immunotherapy | Included patients of HCC |
|---|---|---|---|---|---|
| NCT01522820 | Completed | 2012 | I | DEC-205/NY-ESO-1 fusion protein CDX-1401 with sirolimus | After resection or TACE |
| NCT01853618 | Completed | 2013 | I/II | Tremelimumab with TACE, RFA, SBRT or Cryoablation | BCLC stage B/C |
| NCT01821482 | Recruiting | 2013 | II | DC-CIK | After complete resection or TACE |
| NCT02562755 | Recruiting | 2015 | III | Pexastimogene devacirepvec (Pexa Vec) with sorafenib | Advanced HCC (BLCL-C or AASLD-B) |
| NCT02487017 | Recruiting | 2015 | II | DC-CIK with TACE | After TACE treatment |
| NCT02432963 | Active, not recruiting | 2015 | I | Modified vaccinia virus Ankara vaccine expressing p53 and pembrolizumab | Advanced HCC, confirmed p53 involvement, failed to or refusal to standard therapy |
| NCT02821754 | Recruiting | 2016 | II | Durvalumab and tremelimumab with RFA, cryotherapy or TACE | Multiple HCC technically amenable to ablative therapy |
| NCT02837029 | Recruiting | 2016 | I | Nivolumab with Yttrium Y 90 glass microspheres | Stage III/IV |
| NCT02795429 | Recruiting | 2016 | I/II | PDR001 with or without INC280 | Advanced, recurrent or metastatic HCC |
| NCT02886897 | Recruiting | 2016 | I/II | DC-CIK and anti-PD-1 antibody | Advanced HCC |
| NCT03259867 | Recruiting | 2017 | IIA | Nivolumab or pembrolizumab with trans-arterial tirapazamine embolization | Advanced HCC (BCLC-C), progressive disease (PD) on, intolerant of or refusing sorafenib |
| NCT03380130 | Recruiting | 2017 | II | Nivolumab with selective internal radiation therapy | Candidates for locoregional therapy using selective internal radiation-spheres |
| NCT03277352 | Recruiting | 2017 | I/II | INCAGN01876, pembrolizumab and epacadostat | Locally advanced or metastatic disease |
| NCT03241173 | Recruiting | 2017 | I/II | INCAGN01949, nivolumab and/or ipilimumab | Locally advanced or metastatic disease |
| NCT03126110 | Recruiting | 2017 | I/II | INCAGN01876, nivolumab and/or ipilimumab | Locally advanced or metastatic disease |
| NCT03095781 | Recruiting | 2017 | I | Hsp90 inhibitor XL888 and pembrolizumab | Stage IV or locally advanced unresectable gastrointestinal adenocarcinomas |
| NCT03203005 | Recruiting | 2017 | I/II | A new cancer vaccine called IMA970A and CV8102 with cyclophosphamide | BLCL stage 0/A/B following any standard treatment |
| NCT03067493 | Recruiting | 2017 | II | Neo-MASCT (antigen-pulsed DC, autologous specific cytotoxic T-cells) | Primary HCC with previous RFA or resection |
| NCT03071094 | Recruiting | 2017 | I/IIA | Pexastimogene devacirepvec (Pexa Vec) and nivolumab | Advanced HCC per EASL-EORTC |
| NCT03482102 | Recruiting | 2018 | II | Tremelimumab and durvalumab with radiation | Locally advanced/unresectable or metastatic disease |
| NCT03439891 | Recruiting | 2018 | II | Nivolumab with sorafenib | Unresectable, locally advanced and/or metastatic HCC |
| NCT03511222 | Not yet recruiting | 2018 | I | Vorolanib and pembrolizumab | A solid tumor that can be treated with either pembrolizumab or nivolumab as part of standard of care |
Although immunotherapy is not recommended for the clinical management of HCC patients under current guidelines, several different immunotherapy vaccine strategies have been investigated in the last decade for HCC[15]. Moreover, significantly lower numbers of CD83+ DCs (mature and activated DCs) have been found in liver tissue of patients with HCC compared with liver cirrhosis patients[60].
Many of the HCC clinical studies on therapeutic cancer vaccines have focused on AFP-based vaccinations since the majority of human HCCs overexpress AFP[15]. CD8+ T cell epitopes derived from AFP peptides were used to carry on the first HCC vaccine clinical trial. AFP positive HCC patients received three biweekly intradermal injections of the AFP peptides. All of the patients (n = 6) developed the AFP-specific T cell responses, clearly proving the immunogenicity of AFP even in the environment of high circulating levels of AFP in HCC patients[61]. Subsequently, the authors conducted another phase I/II trial. This time, they immunized AFP positive HCC patients with autologous DCs ex vivo pulsed by AFP epitopes. DCs were prepared from PBMCs cultured with granulocyte-macrophage colony-stimulating factor and IL-4 for 7 days[62]. In this study, AFP-specific T cell response and increased IFN-γ production were shown. Despite this immune response, clinical response was not observed. The authors found the reason for it in a subsequent study that CD4+ T cell help was lacking, which resulted in non-functional AFP-specific CD8+ T cells[63]. Unfortunately, a limited number of clinical trials for HCC have been conducted based on therapeutic vaccine immunotherapy.
Meanwhile, the bioactivity and beneficial effects of DC infusion were evaluated in HCC patients following trans-catheter hepatic arterial embolization (TAE). In this study, tumor recurrence was not completely prevented in patients with TAE and DC infusion than in those with TAE alone. However, TAE with DC infusion enhanced the tumor-specific immune responses more effectively than TAE alone. The authors demonstrated that combination therapy using TAE together with DC infusion is safe for patients with cirrhosis and HCC[64].
In another phase II study, the safety and efficacy of vaccination with mature autologous DCs pulsed with a liver tumor cell line lysate (HepG2) have been investigated in patients with advanced HCC and not suitable for radical or loco-regional therapies[65]. The authors showed that autologous DC vaccination in patients with HCC is safe and well tolerated with evidence of antitumor efficacy with generation of antigen-specific immune responses in some cases. More recent study, reported in 2013, also showed similar results. The safety and efficacy of the autologous pulsed DC vaccine was compared to supportive treatment in advanced HCC patients. They showed that autologous DC vaccination in advanced HCC patients was safe and well tolerated. Additionally, both CD8+ CTL and serum IFN-γ were elevated after DC vaccine[66].
Actually, to date, no vaccine has been approved so far for HCC treatment[19]. Further investigations and improvements of therapeutic cancer vaccines will be required to achieve better efficacies in HCC patients.
Immune checkpoint blockades in HCC
During the last decade, new immuno-oncological treatments were introduced for diverse cancers, eventually leading to the clinical breakthrough of immune checkpoint blockades targeting CTLA-4, PD-1, PD-L1 and PD-L2[67,68]. Under physiological conditions these checkpoint molecules resolve T cell activation to maintain inflammatory homeostasis, also limit collateral tissue damage and prevent unwanted auto-immunity, as observed in response to chronic viral hepatitis[26,27]. Meta-analysis data on solid tumors have suggested that overexpression of PD-L1 in tumor cells, as well as in APCs of tumor microenvironment, is associated with poor prognosis in patients with malignant tumors including HCC[21,69]. The subsequent PD-1/PD-L1 interaction results in T-cell exhaustion and immune evasion by cancer cells[70]. The inhibitory effects of the PD-1/PD-L1 pathway on T cell-mediated antitumor immunity are commonly reported regarding HCC carcinogenesis, and the PD-L1 is over-activated in HCC[9,71]. Also the PD-1/PD-L1 interaction is known to be associated with persistent HBV and HCV viremia, or the progression of HCC, by suppressing specific T-cell immunity and thereby inducing immune tolerance or immune escape of cancer cells[8,27].
Notably, immune checkpoint inhibitors have proven effective in patients who are refractory to tyrosine kinase inhibitors (TKIs) such as sorafenib, and recently several blocking antibodies targeting PD-1 or CTLA-4 have shown promising results in advanced HCC patients who received previous treatment with sorafenib[20,28,72]. Compared to TKIs, immunotherapy has several advantages for the treatment of cancer, as its effects are not hampered by common mutations or neoantigen heterogeneity of tumor cells[28,73]. Therefore, immuno-oncology agent is effective regardless of the response to prior therapies, and also a durable response can be expected due to adaptive immunity to the cancer cells[74]. However, the profile of AEs is completely different from those of other cytotoxic and molecular targeting agents[28]. The tolerability of immuno-oncology agents generally depends on the severity of immune-related AEs (irAEs), although the majority of irAEs are mild and manageable[20,75].
Different clinical trials are currently underway to investigate the safety and efficacy of checkpoint inhibitors for HCC immunotherapy as in monotherapy or in combination [Table 3][28].
The PD-1/PD-L1 pathway
Higher intra-tumoral expression of PD-1/PD-L1 had been associated with significantly poorer PFS and OS after hepatectomy as well as postoperative recurrence in HCC[76]. It was shown that PD-1 immune checkpoint inhibitor therapies have a strong therapeutic effect on patients with high levels of PD-L1 expression[77]. This could be due to the ability of the PD-1/PD-L1 pathway to act as an anti-apoptotic receptor on cancer cells [Figure 1][23,69].
To date, two kinds of anti-PD-1 (nivolumab and pembrolizumab) and anti-PD-L1 (durvalumab, avelumab) antibodies have been applied for clinical trials in HCC and nivolumab, pembrolizumab, and avelumab are in development as monotherapy[20,28]. Two phase III studies are currently ongoing: a comparison of nivolumab and sorafenib in the first line setting for advanced HCC (CheckMate 459), and a comparison of pembrolizumab and a placebo in the second line setting for patients with advanced HCC who progressed on sorafenib (KEYNOTE 240)[20,78].
Nivolumab, a fully human IgG4 anti-PD-1 monoclonal antibody, was granted accelerated approval from U.S. FDA on September 2017 for treatment of HCC patients who were previously treated with sorafenib. Approval was based on findings in a phase I/II, open-label, non-comparative, dose escalation and expansion trial (CheckMate 040) consisting of patients with HCC and Child-Pugh A cirrhosis[78]. Between November 2012 and August 2016, 262 eligible patients were treated (48 patients in the dose-escalation phase and 214 in the dose-expansion phase). At the American Society of Clinical Oncology (ASCO) meeting in 2015, results of the dose-escalation trial of CheckMate 040 were presented; 68% of patients had drug-related AEs, the complete response (CR) rate was 5%, and the partial response (PR) rate 14%. The safety profile of nivolumab is generally consistent with what was previously-reported in other tumor types. Twelve (25%) of 48 patients in the dose-escalation phase had grade 3/4 treatment-related AEs. Autoimmune disease and hepatic dysfunction, which were the AEs of initial concern, were not observed[20]. In the 2017 ASCO meeting, final results of the phase I/II CheckMate 040 study with nivolumab in advanced HCC showed favorable results with objective response rate (ORR) 20% and disease control rate (DCR) 64%[78]. The OS rate of the fixed dose of 3 mg/kg nivolumab group at 12 months was 62%. Considering that a high proportion (66%) progressed on sorafenib treatment, these outcomes appear to be extremely good. In addition, nivolumab was effective regardless of prior sorafenib administration and viral status, indicating that nivolumab could be effective even in cases refractory to sorafenib[20,28,78]. However, the ORR of HBV-positive cases was lower (14%) compared to non-HBV cases (20%-23%)[28,78]. There was no significant association between PD-L1 expression in HCC and the response to nivolumab[20,78].
Another anti-PD-1 antibody, pembrolizumab, was associated with PR and prolongation of survival in a patient with progressive metastatic HCC while being treated with sorafenib[79]. The randomized, placebo-controlled phase III KEYNOTE 240 study (NCT02702401) to compare the efficacy and safety of the pembrolizumab with best supportive care for the treatment of advanced HCC after failure to sorafenib is ongoing. Recently, findings from the KEYNOTE 224 study (NCT02702414), open-label phase II trial investigating pembrolizumab monotherapy in patients with advanced HCC who were previously treated with sorafenib, were presented at the 2018 Gastrointestinal Cancers Symposium. Results showed the ORR of 16.3% (95% CI, 9.8%-24.9%; n = 17/104) with CR of 1% (95% CI, 0.0%-5.2%) and PR of 15.4% (95% CI, 9.1%-23.8%). The DCR was 61.5% (95% CI, 51.5%-70.9%; n = 64/104) and median PFS time was 4.8 months (95% CI, 3.4-6.6 months), with a 6-month PFS rate of 43% and 6-month OS rate of 78%.
Furthermore, a clinical trial of monotherapy agents targeting PD-L1, such as avelumab, has also been conducted in advanced HCC patients (NCT03389126)[28].
The CTLA-4 pathway
Tremelimumab is an IgG2 type anti-CTLA-4 antibody that was evaluated in a phase II clinical trial (NCT01008358) investigating the tremelimumab monotherapy in 21 patients with HCV-related HCC[80]. This study with tremelimumab in HCV infected HCC patients has shown a good safety profile along with a promising PR rate of 17.6% and a time-to-progression (TTP) of 6.5 months[80]. In this CTLA-4 trial, a transient complete virologic response or decrease in HCV viral load was also observed in most patients with the DCR of 76.4%[28,80]. The trial demonstrated efficacy of tremelimumab monotherapy in HCC patients and the anti-tumoral and antiviral effects that warrant further investigation.
Notably, the feasibility of combined locoregional therapies and tremelimumab administration was investigated in patients with liver cirrhosis and HCC[81]. The use of tremelimumab plus RFA, cryoablation, or TACE in patients with BCLC B or C HCC was associated with ORR of 26.3% in areas outside of the ablation zone, and median TTP was 7.4 months. The combination of tremelimumab with local tumor ablation is a smart synergistic mechanism, because, in patients responding to local ablative therapy, prolonged TTP gives time for immunotherapy to unfold[72]. In addition, local tumor ablation releases TAA from apoptotic or necrotic HCC tissue, which in turn accelerates tumor specific APCs and CTLs activation, resulting in immunological synergy evolving from the combination of both treatment modalities [Figure 1]. Also, in this study, 12 of 14 patients with quantifiable HCV experienced a marked reduction in viral load, especially in the patients with PR. Studies have shown that tremelimumab in combination with tumor ablation is a potential new treatment for patients with advanced HCC[81]. Particularly, the positive antiviral immune responses may act as a surrogate for disease control in HCC immunotherapy[72].
In summary, immune checkpoint blockade therapy (anti-PD-1 and anti-CTLA-4) had a favorable safety profile in patients with HCC[20]. It can be used safely in patients with HBV and HCV infection, and its high ORR was a great achievement compared to the rates achievable with other types of immunotherapy[28].
Other immune checkpoint pathways
Although anti-PD-1/PD-L1 antibody is a promising agent for the treatment of HCC, a considerable percentage of HCC patients could not attain satisfactory tumor control, likely due to the immune suppressive cellular components, humoral mediators, and diverse inhibitory checkpoint molecules[28,82]. Their crosstalk becomes more complex during tumor progression. Also, the continuous production of cytokines and chemokines in the inflamed liver and solid immunosuppressive stroma of HCC could induce the production of many types of suppressive checkpoint molecules[83,84].
Cellular components including MDSCs, TAMs, Tregs and type 2 helper T cells might facilitate the immune evasion of HCC tumor cells[20,83]. MDSCs also produce transforming growth factor (TGF)-β and IL-10 that lead to the suppression of CD56+ NK cell and CD8+ CTL activities[85]. TGF-β from MDSCs induces the expression of T-cell immunoglobulin and mucin-containing protein-3 (TIM-3) on TAMs, which is associated with galectin-9 and further facilitates the M2 polarization of macrophages in tumors[86]. Galectin-9, which is a ligand of TIM-3, also induces Treg stimulation and T cell exhaustion[83]. The TIM-3/galectin-9 signaling pathway reportedly mediates T-cell dysfunction in HBV-associated HCC, which might explain the poor ORR of HBV-associated HCC compared with that of non-HBV-associated HCC during the anti-PD-1 antibody administration[78,83,87].
Galectin-3 interacts with lymphocyte activation gene-3 (LAG-3) and inhibits CD8+ T cell and NK cell functions[83]. LAG-3 expression on TILs, along with PD-L1 on tumor cells, is also reported in HCC[88]. As in the PD-1/PD-L1 in HCC tumor, the galectin-3/LAG-3 expression is also associated with a poor prognosis in HCC patients[89]. Another report also showed up-regulated LAG-3 expression and impaired effector function of CD8+ CTLs in HBV-positive HCC patients[90]. Taken together, TIM-3, LAG-3, and PD-1 act synergistically and facilitate the HCC immune evasion resulting in worse prognosis[88]. According to these findings, TIM-3 and LAG-3, checkpoint molecules expressed on the effector T cell, could mediate resistance to the PD-1/PD-L1 blockade[86,88]. Given the fact that there are multiple players in the establishment of immune escape in HCC, anti-PD-1/PD-L1 therapy is being paired with agents targeting TIM-3 (NCT03099109) and LAG-3 (NCT01968109), respectively[83].
HCC immunotherapies in combination with other cancer treatments
While the present adoptive immunotherapy has been restricted to the patients with small tumor burdens so far, treatments using these engineered immune cells have generated some remarkable responses in patients with advanced cancer by combinational immunotherapy[91]. Ongoing investigations of adoptive immunotherapy combined with traditional HCC treatments, including surgery, locoregional interventions, and systemic chemotherapy, may achieve the best objective responses in various stages of HCCs[92,93]. In 2013, a retrospective study was conducted in 174 HCC patients from January 1999 to April 2012. Among them, 85 patients were given CIK cell infusion after treatment with TACE and RFA alone. The results demonstrated that CIK cell infusion significantly prolonged the PFS in patients compared to TACE or RFA monotherapy[94]. A different approach is pretreatment of HCC with TACE, RFA, or RT to induce inflammation of cancer cells, thereby creating conditions that favor tumor neoantigen generation prior to the initiation of immunotherapy[84].
Although the efficacy of immune checkpoint inhibitors in HCC is promising, the majority of the patients remain refractory, due to the immunosuppressive mechanisms of HCC comprising multiple humoral mediators and suppressive checkpoint molecules[82,83]. To enhance the anti-tumor activity, several studies on combined immune checkpoint blockades are being conducted [Table 3]. The most relevant combination is a CTLA-4 and PD-1/PD-L1 blockade[28]. The rationale of this strategy is based on the idea that if CD8+ CTL do not exist in cancer tissue, blockade of the PD-1/PD-L1 pathway cannot be expected to be efficacious. Therefore, blocking CTLA-4 may be an effective strategy to increase the number of activated effector T cells that infiltrate the tumor tissue[20]. Durvalumab, a monoclonal antibody to PD-L1, is currently evaluated in combination with an anti-CTLA-4 antibody (tremelimumab) for sorafenib-experienced HCC patients in a phase II trial (NCT02519348)[83]. Another anti-CTLA-4 antibody, ipilimumab, is also being analyzed in combination with the anti-PD-1 antibody, nivolumab, for evaluation of the safety and efficacy in HCC patients (NCT01658878, NCT03222076)[20].
Given the fact that molecular target agents could collectively block the signaling from various growth factors and affect immune effectors and the vasculature, the combination of TKIs and immune checkpoint inhibitors could reactivate the immune response to HCC[28,84]. Several early phase studies are currently underway to explore the safety and tolerability of TKIs such as sorafenib (NCT03211416, NCT01658878, NCT02988440), lenvatinib (NCT03418922, NCT03006926), cabozantinib (NCT03299946, NCT01658878), axitinib (NCT03289533), and capmatinib (NCT02795429) in combination with immune checkpoint inhibitors[83]. Of note, current clinical trials are focusing on how immunosuppressive conditions in HCC might be overcome using immune checkpoint inhibitors in combination with different types of immune checkpoint blockades, TKIs, and other conventional treatments[83]. To improve the HCC immunotherapy strategies as well as immune stimulatory approaches, identification of TAAs and neoantigens specific to HCC and testing the potential benefits of combinatorial immunotherapies will achieve the most beneficial effect for HCC patients[59].
Conclusions and future perspectives
The journal Science selected cancer immunotherapy as its “Breakthrough of the Year” in 2013, and especially the use of immune checkpoint blockade in cancer therapy is making a paradigm shift in cancer treatment[20,95]. Of note, immunotherapy has the potential to achieve complete, long-lasting remissions and cancer cures, representing the most promising new cancer treatment approach with few side effects[74]. Although disease progression is sometimes observed immediately after initiation of immunotherapy, some responders require longer duration of immunotherapy to achieve tumor response[20]. Therefore, the biomarkers of immunotherapy to predict response are urgently needed, both from the perspective of the effective use of medical resources and to prevent adverse effects caused by unnecessary treatment[84]. There are several highly promising candidate predictors of the cancer immunotherapy: PD-L1 expression in tumor tissue, TAA related mutanome analyses including next-generation sequencing, and the immunome analyses, which employ T cell repertoire analysis and proteomic analysis[96]. Also, additional questions still remain regarding the most effective combination of therapeutic modalities and biomarkers to predict long term treatment outcomes in HCC immuno-oncology.
Notably, to date, very promising published evidence with checkpoint inhibitors in HCC has been reported in the clinical trials of anti-CTLA-4 agent tremelimumab and a large phase II trial with anti-PD-1 agent nivolumab[56,78,80]. Further investigations of immuno-oncology potentially spread the applications of immunotherapy in the various stages of HCCs, and thus immune-based therapies will bring about a paradigm shift of anti-cancer treatment for HCC. We hope the immunotherapy will play a key role in HCC treatment in the near future.
Declarations
Authors’ contributionsMade substantial contributions to conception and design of the study and performed data analysis and interpretation: Lee JH, Nishida N
Performed data acquisition, as well as provided administrative, technical, and material support: Oh SY, Kim JY
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis study was supported by a grant of the Korea Healthcare technology R&D project, Ministry for Health, Welfare & Family Affairs, Republic of Korea (HI13C1398).
Conflicts of interestThe authors declare there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Schutte K, Bornschein J, Malfertheiner P. Hepatocellular carcinoma--epidemiological trends and risk factors. Dig Dis 2009;27:80-92.
2. Gosalia AJ, Martin P, Jones PD. Advances and future directions in the treatment of hepatocellular carcinoma. Gastroenterol Hepatol (N Y) 2017;13:398-410.
3. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015;136:E359-86.
4. Bruix J, Sherman M; American Association for the Study of Liver Disease. Management of hepatocellular carcinoma: an update. Hepatology 2011;53:1020-2.
5. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359:378-90.
6. cancer European organisation for research and treatment of; European association for the study of the liver. EASL-EORTC clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol 2012;56:908-43.
7. Woo HY, Yoo SY, Heo J. New chemical treatment options in second-line hepatocellular carcinoma: what to do when sorafenib fails? Expert Opin Pharmacother 2017;18:35-44.
8. Pardee AD, Butterfield LH. Immunotherapy of hepatocellular carcinoma: unique challenges and clinical opportunities. Oncoimmunology 2012;1:48-55.
9. Miamen AG, Dong H, Roberts LR. Immunotherapeutic approaches to hepatocellular carcinoma treatment. Liver Cancer 2012;1:226-37.
10. Childs RW, Carlsten M. Therapeutic approaches to enhance natural killer cell cytotoxicity against cancer: the force awakens. Nat Rev Drug Discov 2015;14:487-98.
11. Guo Y, Han W. Cytokine-induced killer (CIK) cells: from basic research to clinical translation. Chin J Cancer 2015;34:99-107.
12. Lim O, Jung MY, Hwang YK, Shin EC. Present and future of allogeneic natural killer cell therapy. Front Immunol 2015;6:286.
13. Zamarron BF, Chen W. Dual roles of immune cells and their factors in cancer development and progression. Int J Biol Sci 2011;7:651-8.
14. Breous E, Thimme R. Potential of immunotherapy for hepatocellular carcinoma. J Hepatol 2011;54:830-4.
15. Schmidt N, Neumann-Haefelin C, Thimme R. Cellular immune responses to hepatocellular carcinoma: lessons for immunotherapy. Dig Dis 2012;30:483-91.
16. Chow AK, Ng L, Lam CS, Wong SK, Wan TM, Cheng NS, Yau TC, Poon RT, Pang RW. The enhanced metastatic potential of hepatocellular carcinoma (HCC) cells with sorafenib resistance. PLoS One 2013;8:e78675.
17. Lee HJ, Kim SK, Cho D, Lee JJ. Cellular immunotherapy as a beacon of hope for hematological malignancies. Blood Res 2015;50:126-8.
18. Kalinski P. Dendritic cells in immunotherapy of established cancer: roles of signals 1, 2, 3 and 4. Curr Opin Investig Drugs 2009;10:526-35.
19. Streba LA, Streba CT, Sandulescu L, Vere CC, Mitrut P, Cotoi BV, Popescu LN, Ion DA. Dendritic cells and hepatocellular carcinoma. Rom J Morphol Embryol 2014;55:1287-93.
20. Kudo M. Immune checkpoint inhibition in hepatocellular carcinoma: basics and ongoing clinical trials. Oncology 2017;92 Suppl 1:50-62.
21. Moehler M, Delic M, Goepfert K, Aust D, Grabsch HI, Halama N, Heinrich B, Julie C, Lordick F, Lutz MP, Mauer M, Alsina Maqueda M, Schild H, Schimanski CC, Wagner AD, Roth A, Ducreux M. Immunotherapy in gastrointestinal cancer: recent results, current studies and future perspectives. Eur J Cancer 2016;59:160-70.
22. Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol Rev 2008;224:166-82.
23. Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, Azuma M, Krummel MF, Bluestone JA. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol 2009;10:1185-92.
24. Calderaro J, Rousseau B, Amaddeo G, Mercey M, Charpy C, Costentin C, Luciani A, Zafrani ES, Laurent A, Azoulay D, Lafdil F, Pawlotsky JM. Programmed death ligand 1 expression in hepatocellular carcinoma: relationship with clinical and pathological features. Hepatology 2016;64:2038-46.
25. Kassel R, Cruise MW, Iezzoni JC, Taylor NA, Pruett TL, Hahn YS. Chronically inflamed livers up-regulate expression of inhibitory B7 family members. Hepatology 2009;50:1625-37.
26. Ye B, Liu X, Li X, Kong H, Tian L, Chen Y. T-cell exhaustion in chronic hepatitis B infection: current knowledge and clinical significance. Cell Death Dis 2015;6:e1694.
27. Boonstra A, Woltman AM, Janssen HL. Immunology of hepatitis B and hepatitis C virus infections. Best Pract Res Clin Gastroenterol 2008;22:1049-61.
28. Nishida N, Kudo M. Role of immune checkpoint blockade in the treatment for human hepatocellular carcinoma. Dig Dis 2017;35:618-22.
30. Liu D, Staveley-O'Carroll KF, Li G. Immune-based therapy clinical trials in hepatocellular carcinoma. J Clin Cell Immunol 2015;6:376.
31. Onishi S, Saibara T, Fujikawa M, Sakaeda H, Matsuura Y, Matsunaga Y, Yamamoto Y. Adoptive immunotherapy with lymphokine-activated killer cells plus recombinant interleukin 2 in patients with unresectable hepatocellular carcinoma. Hepatology 1989;10:349-53.
32. Une Y, Kawata A, Uchino J. Adopted immunochemotherapy using IL-2 and spleen LAK cell--randomized study. Nihon Geka Gakkai Zasshi 1991;92:1330-3.
33. Kawata A, Une Y, Hosokawa M, Wakizaka Y, Namieno T, Uchino J, Kobayashi H. Adjuvant chemoimmunotherapy for hepatocellular carcinoma patients. Adriamycin, interleukin-2, and lymphokine-activated killer cells versus adriamycin alone. Am J Clin Oncol 1995;18:257-62.
34. Takayama T, Makuuchi M, Sekine T, Terui S, Shiraiwa H, Kosuge T, Yamazaki S, Hasegawa H, Suzuki K, Yamagata M. Distribution and therapeutic effect of intraarterially transferred tumor-infiltrating lymphocytes in hepatic malignancies. A preliminary report. Cancer 1991;68:2391-6.
35. Wang Y, Chen H, Wu M, Bao J, Cong W, Wang H. Postoperative immunotherapy for patients with hepatocarcinoma using tumor-infiltrating lymphocytes. Chin Med J (Engl) 1997;110:114-7.
36. Lee JH, Lee JH, Lim YS, Yeon JE, Song TJ, Yu SJ, Gwak GY, Kim KM, Kim YJ, Lee JW, Yoon JH. Adjuvant immunotherapy with autologous cytokine-induced killer cells for hepatocellular carcinoma. Gastroenterology 2015;148:1383-91.e6.
37. Takayama T, Sekine T, Makuuchi M, Yamasaki S, Kosuge T, Yamamoto J, Shimada K, Sakamoto M, Hirohashi S, Ohashi Y, Kakizoe T. Adoptive immunotherapy to lower postsurgical recurrence rates of hepatocellular carcinoma: a randomised trial. Lancet 2000;356:802-7.
38. Wang X, Yu W, Li H, Yu J, Zhang X, Ren X, Cao S. Can the dual-functional capability of CIK cells be used to improve antitumor effects? Cell Immunol 2014;287:18-22.
39. Shi M, Zhang B, Tang ZR, Lei ZY, Wang HF, Feng YY, Fan ZP, Xu DP, Wang FS. Autologous cytokine-induced killer cell therapy in clinical trial phase I is safe in patients with primary hepatocellular carcinoma. World J Gastroenterol 2004;10:1146-51.
40. Hui D, Qiang L, Jian W, Ti Z, Da-Lu K. A randomized, controlled trial of postoperative adjuvant cytokine-induced killer cells immunotherapy after radical resection of hepatocellular carcinoma. Dig Liver Dis 2009;41:36-41.
41. Pan CC, Huang ZL, Li W, Zhao M, Zhou QM, Xia JC, Wu PH. Serum alpha-fetoprotein measurement in predicting clinical outcome related to autologous cytokine-induced killer cells in patients with hepatocellular carcinoma undergone minimally invasive therapy. Chin J Cancer 2010;29:596-602.
42. Schmidt S, Tramsen L, Rais B, Ullrich E, Lehrnbecher T. Natural killer cells as a therapeutic tool for infectious diseases - current status and future perspectives. Oncotarget 2018;9:20891-907.
43. Sutlu T, Alici E. Natural killer cell-based immunotherapy in cancer: current insights and future prospects. J Intern Med 2009;266:154-81.
44. Jinushi M, Takehara T, Tatsumi T, Hiramatsu N, Sakamori R, Yamaguchi S, Hayashi N. Impairment of natural killer cell and dendritic cell functions by the soluble form of MHC class I-related chain A in advanced human hepatocellular carcinomas. J Hepatol 2005;43:1013-20.
45. Ghiringhelli F, Menard C, Terme M, Flament C, Taieb J, Chaput N, Puig PE, Novault S, Escudier B, Vivier E, Lecesne A, Robert C, Blay JY, Bernard J, Caillat-Zucman S, Freitas A, Tursz T, Wagner-Ballon O, Capron C, Vainchencker W, Martin F, Zitvogel L. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J Exp Med 2005;202:1075-85.
46. Chew V, Chen J, Lee D, Loh E, Lee J, Lim KH, Weber A, Slankamenac K, Poon RT, Yang H, Ooi LL, Toh HC, Heikenwalder M, Ng IO, Nardin A, Abastado JP. Chemokine-driven lymphocyte infiltration: an early intratumoural event determining long-term survival in resectable hepatocellular carcinoma. Gut 2012;61:427-38.
47. Ohira M, Nishida S, Tryphonopoulos P, Tekin A, Selvaggi G, Moon J, Levi D, Ricordi C, Ishiyama K, Tanaka Y, Ohdan H, Tzakis AG. Clinical-scale isolation of interleukin-2-stimulated liver natural killer cells for treatment of liver transplantation with hepatocellular carcinoma. Cell Transplant 2012;21:1397-406.
48. Johnson LA, June CH. Driving gene-engineered T cell immunotherapy of cancer. Cell Res 2017;27:38-58.
49. Maus MV, Grupp SA, Porter DL, June CH. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood 2014;123:2625-35.
50. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood 2016;127:3321-30.
51. Klebanoff CA, Rosenberg SA, Restifo NP. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med 2016;22:26-36.
52. Koh S, Shimasaki N, Suwanarusk R, Ho ZZ, Chia A, Banu N, Howland SW, Ong AS, Gehring AJ, Stauss H, Renia L, Sallberg M, Campana D, Bertoletti A. A practical approach to immunotherapy of hepatocellular carcinoma using T cells redirected against hepatitis B virus. Mol Ther Nucleic Acids 2013;2:e114.
53. Gehring AJ, Xue SA, Ho ZZ, Teoh D, Ruedl C, Chia A, Koh S, Lim SG, Maini MK, Stauss H, Bertoletti A. Engineering virus-specific T cells that target HBV infected hepatocytes and hepatocellular carcinoma cell lines. J Hepatol 2011;55:103-10.
54. Qasim W, Brunetto M, Gehring AJ, Xue SA, Schurich A, Khakpoor A, Zhan H, Ciccorossi P, Gilmour K, Cavallone D, Moriconi F, Farzhenah F, Mazzoni A, Chan L, Morris E, Thrasher A, Maini MK, Bonino F, Stauss H, Bertoletti A. Immunotherapy of HCC metastases with autologous T cell receptor redirected T cells, targeting HBsAg in a liver transplant patient. J Hepatol 2015;62:486-91.
55. Curran KJ, Brentjens RJ. Chimeric antigen receptor T cells for cancer immunotherapy. J Clin Oncol 2015;33:1703-6.
56. Sim HW, Knox J. Hepatocellular carcinoma in the era of immunotherapy. Curr Probl Cancer 2018;42:40-8.
57. Gao H, Li K, Tu H, Pan X, Jiang H, Shi B, Kong J, Wang H, Yang S, Gu J, Li Z. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin Cancer Res 2014;20:6418-28.
58. Klingemann H, Boissel L, Toneguzzo F. Natural killer cells for immunotherapy - advantages of the NK-92 cell line over blood NK cells. Front Immunol 2016;7:91.
59. Tagliamonte M, Petrizzo A, Tornesello ML, Ciliberto G, Buonaguro FM, Buonaguro L. Combinatorial immunotherapy strategies for hepatocellular carcinoma. Curr Opin Immunol 2016;39:103-13.
60. Chen S, Akbar SM, Tanimoto K, Ninomiya T, Iuchi H, Michitaka K, Horiike N, Onji M. Absence of CD83-positive mature and activated dendritic cells at cancer nodules from patients with hepatocellular carcinoma: relevance to hepatocarcinogenesis. Cancer Lett 2000;148:49-57.
61. Butterfield LH, Ribas A, Meng WS, Dissette VB, Amarnani S, Vu HT, Seja E, Todd K, Glaspy JA, McBride WH, Economou JS. T-cell responses to HLA-A*0201 immunodominant peptides derived from alpha-fetoprotein in patients with hepatocellular cancer. Clin Cancer Res 2003;9:5902-8.
62. Butterfield LH, Ribas A, Dissette VB, Lee Y, Yang JQ, De la Rocha P, Duran SD, Hernandez J, Seja E, Potter DM, McBride WH, Finn R, Glaspy JA, Economou JS. A phase I/II trial testing immunization of hepatocellular carcinoma patients with dendritic cells pulsed with four alpha-fetoprotein peptides. Clin Cancer Res 2006;12:2817-25.
63. Butterfield LH, Ribas A, Potter DM, Economou JS. Spontaneous and vaccine induced AFP-specific T cell phenotypes in subjects with AFP-positive hepatocellular cancer. Cancer Immunol Immunother 2007;56:1931-43.
64. Mizukoshi E, Nakamoto Y, Arai K, Yamashita T, Mukaida N, Matsushima K, Matsui O, Kaneko S. Enhancement of tumor-specific T-cell responses by transcatheter arterial embolization with dendritic cell infusion for hepatocellular carcinoma. Int J Cancer 2010;126:2164-74.
65. Palmer DH, Midgley RS, Mirza N, Torr EE, Ahmed F, Steele JC, Steven NM, Kerr DJ, Young LS, Adams DH. A phase II study of adoptive immunotherapy using dendritic cells pulsed with tumor lysate in patients with hepatocellular carcinoma. Hepatology 2009;49:124-32.
66. El Ansary M, Mogawer S, Elhamid SA, Alwakil S, Aboelkasem F, Sabaawy HE, Abdelhalim O. Immunotherapy by autologous dendritic cell vaccine in patients with advanced HCC. J Cancer Res Clin Oncol 2013;139:39-48.
67. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J 1992;11:3887-95.
68. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996;271:1734-6.
69. Wu P, Wu D, Li L, Chai Y, Huang J. PD-L1 and survival in solid tumors: a meta-analysis. PLoS One 2015;10:e0131403.
70. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012;12:252-64.
71. Gao Q, Wang XY, Qiu SJ, Yamato I, Sho M, Nakajima Y, Zhou J, Li BZ, Shi YH, Xiao YS, Xu Y, Fan J. Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin Cancer Res 2009;15:971-9.
72. Sprinzl MF, Galle PR. Current progress in immunotherapy of hepatocellular carcinoma. J Hepatol 2017;66:482-4.
73. Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, Lin YJ, Wojtkiewicz G, Iwamoto Y, Mino-Kenudson M, Huynh TG, Hynes RO, Freeman GJ, Kroemer G, Zitvogel L, Weissleder R, Pittet MJ. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity 2016;44:343-54.
74. Liu J, Blake SJ, Yong MC, Harjunpaa H, Ngiow SF, Takeda K, Young A, O'Donnell JS, Allen S, Smyth MJ, Teng MW. Improved efficacy of neoadjuvant compared to adjuvant immunotherapy to eradicate metastatic disease. Cancer Discov 2016;6:1382-99.
75. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Duffy SM, Goldberg RM, de la Chapelle A, Koshiji M, Bhaijee F, Huebner T, Hruban RH, Wood LD, Cuka N, Pardoll DM, Papadopoulos N, Kinzler KW, Zhou S, Cornish TC, Taube JM, Anders RA, Eshleman JR, Vogelstein B, Diaz LA Jr. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015;372:2509-20.
76. Shi F, Shi M, Zeng Z, Qi RZ, Liu ZW, Zhang JY, Yang YP, Tien P, Wang FS. PD-1 and PD-L1 upregulation promotes CD8(+) T-cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int J Cancer 2011;128:887-96.
77. Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller TL, Gilson MM, Wang C, Selby M, Taube JM, Anders R, Chen L, Korman AJ, Pardoll DM, Lowy I, Topalian SL. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol 2010;28:3167-75.
78. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, Kim TY, Choo SP, Trojan J, Welling THR, Meyer T, Kang YK, Yeo W, Chopra A, Anderson J, Dela Cruz C, Lang L, Neely J, Tang H, Dastani HB, Melero I. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017;389:2492-502.
79. Truong P, Rahal A, Kallail KJ. Metastatic hepatocellular carcinoma responsive to pembrolizumab. Cureus 2016;8:e631.
80. Sangro B, Gomez-Martin C, de la Mata M, I-arrairaegui M, Garralda E, Barrera P, Riezu-Boj JI, Larrea E, Alfaro C, Sarobe P. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol 2013;59:81-8.
81. Duffy AG, Ulahannan SV, Makorova-Rusher O, Rahma O, Wedemeyer H, Pratt D, Davis JL, Hughes MS, Heller T, ElGindi M, Uppala A, Korangy F, Kleiner DE, Figg WD, Venzon D, Steinberg SM, Venkatesan AM, Krishnasamy V, Abi-Jaoudeh N, Levy E, Wood BJ, Greten TF. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J Hepatol 2017;66:545-51.
82. Prieto J, Melero I, Sangro B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 2015;12:681-700.
83. Nishida N, Kudo M. Immune checkpoint blockade for the treatment of human hepatocellular carcinoma. Hepatol Res 2018; doi: 10.1111/hepr.13191.
84. Kudo M. Immune checkpoint blockade in hepatocellular carcinoma: 2017 update. Liver Cancer 2016;6:1-12.
85. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol 2009;182:240-9.
86. Yan W, Liu X, Ma H, Zhang H, Song X, Gao L, Liang X, Ma C. Tim-3 fosters HCC development by enhancing TGF-beta-mediated alternative activation of macrophages. Gut 2015;64:1593-604.
87. Li H, Wu K, Tao K, Chen L, Zheng Q, Lu X, Liu J, Shi L, Liu C, Wang G, Zou W. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology 2012;56:1342-51.
88. Yarchoan M, Xing D, Luan L, Xu H, Sharma RB, Popovic A, Pawlik TM, Kim AK, Zhu Q, Jaffee EM, Taube JM, Anders RA. Characterization of the immune microenvironment in hepatocellular carcinoma. Clin Cancer Res 2017;23:7333-9.
89. Matsuda Y, Yamagiwa Y, Fukushima K, Ueno Y, Shimosegawa T. Expression of galectin-3 involved in prognosis of patients with hepatocellular carcinoma. Hepatol Res 2008;38:1098-111.
90. Li FJ, Zhang Y, Jin GX, Yao L, Wu DQ. Expression of LAG-3 is coincident with the impaired effector function of HBV-specific CD8(+) T cell in HCC patients. Immunol Lett 2013;150:116-22.
91. Hui Z, Zhang X, Ren B, Li R, Ren X. Rapid response of advanced squamous non-small cell lung cancer with thrombocytopenia after first-line treatment with pembrolizumab plus autologous cytokine-induced killer cells. Front Immunol 2015;6:633.
92. Niu Q, Wang W, Li Y, Qin S, Wang Y, Wan G, Guan J, Zhu W. Cord blood-derived cytokine-induced killer cells biotherapy combined with second-line chemotherapy in the treatment of advanced solid malignancies. Int Immunopharmacol 2011;11:449-56.
93. Cui J, Wang N, Zhao H, Jin H, Wang G, Niu C, Terunuma H, He H, Li W. Combination of radiofrequency ablation and sequential cellular immunotherapy improves progression-free survival for patients with hepatocellular carcinoma. Int J Cancer 2014;134:342-51.
94. Huang ZM, Li W, Li S, Gao F, Zhou QM, Wu FM, He N, Pan CC, Xia JC, Wu PH, Zhao M. Cytokine-induced killer cells in combination with transcatheter arterial chemoembolization and radiofrequency ablation for hepatocellular carcinoma patients. J Immunother 2013;36:287-93.
96. Clifford RJ, Zhang J, Meerzaman DM, Lyu MS, Hu Y, Cultraro CM, Finney RP, Kelley JM, Efroni S, Greenblum SI, Nguyen CV, Rowe WL, Sharma S, Wu G, Yan C, Zhang H, Chung YH, Kim JA, Park NH, Song IH, Buetow KH. Genetic variations at loci involved in the immune response are risk factors for hepatocellular carcinoma. Hepatology 2010;52:2034-43.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Lee JH, Oh SY, Kim JY, Nishida N. Cancer immunotherapy for hepatocellular carcinoma. Hepatoma Res 2018;4:51. http://dx.doi.org/10.20517/2394-5079.2018.78
AMA Style
Lee JH, Oh SY, Kim JY, Nishida N. Cancer immunotherapy for hepatocellular carcinoma. Hepatoma Research. 2018; 4: 51. http://dx.doi.org/10.20517/2394-5079.2018.78
Chicago/Turabian Style
Lee, Joo-Ho, Soo-Yeon Oh, Jin Yong Kim, Naoshi Nishida. 2018. "Cancer immunotherapy for hepatocellular carcinoma" Hepatoma Research. 4: 51. http://dx.doi.org/10.20517/2394-5079.2018.78
ACS Style
Lee, J.H.; Oh S.Y.; Kim JY.; Nishida N. Cancer immunotherapy for hepatocellular carcinoma. Hepatoma. Res. 2018, 4, 51. http://dx.doi.org/10.20517/2394-5079.2018.78
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 6 clicks
Cite This Article 6 clicks

 Like This Article 27
likes
Like This Article 27
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.