
Fat and hepatocellular carcinoma


Abstract
Obesity and diabetes are associated with the onset of hepatocellular carcinoma (HCC). These two illnesses correlate also with the development of non-alcoholic fatty liver disease (NAFLD). Currently, NAFL is considered the leading form of chronic liver disease in the Western industrialized countries. Insulin resistance is the common pathogenic factor among these three pathologies. NAFL is characterized by fat accumulation in the liver that involves greater than 5% of the liver parenchyma with no evidence of hepatocyte injury. However, NAFL may progress toward non-alcoholic steatohepatitis that in turn may lead to advanced fibrosis, cirrhosis and HCC. It is alarming that NAFLD related HCC has been, at present, considered as a growing burden worldwide, and its prevalence is tending to further increase together with the increasing incidence of obesity and diabetes. Worthy of note is that in the presence of chronic accumulation of fat in the liver it has been reported the emergence of HCC during chronic liver disease in absence of liver cirrhosis, usually the major risk factor for the development of HCC. Thus, in the future NAFLD related HCCs will place a growing strain on health-care systems from the need for their management. Unfortunately, most of the NAFLD related HCC patients are diagnosed at advanced stages and are characterized by a poor prognosis, because they are ineligible to radical treatments. Thus, it is urgent to boost up new screening policies to make early diagnoses, as well as to develop preventive-therapeutic strategies.
Keywords
Introduction
In Western countries the growing epidemics of obesity and type 2 diabetes are associated with increasing incidence of hepatocellular carcinoma (HCC)[1]. These two conditions are strictly associated with the development of non-alcoholic fatty liver disease (NAFLD) and considered the leading forms of chronic liver disease in the Western industrialized countries[2]. NAFLD has a wide geographic distribution to take on epidemic proportions: prevalence of NAFLD has been reported to be 25% worldwide. The highest prevalence is reported from South America (31%) and Middle East (32%), followed by Asia (27%) and USA (24%), while the prevalence is lowest in Africa (14%). In Europe the median prevalence is 23%-26% with variations in different European populations[3].
NAFLD is caused by an insulin resistance and, as reported above, often occurs with the presence of diabetes, obesity, and metabolic syndrome; therefore, liver can be considered the alarm bell of all of these pathologies[4]. The progressive form of NAFLD is non-alcoholic steatohepatitis (NASH) that can lead to advanced fibrosis, cirrhosis and HCC. It is worrying that NAFLD related HCC has been at present recognized as a growing burden worldwide, and its impact is expected to further grow together with the increasing incidence of obesity and diabetes[4].
Of particular interest is the emergence of HCC during chronic liver disease in absence of liver cirrhosis, that is known as the major risk factor for HCC development[5,6].
HCC development requires decades and is characterized by a gradual transition through a dysplastic to transformed liver tissue[7]. Liver transformation is the result of uncontrolled cell growth that results in the accumulation of genomic alterations occurring during cell division thus becoming the driving force for tumorigenesis. Five mechanisms are involved in maintaining genomic stability during cell division: (1) high-fidelity of DNA replication in S-phase; (2) precise distribution of chromosomes in daughter cells during mitosis; (3) DNA repair throughout the cell cycle; (4) cell cycle checkpoints; and (5) induction of apoptosis or senescence in case of genomic instability[8]. On the other hand, there are multiple oncogenic mechanisms that participate in genomic instability: alterations in the DNA-damage-response pathways, telomere erosion, chromosome segregation defects[9].
Even if several pathogenic mechanisms, such as obesity-mediated chronic inflammation and diabetes, have been described to be involved in NAFLD related HCC till now, as extensively reported below, we do not have clear ideas on the pathogenic mechanisms driving transformation of the cell during NAFLD[10,11]. In this context, low grading chronic inflammation has indubitably a crucial role in NAFLD disease progression toward HCC[12]. During low grade inflammation, the overproduction of reactive oxygen species (ROS) induces the output of advanced glycation end-products (AGEs), advanced lipoxidation end-products (ALEs) and protein oxidation products (PrOPs) in tissues[13,14], inducing pro-inflammatory cascades and increasing the risk of liver tissue transformation. Thus, to better understand the pathogenic mechanisms underlying NAFLD related HCC, first of all, we should better know all the biological factors involved in promoting inflammation that consequently participate in hepatocarcinogenesis.
Further studies should be performed to highlight new insights in the pathogenesis of HCC during NAFLD. However, scientific consensus exists a on the concept that the progression of NAFLD toward HCC is surely linked not only to environmental but also genetic factors. Accordingly, genome-wide association studies highlighted several single nucleotide polymorphisms (SNPs) associated with the pathology of NAFLD [Table 1]. Furthermore, induction of epigenetic alterations due to unhealthy diet and/or other environmental factors are surely involved in NAFLD related HCC.
Single nucleotide polymorphisms associated with the pathology of nonalcoholic fatty liver disease
| Gene | SNP | Region | Location | Functional class | Total allele frequency (Gnomad) |
|---|---|---|---|---|---|
| PNPLA3 | rs738409-G | 22q13.31 | 22:43928847 | missense_variant | 0.2709 |
| PNPLA3 | rs2896019-G | 22q13.31 | 22:43937814 | intron_variant | 0.1981 |
| SAMM50 | rs738491-T | 22q13.31 | 22:43958231 | intron_variant | 0.356 |
| SAMM50 | rs2143571-A | 22q13.31 | 22:43995806 | intron_variant | 0.2495 |
| GCKR | rs1260326-T | 2p23.3 | 2:27508073 | missense_variant | 0.6381 |
| GCKR | rs780094-T | 2p23.3 | 2:27518370 | intron_variant | 0.6702 |
| GATAD2A | rs4808199-A | 19p13.11 | 19:19434290 | intron_variant | 0.1817 |
| COL13A1 | rs1227756-G | 10q22.1 | 10:69828748 | intron_variant | 0.4676 |
| FDFT1 | rs2645424-A | 8p23.1 | 8:11826954 | intron_variant | 0.5508 |
| CRACR2A | rs887304-A | 12p13.32 | 12:3648382 | 3_prime_UTR_variant | 0.764 |
| SAMM50 - PARVB | rs2073080-T | 22q13.31 | 22:43998522 | intron_variant | 0.2017 |
| EHBP1L1 | rs6591182-A | 11q13.1 | 11:65582285 | missense_variant | 0.4756 |
| KLRG1 | rs6487679-G | 12p13.31 | 12:9218736 | intergenic_variant | 0.8025 |
| ZNF512 | rs1881396-T | 2p23.3 | 2:27621734 | 3_prime_UTR_variant | 0.2063 |
| MUM1 | rs2668423-T | 19p13.3 | 19:1370527 | intron_variant | 0.7159 |
| ACTR5 | rs6128907-C | 20q11.23 | 20:38759219 | intron_variant | 0.1645 |
| KHDRBS3 - RNU1-35P | rs4243849-G | 8q24.23 | 8:135700894 | intergenic_variant | 0.3522 |
| FARP1 | rs9584805-G | 13q32.2 | 13:98341776 | intron_variant | 0.3288 |
| LOC643381 - CNTN5 | rs4237591-G | 11q22.1 | 11:98595538 | intergenic_variant | 0.3955 |
| SLC38A8 | rs11864146-A | 16q23.3 | 16:84013110 | intron_variant | 0.169 |
| SLC9A9 | rs2800-G | 3q24 | 3:143705980 | intron_variant | 0.6618 |
| FDFT1 | rs2645424-A | 8p23.1 | 8:11826954 | intron_variant | 0.5508 |
| LCP1 | rs7324845-A | 13q14.13 | 13:46129007 | intron_variant | 0.8398 |
| ST8SIA1 | rs2216228-G | 12p12.1 | 12:22212901 | intron_variant | 0.1949 |
| SLC9A9 | rs7632299-A | 3q24 | 3:143337625 | intron_variant | 0.2716 |
| ETS1 | rs3935794-G | 11q24.3 | 11:128520782 | intron_variant | 0.07114 |
| RNA5SP489 - RPL13AP7 | rs9977253-G | 21q21.2 | 21:25272769 | intron_variant | 0.7688 |
| EEF1A1P20 - MTCYBP22 | rs10067427-G | 5q21.1 | 5:100006343 | intergenic_variant | 0.4205 |
| YIPF1 | rs11206226-A | 1p32.3 | 1:53854664 | intron_variant | 0.03217 |
| SDK1 | rs688020-C | 7p22.2 | 7:4188921 | intron_variant | 0.4197 |
| MACROD2 | rs6079395-A | 20p12.1 | 20:14347253 | intron_variant | 0.5135 |
| CACNA2D1 | rs10954668-A | 7q21.11 | 7:82218335 | intron_variant | 0.2566 |
| COL13A1 | rs7077164-A | 10q22.1 | 10:69823442 | intron_variant | 0.35 |
| TEX36 | rs10510146-A | 10q26.13 | 10:125607576 | intron_variant | - |
| SEL1L3 | rs959903-A | 4p15.2 | 4:25808474 | intron_variant | 0.2551 |
| NGF - TCEB1P20 | rs7552722-A | 1p13.2 | 1:115378734 | intergenic_variant | 0.6805 |
| CDH2 - ARIH2P1 | rs11083271-A | 18q12.1 | 18:28346095 | intergenic_variant | 0.2673 |
| SDR42E1P5 - IL18RAP | rs11465670-C | 2q12.1 | 2:102417980 | upstream_gene_variant | 0.1239 |
| SLC46A3 | rs1305088-A | 13q12.3 | 13:28704313 | non_coding_transcript_exon_variant | 0.854 |
| RAB37 | rs12942311-C | 17q25.1 | 17:74714657 | intron_variant | 0.2134 |
Perspective studies are needed to implement screening strategies and preventive approaches for NAFLD-related HCC development, particularly in the non-cirrhotic population. The notions reported in this review, describing several NAFLD-related molecular target pathways, will be useful to clinicians to outline diagnostic and prognostic profiles of these complex and heterogeneous patients.
Epidemiology
HCC causes more than 700,000 deaths/year worldwide and accounts for 70%-85% of cases of liver cancers. HCC is the fourth most often diagnosed cancer in males (70% occur over age 50) and the seventh in females[15]; moreover, it represents the overall second cause of cancer deaths[16,17]. These statistics reflect the poor prognosis of liver cancer worldwide.
About 80% of HCC cases occur in less developed countries and are typically associated with alcohol, chronic hepatitis B (HBV) and C (HCV) infections: importantly, the incidence in these countries is decreasing[18,19]. On the other hand, in western countries the HCC incidence is increasing, ranging from 2.4% over 7 years to 12.8% over 3.2 years of median follow-up period, following the geographic distribution of obesity[4]. In particular, 10 year annual cumulative risks of HCC in alcohol, HCV or NAFLD are 1.1%, 2.9% and 3.1%, respectively[20]. Accordingly, an increasing number of HCC has been reported in the setting of obesity and diabetes[15,21] and it has been associated with an increased relative risk of dying for HCC[22].
Unfortunately, even if consistent epidemiological data concerning viral and alcoholic hepatitis have been reported, there is a lack of strong epidemiological results regarding the incidence and prevalence of NAFLD-related HCC. The problem is mainly due to the absence of a correct and clear definition of NAFL/NAFLD/NASH. Thus, so far we cannot evaluate the real dimension of NAFLD-related HCC and how to lower and prevent its appearance.
A few longitudinal outcome studies reveal that the cumulative mortality in NAFL/NASH, in a follow-up period between 5.6 and 21 years, vary from 0% to 3%[23], but we have to take into account that there are 400,000 and 40,000-80,000 new cases/year of NAFL and NASH, respectively.
Finally, the unquestionable evidence showing the increased risk of HCC in patients with NAFLD, and mainly its appearance in non-cirrhotic patients, is in close association with the alarming and more rapidly increasing indication for liver transplantation in respect to any other liver disease[24].
Pathogenesis
The aberrant activation of immune response and inflammation signaling observed in NAFLD have a key role in the pathogenesis and progression of this liver disease.
The accumulation of lipids in patients with NAFLD may induce an intracellular chronic status of oxidative stress that, in turn, leads to the activation of low-grade inflammation. The enlargement of adipocytes may lead over time to the rupture of these cells. As a consequence, macrophages are recruited in the site of inflammation and M1/M2 macrophage polarization is induced. The activation of macrophages stimulates the production of adipose tissue related adipocytokines, that, once released in the systemic circulation, reach different organs, including liver[25,26]. The inversion of M1/M2 ratio is due to the increase of M1 macrophages and reduction of M2 macrophages[26]. The higher number of M1 cells cause an over production of several pro-inflammatory cytokines, such as IL-1b, IL-6, IL-8, IL-12, and TNF-a.
Consequently, the serum of NAFLD patients is characterized by the presence of high levels of TNF-α and IL6, that in turn are correlated with a higher risk of progression to NASH[27-30]. Moreover, higher levels of TNF-α induce insulin resistance[31-33] and contribute to exacerbate the liver damage through the activation of nuclear factor-kappa-B (NFκB) inflammatory pathways[34]. Furthermore, NFκB protein has been recently found to be involved in the regulatory feedback of two important chemokine receptors: C-X-C chemokine receptor type 4 and 7 (CXCR4/7)[35]. The NFκB-CXCR4/7 axis mediates the signaling of toll-like receptors, TLR3 and TLR4, promote, in this way, the progression of NASH towards HCC[36].
Thus, deeply understanding the role of chronic inflammation as underlying the cause of liver transformation will improve the prevention and cure of this cancer.
An incorrect lifestyle is currently considered the main predisposing factor of NAFLD-related HCC. In fact, the development of HCC in NAFLD includes low-grade chronic inflammatory response (NASH) associated with genetic alterations, oxidative stress, obesity, insulin resistance and alteration of gut microbiota [Figure 1]. The pathogenic mechanisms involved in the progression of NAFL toward NASH are characterized by two hits: excess accumulation of triglyceride (TG) in the hepatocyte and, in a second moment, induction of oxidative stress and inflammation by several factors, such as free radicals[37]. In line with these findings, more and more researchers are recognizing the central role of low-grade inflammation in inducing all of the NAFLD-related comorbidities, such as insulin resistance, diabetes, and cardiovascular disease. Accordingly, NASH is a recognized cause of cirrhosis and is associated with development of HCC[38].
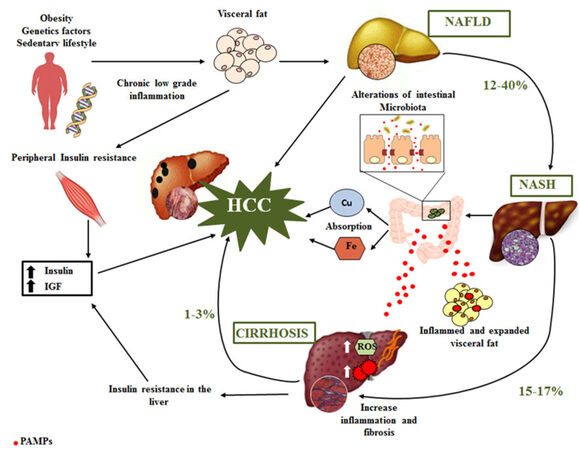
Figure 1. Pathogenic mechanisms involved in the development of HCC. HCC: hepatocellular carcinoma; NAFLD: nonalcoholic fatty liver disease; NASH: nonalcoholic steatohepatitis; IGF: insulin-like growth factor; ROS: reactive oxygen species; PAMPs: pathogen-associated molecular patterns
In this context, it is worth mentioning that multiple additional mechanisms may be implicated in the progression from NAFLD to NASH and HCC. In fact, keeping in mind that there are a growing number of patients who can progress from NAFLD to advanced fibrosis in the absence of significant inflammation, the alterations in immunologic, endocrine and metabolic pathways have a key role in the progression of NASH toward HCC.
Accordingly, despite the few data on NAFLD-related hepatocarcinogenesis, it has been highlighted that the phosphoinositide 3-kinase (PI3K)-AKT-mTOR pathway, implicated in the control of cellular energetic homeostasis, is deregulated in over 50% of NAFLD-related HCCs[39].
The β-catenin/WNT signaling, that has a crucial role in cell proliferation, stem cell self-renewal and cell migration, was found affected by somatic mutation in > 37% of NAFLD-related HCC[39].
Below we reported a detailed description of some factors involved in HCC development in patients with NAFLD.
Regarding the genetic factors involved in the progression from NAFLD to HCC, recent genome-wide studies have highlighted genetic heterogeneity of liver cancers. Of note, some SNPs, such as Patatin-like phospholipase domain-containing 3 (PNPLA3) gene variant I148M, have been related to the development and progression of NAFLD, NASH and NAFLD-related HCC, whereas others, such as the transmembrane 6 superfamily member 2 (TM6SF2) gene variant E167K, have been mainly correlated with the development of cardiovascular diseases[5,40,41]. In this context, the most recent findings from genomic profiling let us better understand that different pathways are involved in the initiation and progression of liver cancer[42], as shown in Figure 1.
In addition, altered transcriptional gene expression might be linked to inappropriate microRNAs (miRNAs)-guided transcriptional control. The human genome is envisaged to encode approximately 1000 miRNAs[43], which are a perfect class of blood-based biomarkers for cancer detection[44]. MiRNAs are endogenous 19-24 nucleotides noncoding single-stranded RNAs, which control, at post-transcriptional level, many complementary target mRNAs implicated in several pathophysiological processes, such as cell proliferation, differentiation, metabolism, apoptosis and cancer[45]. Lack of miRNA processing enzymes in cancer cells promotes tumor invasiveness and more aggressive phenotypes, revealing their main role in controlling tumor- and metastasis-initiating events[46-48]. Accordingly, different sets of miRNAs have been specifically correlated with NAFLD, NASH and HCC [Table 2][49-67]. Among the miRNAs recently identified in NAFLD patients, it is worth mentioning the up-regulation of miR-146b-5p, miR-181b and miR-375, and the down-regulation of miR-29a, miR30b-5p, miR-122-5p, miR-139-5p, miR-155 and miR-422a[49,53-56]. In addition, in NASH it has been reported the up-regulation of miR-33a and miR-144 and the down-regulation of miR-451[58,60]. Finally, in NAFLD-related HCC the up-regulation of miR-10a, miR-33a, miR-144, miR-155, miR-183, miR-375 and miR-423 and the down-regulation of miR-229 and miR-7706 were found[65-67].
Summary of miRNAs significantly associated with NAFLD, NASH and HCC patients
| Disease | Upregulated miRNAs | Downregulated miRNAs | References |
|---|---|---|---|
| NAFLD | miR-21, miR-34a, miR-122 (serum), miR-146b-5p (tisuue), miR-181b, miR-451 | miR-29a, miR-139-5p, miR-30b-5p, miR-122-5p (tissue), miR-155, miR-422a, miR-181d, miR-99a, miR-197, miR-146b (serum) | [49-56] |
| NASH | miR-21, miR-33a, miR-34a, miR-122, miR-144, miR-192, pri-miR-7-1, pri-miR-26a-1/2 | miR-125b, miR-451 | [50,51,57-60] |
| HCC | miR-10a, miR-21 (tissue), miR23a, miR-31, miR-34a-5p, miR-93-5p, miR-122, miR-155, miR-183, miR-221-3p, miR-222-3p, miR-375, miR-423 | let-7f, miR-16, miR-21 (serum), miR-24, miR-30e, miR-99a, miR-106b, miR-125b, miR-145, miR-146a, miR-148a, miR-155, miR-183, miR-199a, miR-199a3p, miR-200c, miR-215, miR-223, miR-229, miR-7706 | [50,61-67] |
MiRNA analyses, in combination with other clinical parameters and standard liver examinations, may be extremely useful to predict the possible progression of NAFLD toward HCC, and for monitoring the response to treatments[50]. However, despite the association between definite miRNA signatures and pathogenesis of NAFLD-related HCC, the expression levels of specific hepatic miRNAs during liver tissue transformation are still controversial. Further studies are needed to shed light on their function in the context of NAFLD-related HCC.
Regarding oxidative stress, reactive oxygen species (ROS) have a central role in HCC onset. ROS, in fact, are noticeable factors playing an essential role in regulating cell homeostasis. In this regard, our group has recently highlighted the role of altered systemic biometals distribution in NAFL/NASH patients and the associated increasing levels of ROS[68]. Accordingly, toxic biometal accumulation is a common feature in many cancers. Moreover, perturbations of mechanisms that control transcripts encoding proteins that regulate biometals have been described in cancer cells, including differences in epigenetic control (methylation and acetylation), miRNAs expression and protein activities[68,69].
In particular, biometals have the ability to catalyze oxidation-reduction reactions, which can lead to the production of ROS, thus their tight homeostatic regulation should be always present in the body. Accordingly, although the mechanisms are at present still unclear, a contribution of iron (Fe), copper (Cu) and zinc (Zn), in the development of HCC, has been often suggested[68,70-72]. In fact, dysregulation of Fe, Cu or Zn homeostasis stimulate proliferation, modulate the expression of epithelial mesenchimal transition (EMT) related proteins, glycolysis, and antioxidant molecules, such as SOD1 and 2, HIF1, GSH, in various cancer cells and human tumors[68,69,73-75].
Accordingly, NASH and NAFL patients display higher iron absorption after the administration of an oral iron absorption test and its deposition has been related to HCC development in NAFLD-cirrhosis[76]. The underlying mechanisms are not already clear, but might be related to oxidative DNA damage[77]. Interestingly, a recent meta-analysis highlighted that HFE mutations C282Y and H63D, associated in homozygosity with hemochromatosis, were characterized by a higher risk of HCC in NAFLD patients[78]. In addition, our group highlighted that a statistical significant enhancement of serum copper levels has been reported in NAFLD-cirrhotic patients and the altered homeostasis of this biometal was even more evident in HCC patients. In the presence of higher concentrations of extracellular copper liver cells are sensitized to transformation. The pathogenic copper-related pro-oncogenic mechanism seems to be, at least in part, managed by MYC, which is able to directly bind a specific region of the CTR1 promoter, regulating its transcription[68]. In this regard, it is really interesting the recent study reporting that Golgi protein 73 (GP73) is an effective and reliable serological marker for the diagnosis of advanced fibrosis and prediction of appearance of cirrhosis[79]. The awareness that copper serves as a limiting factor for multiple aspects of tumor progression, including growth, angiogenesis and metastasis suggests more attention to be paid to the potential and undiscovered role of copper-specific chelators as effective therapeutic agents against HCC.
The prevalence of obesity is increasing worldwide as well as the link between obesity and cancer, becoming an important and accepted risk factor for the development of HCC. As reported above, it is currently accepted that NAFLD is caused by an insulin resistance and often appears in the presence of obesity. The relationship between obesity and HCC was supported by a cohort study in Italy. In this work the odds ratio progressively increased in the patients who have associated metabolic syndrome factors[80]. Obesity is characterized by the excess of adipose tissue and the altered secretion of adipocytokines that correlate with the occurrence of HCC and liver-related death in patients with cirrhosis[81]. In the last decade, it has become evident that obesity-related metabolic inflammation is involved in different aspects of HCC progression and metastatic dissemination, among which: neural regulation, innate immune responses, intestinal immune system and endocrinal regulation. Unfortunately, only few studies have been focusing on long-term mechanisms involved in obesity related HCC development[82], thus prospective studies are needed.
Finally, alterations in intestinal microbiota (or dysbiosis, defined as any change in the composition of the microbiota commonly found in healthy conditions), creating a pro-inflammatory microenvironment in the liver, seem to play a main role in the development of NAFLD-related HCC[83]. Dysbiosis, beyond the known risk factors for NAFLD, promotes the development of chronic liver diseases and HCC, independent of body mass index (BMI) and insulin resistance, producing a large amount of bioactive molecules, which deeply affect physiological and pathological body status[84]. Interestingly, in a mouse model, drugs able to modify the microbiome (e.g., rifaximin) may prevent HCC development. Rifaximin may additionally improve portal hypertension, spontaneous bacterial infection (SBP) risk, liver fibrosis and hepatic encephalopathy[85]. Actually, metabolic alterations have been associated with dysbiosis: ob-ob mice (homozygous for the obese mutation) have an imbalance of the intestinal microbiota with a decrease of Bacteroides and an increase in Firmicutes. This pattern of intestinal bacteria has the increased capacity to harvest energy from diet[86], as well as the microbiota composition described in NAFLD[57]. The altered microbioma (the genetic information genomes of gut microbiota) is characterized by the ability to produce alcohol, which in turn will be increased in the blood promoting hepatic oxidative stress and liver inflammation[87]. As demonstrated in obese patients, the equilibrium can be restored in case of a fat restriction diet[83].
Target/biomarker discovery and “Omic” approaches will help in finding new pro-oncogenic and oncosuppressor to be used as novel biomarkers. The new knowledge on HCC pathogenesis will open new avenues in the diagnosis and design of patient-tailored therapies.
HCC in NAFLD chronic hepatitis
NAFLD has a proportion of HCC, occurring in the absence of cirrhosis, higher than other chronic liver diseases. HCC in NAFLD generally lacks encapsulation and is well differentiated and characterized by large dimensions[88]. Multiple studies described a significant proportion of HCC (from 51% to 65%) that have stage 0-2 fibrosis[89-91], highlighting a specific dangerous behavior of NAFLD chronic hepatitis. Given the high number of patients with non cirrhotic NAFLD, screening for HCC in this population is not practicable[15]. Interestingly, the features of NAFLD-related HCC are similar to those of HCC of obese patients and of non-cirrhotic HCC, independently of the etiology[92,93]. Accordingly, it has been reported that obese patients have a relative risk of liver cancer of 189% relative to the 117% of overweight subjects[94]. Thus, the pathogenic mechanisms of hepatocarcinogenesis in steatosis might be different from the classic mechanisms involved in cirrhosis[95]. In fact, all the NAFLD-related HCC pathogenic mechanisms are independent from fibrosis and this might explain the particular epidemiology of HCC in NASH, where non-cirrhotic HCC is quite frequent relative to other etiological factors.
In the light of what has been reported above, pathophysiological studies are needed to better understand the underlying mechanisms involved in NAFLD-related HCC development. In this context, it is important to note that the EASL evidence based clinical practice guidelines should be improved because the up-to-date version does not exhaustively represent this specific problem.
HCC in NAFLD cirrhosis
Cirrhosis in NAFLD modifies prognosis and management. Increasing age, obesity and diabetes are considered as risk factors for the progression of NAFLD to cirrhosis[96]. Thus, it is well known that a subset of individuals with NAFLD may progress to liver cirrhosis, which in turn could be complicated by liver failure or even HCC, requiring liver transplantation (LT), resection, or loco-regional therapies[97].
However, although NAFLD has begun the most common cause of chronic liver disease worldwide[3,98], even today, a significant amount of patients with NAFLD are already incidentally diagnosed with cirrhotic. Unfortunately, NAFLD patients are asymptomatic, thus, the diagnosis of cirrhosis often occurs incidentally (70%) because it is done during clinical assessments for the investigation of different medical conditions unrelated to liver disease or an unexpected surgical finding. Accordingly, about the 15% of NAFLD patients selected for biopsy have cirrhosis, confirming that the prevalence of cirrhosis in patients with NAFLD is higher than expected[99]. In the presence of liver cirrhosis, the main problem is the occurrence of important complications, such as: liver decompensation, thrombocytopenia, splenomegaly or, sometimes, HCC related with a poor survival[100,101]. Late diagnosis increases the risk to find a late stage HCC, no longer curable with the available treatments, whereas the diagnosis of HCC, if done at the early stage, is associated with better results.
Cirrhosis has to be seen as a prognostic factor predicting negative outcomes in patients. Accordingly, in recent studies, it has been reported in NAFLD cirrhotic patients an overall mortality of 80% and a liver-related mortality of 55%, after 12 years[99].
Early recognition of NAFLD patients with cirrhosis, who have a higher risk of progression toward HCC, is the first crucial aim to reduce NAFLD-related morbidity and mortality. Thus, in patients with NAFLD, an improvement of diagnostic approach alertness is required for underrating the prevalence and the important clinical condition of NAFLD. Clinicians have developed adequate screening[102]. Finally, it is important to underline that ultrasonography (US) is likely inadequate in several subgroups of patients (obese, Child Pugh B or C, alcohol and NASH related cirrhotic) and does not permit the exclusion of the presence of HCC[103].
Declarations
Authors’ contributionsWrote the manuscript: Balsano C
Contributed critical revisions, edited the manuscript, and read and approved the final version of the manuscript: Balsano C, Porcu C, Sideri S, Tavolaro S
Availability of data and materialsNot applicable.
Financial support and sponsorshipWe thank the Francesco Balsano Foundation for financial support.
Conflicts of interestAll authors declare that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Siddique A, Kowdley KV. Insulin resistance and other metabolic risk factors in the pathogenesis of hepatocellular carcinoma. Clin Liver Dis 2011;15:281-96.
2. Araújo AR, Rosso N, Bedogni G, Tiribelli C, Bellentani S. Global epidemiology of non-alcoholic fatty liver disease/non-alcoholic steatohepatitis: what we need in the future. Liver Int 2018;38 Suppl 1:47-51.
3. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016;64:73-84.
4. White DL, Thrift AP, Kanwal F, Davila J, El-Serag HB. Incidence of hepatocellular carcinoma in all 50 United States, from 2000 through 2012. Gastroenterology 2017;152:812-20.e5.
5. Tsukuma H, Hiyama T, Tanaka S, Nakao M, Yabuuchi T, Kitamura T, Nakanishi K, Fujimoto I, Inoue A, Yamazaki H, Kawashima T. Risk factors for hepatocellular carcinoma among patients with chronic liver disease. N Engl J Med 1993;328:1797-801.
6. Ertle J, Dechêne A, Sowa JP, Penndorf V, Herzer K, Kaiser G, Schlaak JF, Gerken G, Syn WK, Canbay A. Non-alcoholic fatty liver disease progresses to hepatocellular carcinoma in the absence of apparent cirrhosis. Int J Cancer 2011;128:2436-43.
7. Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet 2002;31:339-46.
9. Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer 2006;6:674-87.
10. Margini C, Dufour JF. The story of HCC in NAFLD: from epidemiology, across pathogenesis, to prevention and treatment. Liver Int 2016;36:317-24.
11. Karagozian R, Derdák Z, Baffy G. Obesity-associated mechanisms of hepatocarcinogenesis. Metabolism 2014;63:607-17.
12. Rajendran P, Chen YF, Chen YF, Chung LC, Tamilselvi S, Shen CY, Day CH, Chen RJ, Viswanadha VP, Kuo WW, Huang CY. The multifaceted link between inflammation and human diseases. J Cell Physiol 2018;233:6458-71.
13. Sayre LM, Smith MA, Perry G. Chemistry and biochemistry of oxidative stress in neurodegenerative disease. Curr Med Chem 2001;8:721-38.
14. Palma-Duran SA, Kontogianni MD, Vlassopoulos A, Zhao S, Margariti A, Georgoulis M, Papatheodoridis G, Combet E. Serum levels of advanced glycation end-products (AGEs) and the decoy soluble receptor for AGEs (sRAGE) can discriminate non-alcoholic fatty liver disease in age-, sex- and BMI-matched normo-glycemic adults. Metabolism 2018;83:120-7.
15. Said A, Ghufran A. Epidemic of non-alcoholic fatty liver disease and hepatocellular carcinoma. World J Clin Oncol 2017;8:429-36.
16. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011;61:69-90.
17. GLOBOCAN. Estimated cancer incidence: mortality andprevalence worldwide 2012. Available from: http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx [Last accessed on 17 Jul 2018].
19. Ringelhan M, McKeating JA, Protzer U. Viral hepatitis and liver cancer. Philos Trans R Soc Lond B Biol Sci 2017;372:20160274.
20. Marot A, Henrion J, Knebel JF, Moreno C, Deltenre P. Alcoholic liver disease confers a worse prognosis than HCV infection and non-alcoholic fatty liver disease among patients with cirrhosis: an observational study. PLoS One 2017;12:e0186715.
21. Reeves HL, Zaki MY, Day CP. Hepatocellular carcinoma in obesity, type 2 diabetes, and NAFLD. Dig Dis Sci 2016;61:1234-45.
22. Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med 2003;348:1625-38.
23. Calzadilla Bertot L, Adams LA. The natural course of non-alcoholic fatty liver disease. Int J Mol Sci 2016;17:E774.
24. Cholankeril G, Ahmed A. Alcoholic liver disease replaces HCV infection as the leading indication for liver transplantation in the United States. Clin Gastroenterol Hepatol 2017; doi: 10.1016/j.cgh.2017.11.045.
25. Musso G, Paschetta E, Gambino R, Cassader M, Molinaro F. Interactions among bone, liver, and adipose tissue predisposing to diabesity and fatty liver. Trends Mol Med 2013;19:522-35.
26. Alisi A, Carpino G, Oliveira FL, Panera N, Nobili V, Gaudio E. The role of tissue macrophage-mediated inflammation on NAFLD pathogenesis and its clinical implications. Mediators Inflamm 2017;2017:8162421.
27. Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol 2011;11:85-97.
28. Seo YY, Cho YK, Bae JC, Seo MH, Park SE, Rhee EJ, Park CY, Oh KW, Park SW, Lee WY. Tumor necrosis factor-α as a predictor for the development of nonalcoholic fatty liver disease: a 4-year follow-up study. Endocrinol Metab (Seoul) 2013;28:41-5.
29. Hui JM, Hodge A, Farrell GC, Kench JG, Kriketos A, George J. Beyond insulin resistance in NASH: TNF-alpha or adiponectin? Hepatology 2004;40:46-54.
30. Abiru S, Migita K, Maeda Y, Daikoku M, Ito M, Ohata K, Nagaoka S, Matsumoto T, Takii Y, Kusumoto K, Nakamura M, Komori A, Yano K, Yatsuhashi H, Eguchi K, Ishibashi H. Serum cytokine and soluble cytokine receptor levels in patients with non-alcoholic steatohepatitis. Liver Int 2006;26:39-45.
31. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 1993;259:87-91.
32. Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-α function. Nature 1997;389:610-4.
33. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 2003;112:1821-30.
34. Onyekwere CA, Ogbera AO, Samaila AA, Balogun BO, Abdulkareem FB. Nonalcoholic fatty liver disease: synopsis of current developments. Niger J Clin Pract 2015;18:703-12.
35. Huang CY, Lee CY, Chen MY, Yang WH, Chen YH, Chang CH, Hsu HC, Fong YC, Tang CH. Stromal cell-derived factor-1/CXCR4 enhanced motility of human osteosarcoma cells involves MEK1/2, ERK and NF-kappaB-dependent pathways. J Cell Physiol 2009;221:204-12.
36. Liu H, Li J, Tillman B, Morgan TR, French BA, French SW. TLR3/4 signaling is mediated via the NFκB-CXCR4/7 pathway in human alcoholic hepatitis and non-alcoholic steatohepatitis which formed Mallory-Denk bodies. Exp Mol Pathol 2014;97:234-40.
38. Ascha MS, Hanouneh IA, Lopez R, Tamimi TA, Feldstein AF, Zein NN. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology 2010;51:1972-8.
39. Zoller H, Tilg H. Nonalcoholic fatty liver disease and hepatocellular carcinoma. Metabolism 2016;65:1151-60.
40. Del Ben M, Polimeni L, Brancorsini M, Di Costanzo A, D'Erasmo L, Baratta F, Loffredo L, Pastori D, Pignatelli P, Violi F, Arca M, Angelico F. Non-alcoholic fatty liver disease, metabolic syndrome and patatin-like phospholipase domain-containing protein3 gene variants. Eur J Intern Med 2014;25:566-70.
41. Seko Y, Yamaguchi K, Itoh Y. The genetic backgrounds in nonalcoholic fatty liver disease. Clin J Gastroenterol 2018;11:97-102.
42. Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M, Degos F, Clément B, Balabaud C, Chevet E, Laurent A, Couchy G, Letouzé E, Calvo F, Zucman-Rossi J. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet 2012;44:694-8.
44. Qu H, Xu W, Huang Y, Yang S. Circulating miRNAs: promising biomarkers of human cancer. Asian Pac J Cancer Prev 2011;12:1117-25.
46. Khoshnaw SM, Rakha EA, Abdel-Fatah TM, Nolan CC, Hodi Z, Macmillan DR, Ellis IO, Green AR. Loss of Dicer expression is associated with breast cancer progression and recurrence. Breast Cancer Res Treat 2012;135:403-13.
47. Zeng S, Yang J, Zhao J, Liu Q, Rong M, Guo Z, Gao W. Silencing Dicer expression enhances cellular proliferative and invasive capacities in human tongue squamous cell carcinoma. Oncol Rep 2014;31:867-73.
48. Zhu Y, Cai L, Guo J, Chen N, Yi X, Zhao Y, Cai J, Wang Z. Depletion of Dicer promotes epithelial ovarian cancer progression by elevating PDIA3 expression. Tumour Biol 2016;37:14009-23.
49. Lei L, Zhou C, Yang X, Li L. Down-regulation of microRNA-375 regulates adipokines and inhibits inflammatory cytokines by targeting AdipoR2 in non-alcoholic fatty liver disease. Clin Exp Pharmacol Physiol 2018; doi: 10.1111/1440-1681.12940.
50. Gori M, Arciello M, Balsano C. MicroRNAs in nonalcoholic fatty liver disease: novel biomarkers and prognostic tools during the transition from steatosis to hepatocarcinoma. Biomed Res Int 2014;2014:741465.
51. Becker PP, Rau M, Schmitt J, Malsch C, Hammer C, Bantel H, Müllhaupt B, Geier A. Performance of serum microRNAs -122, -192 and -21 as biomarkers in patients with non-alcoholic steatohepatitis. PLoS One 2015;10:e0142661.
52. Yamada H, Suzuki K, Ichino N, Ando Y, Sawada A, Osakabe K, Sugimoto K, Ohashi K, Teradaira R, Inoue T, Hamajima N, Hashimoto S. Associations between circulating microRNAs (miR-21, miR-34a, miR-122 and miR-451) and non-alcoholic fatty liver. Clin Chim Acta 2013;424:99-103.
53. Wang Y, Zhu K, Yu W, Wang H, Liu L, Wu Q, Li S, Guo J. MiR-181b regulates steatosis in nonalcoholic fatty liver disease via targeting SIRT1. Biochem Biophys Res Commun 2017;493:227-32.
54. Jampoka K, Muangpaisarn P, Khongnomnan K, Treeprasertsuk S, Tangkijvanich P, Payungporn S. Serum miR-29a and miR-122 as potential biomarkers for non-alcoholic fatty liver disease (NAFLD). Microrna 2018; doi: 10.2174/2211536607666180531093302.
55. Latorre J, Moreno-Navarrete JM, Mercader JM, Sabater M, Rovira ò, Gironès J, Ricart W, Fernández-Real JM, Ortega FJ. Decreased lipid metabolism but increased FA biosynthesis are coupled with changes in liver microRNAs in obese subjects with NAFLD. Int J Obes (Lond) 2017;41:620-30.
56. Wang L, Zhang N, Wang Z, Ai DM, Cao ZY, Pan HP. Decreased miR-155 level in the peripheral blood of non-alcoholic fatty liver disease patients may serve as a biomarker and may influence LXR activity. Cell Physiol Biochem 2016;39:2239-48.
57. Cheung O, Puri P, Eicken C, Contos MJ, Mirshahi F, Maher JW, Kellum JM, Min H, Luketic VA, Sanyal AJ. Nonalcoholic steatohepatitis is associated with altered hepatic microRNA expression. Hepatology 2008;48:1810-20.
58. Hur W, Lee JH, Kim SW, Kim JH, Bae SH, Kim M, Hwang D, Kim YS, Park T, Um SJ, Song BJ, Yoon SK. Downregulation of microRNA-451 in non-alcoholic steatohepatitis inhibits fatty acid-induced proinflammatory cytokine production through the AMPK/AKT pathway. Int J Biochem Cell Biol 2015;64:265-76.
59. Sharma H, Estep M, Birerdinc A, Afendy A, Moazzez A, Elariny H, Goodman Z, Chandhoke V, Baranova A, Younossi ZM. Expression of genes for microRNA-processing enzymes is altered in advanced non-alcoholic fatty liver disease. J Gastroenterol Hepatol 2013;28:1410-5.
60. Vega-Badillo J, Gutiérrez-Vidal R, Hernández-Pérez HA, Villamil-Ramírez H, León-Mimila P, Sánchez-Mu-oz F, Morán-Ramos S, Larrieta-Carrasco E, Fernández-Silva I, Méndez-Sánchez N, Tovar AR, Campos-Pérez F, Villarreal-Molina T, Hernández-Pando R, Aguilar-Salinas CA, Canizales-Quinteros S. Hepatic miR-33a/miR-144 and their target gene ABCA1 are associated with steatohepatitis in morbidly obese subjects. Liver Int 2016;36:1383-91.
61. Mohamed AA, Ali-Eldin ZA, Elbedewy TA, El-Serafy M, Ali-Eldin FA, AbdelAziz H. MicroRNAs and clinical implications in hepatocellular carcinoma. World J Hepatol 2017;9:1001-7.
62. De Conti A, Ortega JF, Tryndyak V, Dreval K, Moreno FS, Rusyn I, Beland FA, Pogribny IP. MicroRNA deregulation in nonalcoholic steatohepatitis-associated liver carcinogenesis. Oncotarget 2017;8:88517-28.
63. Ge W, Yu DC, Li QG, Chen X, Zhang CY, Ding YT. Expression of serum miR-16, let-7f, and miR-21 in patients with hepatocellular carcinoma and their clinical significances. Clin Lab 2014;60:427-34.
64. Karakatsanis A, Papaconstantinou I, Gazouli M, Lyberopoulou A, Polymeneas G, Voros D. Expression of microRNAs, miR-21, miR-31, miR-122, miR-145, miR-146a, miR-200c, miR-221, miR-222, and miR-223 in patients with hepatocellular carcinoma or intrahepatic cholangiocarcinoma and its prognostic significance. Mol Carcinog 2013;52:297-303.
65. Ashmawy AM, Elgeshy KM, Abdel Salam ET, Ghareeb M, Kobaisi MH, Amin HAA, Sharawy SK, Abdel Wahab AHA. Crosstalk between liver-related microRNAs and Wnt/β-catenin pathway in hepatocellular carcinoma patients. Arab J Gastroenterol 2017;18:144-50.
66. Wang F, Dai M, Chen H, Li Y, Zhang J, Zou Z, Yang H. Prognostic value of hsa-mir-299 and hsa-mir-7706 in hepatocellular carcinoma. Oncol Lett 2018;16:815-20.
67. An Y, Gao S, Zhao WC, Qiu BA, Xia NX, Zhang PJ, Fan ZP. Novel serum microRNAs panel on the diagnostic and prognostic implications of hepatocellular carcinoma. World J Gastroenterol 2018;24:2596-604.
68. Porcu C, Antonucci L, Barbaro B, Illi B, Nasi S, Martini M, Licata A, Miele L, Grieco A, Balsano C. Copper/MYC/CTR1 interplay: a dangerous relationship in hepatocellular carcinoma. Oncotarget 2018;9:9325-43.
69. Manz DH, Blanchette NL, Paul BT, Torti FM, Torti SV. Iron and cancer: recent insights. Ann N Y Acad Sci 2016;1368:149-61.
70. Tirnitz-Parker JE, Glanfield A, Olynyk JK, Ramm GA. Iron and hepatic carcinogenesis. Crit Rev Oncog 2013;18:391-407.
71. Antonucci L, Porcu C, Iannucci G, Balsano C, Barbaro B. Non-alcoholic fatty liver disease and nutritional implications: special focus on Copper. Nutrients 2017;9:E1137.
72. Tarantino G, Porcu C, Arciello M, Andreozzi P, Balsano C. Prediction of carotid intima-media thickness in obese patients with low prevalence of comorbidities by serum copper bioavailability. J Gastroenterol Hepatol 2018;33:1511-7.
73. Marshall KM, Laval M, Estacio O, Hudson DF, Kalitsis P, Shulkes A, Baldwin GS, Patel O. Activation by zinc of the human gastrin gene promoter in colon cancer cells in vitro and in vivo. Metallomics 2015;7:1390-8.
74. Ranasinghe WK, Baldwin GS, Bolton D, Shulkes A, Ischia J, Patel O. HIF1alpha expression under normoxia in prostate cancer--which pathways to target? J Urol 2015;193:763-70.
75. Alam S, Kelleher SL. Cellular mechanisms of zinc dysregulation: a perspective on zinc homeostasis as an etiological factor in the development and progression of breast cancer. Nutrients 2012;4:875-903.
76. Sorrentino P, D'Angelo S, Ferbo U, Micheli P, Bracigliano A, Vecchione R. Liver iron excess in patients with hepatocellular carcinoma developed on non-alcoholic steato-hepatitis. J Hepatol 2009;50:351-7.
77. Fargion S, Valenti L, Fracanzani AL. Role of iron in hepatocellular carcinoma. Clin Liv Dis 2014;3:108-10.
78. Ye Q, Qian BX, Yin WL, Wang FM, Han T. Association between the HFE C282Y, H63D polymorphisms and the risks of non-alcoholic fatty liver disease, liver cirrhosis and hepatocellular carcinoma: an updated systematic review and meta-analysis of 5,758 cases and 14,741 controls. PLoS One 2016;11:e0163423.
79. Yao M, Wang L, Leung PSC, Li Y, Liu S, Wang L, Guo X, Zhou G, Yan Y, Guan G, Chen X, Bowlus CL, Liu T, Jia J, Gershwin ME, Ma X, Zhao J, Lu F. The clinical significance of GP73 in immunologically mediated chronic liver diseases: experimental data and literature review. Clin Rev Allergy Immunol 2018;54:282-94.
80. Turati F, Talamini R, Pelucchi C, Polesel J, Franceschi S, Crispo A, Izzo F, La Vecchia C, Boffetta P, Montella M. Metabolic syndrome and hepatocellular carcinoma risk. Br J Cancer 2013;108:222-8.
81. Sharma D, Wang J, Fu PP, Sharma S, Nagalingam A, Mells J, Handy J, Page AJ, Cohen C, Anania FA, Saxena NK. Adiponectin antagonizes the oncogenic actions of leptin in hepatocellular carcinogenesis. Hepatology 2010;52:1713-22.
82. Gan L, Liu Z, Sun C. Obesity linking to hepatocellular carcinoma: a global view. Biochim Biophys Acta 2018;1869:97-102.
83. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature 2006;444:1022-3.
84. Da Silva HE, Teterina A, Comelli EM, Taibi A, Arendt BM, Fischer SE, Lou W, Allard JP. Nonalcoholic fatty liver disease is associated with dysbiosis independent of body mass index and insulin resistance. Sci Rep 2018;8:1466.
85. Ponziani FR, Gerardi V, Pecere S, D'Aversa F, Lopetuso L, Zocco MA, Pompili M, Gasbarrini A. Effect of rifaximin on gut microbiota composition in advanced liver disease and its complications. World J Gastroenterol 2015;21:12322-33.
86. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006;444:1027-31.
87. Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, Gill SR. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology 2013;57:601-9.
88. Iannaccone R, Piacentini F, Murakami T, Paradis V, Belghiti J, Hori M, Kim T, Durand F, Wakasa K, Monden M, Nakamura H, Passariello R, Vilgrain V. Hepatocellular carcinoma in patients with nonalcoholic fatty liver disease: helical CT and MR imaging findings with clinical-pathologic comparison. Radiology 2007;243:422-30.
89. Farges O, Ferreira N, Dokmak S, Belghiti J, Bedossa P, Paradis V. Changing trends in malignant transformation of hepatocellular adenoma. Gut 2011;60:85-9.
90. Yasui K, Hashimoto E, Komorizono Y, Koike K, Arii S, Imai Y, ShimaT, Kanbara Y, Saibara T, Mori T, Kawata S, Uto H, Takami S, Sumida Y, Takamura T, Kawanaka M, Okanoue T; Ministry of Health, Labour, and Welfare of Japan. Characteristics of patients with nonalcoholic steatohepatitis who develop hepatocellular carcinoma. Clin Gastroenterol Hepatol 2011;9:428-33.
91. Dyson J, Jaques B, Chattopadyhay D, Lochan R, Graham J, Das D, Aslam T, Patanwala I, Gaggar S, Cole M, Sumpter K, Stewart S, Rose J, Hudson M, Manas D, Reeves HL. Hepatocellular cancer: the impact of obesity, type 2 diabetes and a multidisciplinary team. J Hepatol 2014;60:110-7.
92. Chang CY, Hernandez-Prera JC, Roayaie S, Schwartz M, Thung SN. Changing epidemiology of hepatocellular adenoma in the United States: review of the literature. Int J Hepatol 2013;2013:604860.
93. Schütte K, Schulz C, Poranzke J, Antweiler K, Bornschein J, Bretschneider T, Arend J, Ricke J, Malfertheiner P. Characterization and prognosis of patients with hepatocellular carcinoma (HCC) in the non-cirrhotic liver. BMC Gastroenterol 2014;14:117.
94. Larsson SC, Wolk A. Overweight, obesity and risk of liver cancer: a meta-analysis of cohort studies. Br J Cancer 2007;97:1005-8.
95. Torres DM, Harrison SA. Nonalcoholic steatohepatitis and noncirrhotic hepatocellular carcinoma: fertile soil. Semin Liver Dis 2012;32:30-8.
96. Angulo P, Hui JM, Marchesini G, Bugianesi E, George J, Farrell GC, Enders F, Saksena S, Burt AD, Bida JP, Lindor K, Sanderson SO, Lenzi M, Adams LA, Kench J, Therneau TM, Day CP. The NAFLD fibrosis score:a noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology 2007;45:846-54.
99. Bertot LC, Jeffrey GP, Wallace M, MacQuillan G, Garas G, Ching HL, Adams LA. Nonalcoholic fatty liver disease-related cirrhosis is commonly unrecognized and associated with hepatocellular carcinoma. Hepatol Commun 2017;1:53-60.
100. Sanyal AJ, Banas C, Sargeant C, Luketic VA, Sterling RK, Stravitz RT, Shiffman ML, Heuman D, Coterrell A, Fisher RA, Contos MJ, Mills AS. Similarities and differences in outcomes of cirrhosis due to nonalcoholic steatohepatitis and hepatitis C. Hepatology 2006;43:682-9.
101. Mofrad P, Contos MJ, Haque M, Sargeant C, Fisher RA, Luketic VA, Sterling RK, Shiffman ML, Stravitz RT, Sanyal AJ. Clinical and histologic spectrum of nonalcoholic fatty liver disease associated with normal ALT values. Hepatology 2003;37:1286-92.
102. Wieland AC, Quallick M, Truesdale A, Mettler P, Bambha KM. Identifying practice gaps to optimize medical care for patients with nonalcoholic fatty liver disease. Dig Dis Sci 2013;58:2809-16.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Balsano C, Porcu C, Sideri S, Tavolaro S. Fat and hepatocellular carcinoma. Hepatoma Res 2018;4:38. http://dx.doi.org/10.20517/2394-5079.2018.51
AMA Style
Balsano C, Porcu C, Sideri S, Tavolaro S. Fat and hepatocellular carcinoma. Hepatoma Research. 2018; 4: 38. http://dx.doi.org/10.20517/2394-5079.2018.51
Chicago/Turabian Style
Balsano, Clara, Cristiana Porcu, Silvia Sideri, Simona Tavolaro. 2018. "Fat and hepatocellular carcinoma" Hepatoma Research. 4: 38. http://dx.doi.org/10.20517/2394-5079.2018.51
ACS Style
Balsano, C.; Porcu C.; Sideri S.; Tavolaro S. Fat and hepatocellular carcinoma. Hepatoma. Res. 2018, 4, 38. http://dx.doi.org/10.20517/2394-5079.2018.51
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 15 clicks
Cite This Article 15 clicks

 Like This Article 10
likes
Like This Article 10
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.