
Mitoepigenetics and hepatocellular carcinoma


Abstract
Mitochondria are the center of energy production in eukaryotic cells and are crucial for several cellular processes. Dysfunctional mitochondria have been associated with cancer progression. Mitochondria contain their own circular DNA (mtDNA), which codes for 13 proteins, 2rRNA, 22tRNA and non-coding RNAs. Recent evidence showed the presence of 5-methylcytosine and 5-hydroximethylcytosine in mtDNA suggesting that the level of gene expression could be modulated like a nuclear DNA by direct epigenetic modifications. Mitoepigenetics is a bidirectional phenomenon in the epigenetic regulation of mitochondrial genes encoded in both the nucleus and the mitochondrion. This process is affected by SAM-mediated methylation and hydroxymethylation of mtDNA and by nuclear chromatin modulators from mitochondria, such as Acetyl-CoA and NAD+. There is some information about physiological and pathological methylated profiles, but information is scarce for hepatocellular carcinoma (HCC). The aim of this review is to summarize the mitoepigenetic knowledge in HCC already reported so far, through a keywords search in Medline. In addition, the deregulation of energy intermediaries needed for the mitoepigenetic regulation is described. As this is a new area of study, a rigorous analysis and careful interpretation and integration of results are needed.
Keywords
Introduction
Hepatocellular carcinoma (HCC) is a highly malignant cancer, with high recurrence rate and a poor prognosis. HCC is a complex pathology associated with chronic liver disease, 80% to 90% are originated from cirrhosis of diverse etiology, most frequently infections with hepatitis B virus, hepatitis C virus, the mycotoxin, aflatoxin B1, and the metabolic syndrome[1,2].
Mitoepigenetics events include the interplay between mitochondrial-derived substrates and the nuclear epigenetic landscape. This includes all epigenetic events that affect the expression of the mitochondrial genome and the nuclear-encoded mitochondrial genes[3,4]. The importance of mitoepigenetics lies not only in the functions described for this organelle, but also in the generation of intermediaries that serve to regulate the function of other cellular components, as will be mentioned later.
Mitochondrial dysfunction is involved in several diseases including cancer[5]. The ability of mitochondria to regulate the energetic redox state and the metabolism of cells could result in the production of epigenetic intermediates that participate in normalization of the mitochondrial function. Studies have revealed several metabolic alterations in liver diseases including modification in energy supply[6,7]. A sequential model of cirrhosis-HCC induced by diethylnitrosamine (DEN) revealed that cancer progression is associated with mitochondrial dysfunction[8].
Multiple insults to the mitochondrial genome have been associated with different pathophysiologies, and have been described as one of the most common and consistent phenotypes of cancers[9-13].
Mitochondria are vital for the cell because they are responsible for its metabolic activity, as well as for producing the bulk of the energy requirements in the form of ATP, maintaining calcium homeostasis, and inducing apoptosis[14,15]. In mitochondria, ATP is generated through the process of oxidative phosphorylation (OXPHOS), which occurs via the electron transport chain (ETC). Mitochondria contain their own genome (mitochondrial DNA, mtDNA). Each organelle contains about 1-10 copies of mtDNA[16]. MtDNA is distinctly different from the nuclear DNA (nDNA), the mtDNA is a circular, double-stranded DNA molecule of approximately 16.6 kb in size and it is inherited only through the mother. MtDNA is found associated and packed with proteins in a nucleoid, where an encoded nuclear protein known as mitochondrial transcription factor A (TFAM) is the major protein component[17]. The mtDNA comprises a heavy (H) strand and a light (L) strand, which encode 13 of the polypeptides that constitute the complexes I, III, IV, and V of the ETC. MtDNA also encodes some of its own transcriptional and translational machinery, which includes 22 tRNAs and 2 rRNAs[12,13,18]. The rest of the mitochondrial proteins (~1500), involved in the mitochondrial function, replication, transcription and translation of mtDNA, are encoded by nuclear genes and are targeted to the mitochondrion by a specific transport system[19].
Methods
A bibliographic search was performed of the Medline database (US National Library of Medicine, http://www.ncbi.nlm.nih.gov). The keywords used or combinations of them were: cancer, hepatocellular carcinoma, epigenetics, mitoepigenetics, mitochondria, methylation, hydroxymethylation and miRNA. All the articles that included the terms and/or combinations referring to the metabolic regulation, as well as to mitoepigenetics in HCC were selected.
Epigenome substrates generated by mitochondria
Cellular growth and replication depends on the energetic state through epigenetic modifications in the DNA chromatin structure[20]. This is achieved by coupling modulation of nDNA chromatin structure and function by modification via high energy intermediates: phosphorylation by ATP, acetylation by acetyl-coenzyme A (Ac-CoA), deacetylation by nicotinamide adenine dinucleotide (NAD+), and methylation by S-adenosyl-methionine (SAM)[20]. As afore mentioned, the mitochondrion is responsible for ATP production as part of the energetic metabolism. However, the role of ATP is not just to be the main energy provider, but it also regulates multiple cellular functions through phosphorylation and dephosphorylation reactions (when there are low levels of ATP), as part of what is known as post-translational modifications.
Glucose is the main source of Ac-CoA and is the link between glycolysis and the tricarboxylic acid cycle (TCA). In addition to its energetic role, the Ac-CoA is the substrate for acetylation reactions that are important to modulate gene expression and the function of some proteins[21]. Acetylation of mitochondrial substrates is controlled by a NAD+-dependent deacetylase sirtuin-3 (Sirt-3). Therefore, the mitochondrial redox state must be controlled to maintain the NAD+ availability for the Sirt-3 activity.
On the other hand, SAM is the physiological methyl donor group synthesized from L-methionine and ATP in a reaction catalyzed by the methionine adenosyltransferase (MAT) enzyme[22]. SAM is synthesized in the cytosol and imported to the mitochondrial matrix via the mitochondrial SAM carrier, likely via exchange for its metabolized variant S-adenosyl-homocysteine (SAH)[23]. SAM synthesis is regulated in part by the mitochondrial one-carbon (folate) metabolism[24]. An enzyme that participates in mitochondrial folate metabolism, the mitochondrial bifunctional enzyme (MBE), regulates the change between SAM and the nucleotide synthesis. In proliferative cells, such as embryonic or cancer cells, MBE is expressed, and the one-carbon units are shuttled predominantly towards nucleotide synthesis. Under these conditions, less one-carbon units are available for SAM synthesis and DNA methylation. Conversely, in differentiated cells, MBE is turned off, less mitochondria toward nucleotides synthesis are produced, and one-carbon units are directed through increased SAM synthesis and increased DNA methylation[25]. SAM is important for DNA epigenetic, methylation of phospholipids[26], and proteins; thus, modulating relevant cellular functions. For example, the relationship between methylation and mitochondrial dysfunction being so close that deficiency of SAM may lead to mitochondrial damage and, finally, to insulin resistance[27].
Principal mitoepigenetics processes
mtDNA methylation and hydroxymethylation
DNA methylation is an epigenetic modification of the DNA that is frequently disrupted in nearly all types of cancer. Hypomethylation of the repetitive elements associated with increased genomic instability is frequently observed in cancer cells[28]. The hypermethylation of specific CpG islands in promoter regions of several tumor-suppressor genes is commonly observed to be associated with transcriptional silencing of the gene[29,30]. Epigenetic regulation of the mitochondrial genome was an enigma, until recent studies[31]. For example, there is no evidence of post-translational modifications of TFAM as it happens in nuclear DNA histones, and the most important epigenetic regulation of mtDNA is DNA methylation and hydroxymethylation. DNA methylation is regulated by four DNA methyltransferases (DNMT1, 3A, 3B, and 3L) and three demethylases, that is, ten-eleven translocations (TET1-3)[32].
The mtDNA methylation is accomplished by the mitochondrial DNA methyltransferase (mtDNMT1), a nuclear encoded DNMT1 that contains a mitochondrial targeting sequence[33]. Methylation of nDNA occurs principally in cytosines (5mC) of CpG dinucleotides, but recently it has been shown that mtDNA methylation is found predominantly in non-CpG sites and that it is DNMT independent[4]. In general, mtDNA is undermethylated, with only 1% to 5% of methylated cytosines. Several factors increase mtDNMT1 transcription and translocation to the mitochondria, like p53, oxidative stress-responding transcription factors, nuclear respiratory factor 1 (NRF1), peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC-1α), and p16 cell cycle inhibitor[33]. The methyltransferase DNMT3a may also be involved in the methylation of mtDNA since it has been found in mitochondrial fractions from mouse cell lines and from the human central nervous system[32]. In 2011, Shock et al.[33] reported the presence of 5-hydroxymethylcytosine (5hmC) and 5-methylcytosine (5mC) in mammalian mtDNA, further demonstrating the translocation of methyltransferase 1 (DNMT1) to mitochondria. Alterations in mtDNMT expression affect transcripts of the heavy and light strands of mtDNA. The modulation of mtDNA methylation has been studied in response to oxidative stress, where there seems to be a decrease in this modulation[34], this response can be a compensatory response to mtDNA damage by increasing the expression of residual mtDNA genes. The DNMT enzymes use SAM as the methyl donor. However, the importance of mtDNA in methylation not only lies in the mitochondrial function but it can modify the overall epigenetic state of the cell. In fact, it has been observed that the decrease of mtDNA leads to altered levels of methylation in the genomic DNA, which is normalized once the mtDNA content is reestablished[33]. In 2013, Bellizzi et al.[35] studied the methylation patterns in mitochondrial cytosines in humans and mice and the effect of suppressing DNMT1, DNMT3a, and 3b. In general, a preponderant methylation of CpG dinucleotides and its inhibition was found in knockout mice without affecting methylation in non-CpG sites[35].
The DNA methylation pattern of the human mitochondrial genome remains relatively constant; however, there are some loci that are differentially methylated in different tissues and over time[36]. For example, subunit 6 of NADH dehydrogenase, a crucial subunit for the assembly of complex I, is suppressed due to hypermethylation by an increase in the expression of DNMT1[33]. It is also known that the 12s rRNA gene is methylated by the rRNA methyltransferase-related transcription factor 1 (mt-TFB1)[37]. This epigenetic regulation is important for ribosomal biogenesis and mitochondrial translation and has been related to aging; this modification alters the efficiency of the ETC by hampering the assembly of complex I[38]. In 2016, Liu et al.[39] determined methylation of the human mitochondrial genome in blood and saliva samples; by bisulfite pyrosequencing, 9 regions in human mtDNA were detected including a D-loop, 12S rRNA, 16S rRNA, ND1, COXI, ND3, ND4, ND5, CYTB.
Hydroxymethylation is another important epigenetic modification described for mtDNA, where 5mC is oxidized into 5-hmC by the TET family of methylcytosine dioxygenases. The presence of TET1 and TET2 has been described in neuronal mitochondrial fractions[32,40]. TET proteins are members of the family of 2-oxoglutarate-dependent dioxygenases (2-OGDO) that can oxidize 5mC to generate 5hmC, 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC), mediating DNA demethylation by oxidation in cooperation with the BER repair pathway[40]. The reaction is dependent on the presence of oxygen, 2-oxoglutarate, and Fe2+. 2-Oxoglutarate is a key metabolite in the Krebs cycle that occurs in the matrix of mitochondria; on the other hand, succinate and fumarate, also TCA intermediates, are potent inhibitors of 2-OGDO enzymes, in this manner TCA cycle controls the DNA and histone methylation and, thus, controls gene expression[40].
The kind of hydroxymethylation of cytosines in mitochondria has also been reported[33]. The profile of modifications in the D-loop region (a non-coding region that acts as a promoter for the H and L strands of the mtDNA and contains transcription and replication elements), and the similarity of this profile in cells of similar tissues and how the profile is different in cells of different tissues have been described recently[41].
Given the relevance of mitochondrial DNA and its regulation at different levels, recent studies have also proposed mitochondrial DNA methylation as a potential biomarker[42].
The central role of microRNAs in modulating mitochondria
Other regulators that have recently been studied in mitochondria are microRNAs (miRNAs)[43]. miRNAs are small non-coding RNAs, implicated in gene post-transcriptional regulation and the conformation of genetic expression patterns with physiological relevance. They derive from longer RNAs, primary miRNAs (pri-miRNAs), and are sequentially cleaved by ribonuclease III (RNAse III) enzymes or processed for pre-miRNA splicing and RNA degradation pathways[44].
It has been found that both pre-miRNAs and mature miRNAs can be found in mitochondria, suggesting that this organelle can synthesize them and keep them active in their transcriptional machinery or export them to the cytosol. Likewise, the possibility arises that the miRNAs processed in mitochondria regulate the expression of genes related to the function of the same organelle[45]. On the other hand, to mention some examples, miR-181c-5p regulates mitochondrial energy metabolism through mt-COX1 mRNA; although its origin is nuclear it can be exported to the mitochondria and execute its effect[46,47]. In addition, miR-499 regulates mitochondrial dynamics through mitochondrial fission protein and apoptosis[48].
Epigenetic regulation of nuclear-encoded mitochondrial genes
Mitoepigenetics includes the regulation of nuclear-encoded mitochondrial genes as previously mentioned in its definition; importantly, two of the transcription factors that carried out this modulation are: PGC-1α and NRF-1. However, as we describe in this section, little is known about its epigenetic regulation in HCC, then, it represents an area of study to explore.
PGC-1α
PGC-1α is critical for the expression of genes involved in fatty acid oxidation, as well as in mitochondrial gene expression through the coactivation of major transcription factors, controlling the complex program of mitochondrial biogenesis[49]. It has been suggested that biogenesis induced by PGC-1α is tumor promoting[50]. Sirtuin-1 (Sirt-1), a NAD+-dependent deacetylase, targets several transcription factors, like PGC-1α, both proteins have been found overexpressed in HCC and are related to defective mitochondrial accumulation[51]. Non-CpG methylation of the PGC-1α promoter controls mitochondrial density and has been detected in pathological conditions such as obesity[52]. It may be interesting to obtain an epigenetic PGC-1α pattern in HCC to know if its modulation is carcinogenesis stage-dependent and to establish the accurate way to be pharmacologically controlled.
NRF-1
NRF-1 binds to the cytochrome C promoter and positively regulates nuclear-encoded mitochondrial genes[53]; low-levels of NRF-1 cause mitochondrial dysfunction. The NRF-1 gene sequence has several CpG islands which are susceptible to methylation and demethylation processes; it has been shown that hypermethylation of the promoter region of NRF-1 causes a decrease in its expression[54]. Further studies of the epigenetic regulation of NRF-1 in HCC are needed.
Mitoepigenetics in HCC
One of the most and consistent phenotypes of cancer are defective mitochondria. There is evidence that the loss of mitochondrial function and epigenetic alterations in this organelle are related to the process of carcinogenesis because of their vital role in energy production and contribution to the metabolism of epigenome effectors[1,55,56]. Mitochondrial dysfunctions also lead to resistance to apoptosis. Since Warburg's hypothesis, a number of mitochondrial abnormalities in cancers, both at the genetic and metabolic levels, have been reported[8,9,11].
Moreover, progressive mitochondrial dysfunction has been linked to an enormous variety of diseases, such as mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes syndrome (MELAS), Leber’s hereditary optic neuropathy (LHON), deafness, diabetes, Alzheimer, and Parkinson disease[57,58].
Alterations observed in the epigenetic substrates in HCC
As previously reported, metabolic alterations have been found in liver diseases[6,7] and the HCC is not the exception; the metabolic reprogramming that happens in cancer cells implies a decrease in ATP[8,59]. However, some studies have not found significant changes in acetyl-CoA levels that normally depend on energy metabolism, but which, in HCC, may have a nuclear origin and play a fundamental role in the progression of the cell cycle and DNA replication[8,60].
We have mentioned that the mitochondrial redox state is fundamental for the proper functioning of mitochondria; with respect to HCC, it is known that the NAD+/NADH ratio decreases significantly[8], which could modify the activity of enzymes that depend on the cellular redox state. On the other hand, currently a change in the levels of an enzyme called MAT that catalyzes the formation of SAM has been implicated also. MAT is encoded by two genes, Mat1a and Mat2a. In HCC, the liver decreases the amount of MAT1A and increases MAT2A through epigenetic mechanisms alone; this switch is responsible for the decreasing level of SAM, favoring the development of this pathology[61,62].
Due to the above, it is necessary not only to consider the epigenetic modifications but also the generation of intermediaries by mitochondria that allow for the appropriate epigenetics control of both the mtDNA and the nDNA.
Altered mitoepigenetics in HCC
As afore mentioned, NRF1 and PGC1-α act on nuclear genes encoding respiratory subunits from ETC and are involved in the transcription and replication machinery. An up-regulation of this protein in HCC has been demonstrated[8,63]. An increased PGC1α level has been suggested to be an important inducer for the accumulation of dysfunctional mitochondria[8]. Although the role of pgc1-α and nrf1 genes methylation has not been studied in HCC, this is an interesting area to be investigated. A meta-analysis of DNA methylation in HCC revealed a correlation between several aberrant methylated genes and the risk of HCC, among them p53 was hypermethylated in HCC tumor tissue compared to the adjacent tissue. It is important to consider that, in turn, this gene is implicated in the transcription and translocation of mtDNMT1 to mitochondria, and, in this way, besides of its role as tumor suppressor it could be modulating the methylation status of mitochondrial genes[64].
Under conditions of oxidative stress, which may be a factor for the development of HCC, the transcriptional and mitochondrial DNA replication machinery is altered. Consequently, the ETC loses its functionality and favors the accumulation of reactive oxygen species (ROS). In addition, the mtDNA may suffer injuries because of the accumulation ROS[1,65]. On the other hand, the mitochondrial damage observed in cancer cells can have consequences on the expression of nuclear genes. There are studies that indicate that the removal of mtDNA responds to changes in the nuclear genome[56,66].
A genome-wide mapping of DNA methylation and hydroxymethylation in a study on HBV-related HCC revealed that the metabolic pathways that include glycolysis, gluconeogenesis, oxidative phosphorylation, and TCA contained the largest number of (hydroxy) methylation-altered genes, indicating the crucial roles of metabolic processes that implicate mitochondria in the progression of HCC, which, in turn, are regulated by epigenetic mechanisms. The authors propose that some of the identified (hydroxy) methylation-altered genes may serve as biomarkers for the diagnosis and prognosis of HCC[67]. Among the 5-mC and 5-hmC altered genes related to OXPHOS were the following: NADH dehydrogenase [ubiquinone] 1 subunit C2 (NDUFC2), NADH dehydrogenase [ubiquinone] flavoprotein 1 (NDUFV1), NADH: ubiquinone oxidoreductase subunit S6 (NDUFS6) from complex 1 and succinate dehydrogenase complex flavoprotein subunit A (SDHA) from complex II. Among the TCA genes: succinyl-CoA ligase [GDP-forming] subunit beta (SUCLG2) and pyruvate carboxylase.
Also, in HBV-induced hepatic carcinogenesis, protein X (HBx), encoded by the virus, has been proposed as an epigenetic regulator for tumor suppressor genes, by hypermethylation. It has been suggested that, in hepatomas, NQO1 (NAD(P)H: quinone oxidoreductase 1), which is a cytosolic protein that catalyzes two-electron reduction, can be deregulated by induction of HBx, generating mitochondrial damage and increasing oxidant stress in cells through hypermethylation of the NQO1 promoter[68].
Specifically in mitochondria, some epigenetic modifications have been described in HCC, such as hypermethylation of the Mrps12 (mitochondrial ribosomal protein S12), Mgrap (mitochondria-localized glutamic acid-rich protein), and Tmem70 (transmembrane protein 70) genes[69]. The TMEM70 protein, encoded by the Tmem70 gene, is a protein of the mitochondrial inner membrane that participates in mitochondrial biogenesis and whose mutations can be associated with the deficiency in the synthesis of ATP[70,71]. MRPS12 is a mitoribosomal conserved protein[72] and MGRAP is an important protein for the maintenance of mitochondrial morphology and quantity, as well as for the process of steroidogenesis[73].
Enzymes that catalyze acetylation, methylation, or their loss also regulate epigenetic changes[74,75]. For example, the enzyme LSD1 (lysine-specific demethylase 1) uses the mitochondrial cofactor FAD to carry out the demethylation of modified histones such as H3K4me1 and H3K4me2[76].
LSD1 has been proposed as a regulator of cell proliferation in several cancer types, as well as its metabolic reprogramming[77,78]. In HCC, it has been determined that LSD1 regulates energy production and suppresses mitochondrial respiration[78]. These studies also determined that the demethylase activity by LSD1 represses mitochondrial metabolism genes and induces the expression of glycolytic genes[78].
In HCC, differential expression of different miRNAs has been observed, as well as epigenetic modifications[79].
miR-122 regulation of PGC-1α (peroxisome proliferator activated receptor gamma, coactivator 1 alpha) and SDH (succinate dehydrogenase) subunits A and B is necessary for mitochondrial metabolism. In HCC, this miRNA is scarcely expressed. Studies propose this microRNA as a tumor suppressor, since in primary HCC tumors, in both human and rodents, it is seen at low levels, compared to its healthy controls. Also, in HBV-infected patients, this miRNA is reduced in hepatic tissue[45,80,81]. miR-122 is implicated in the control of lipid metabolism and circadian regulation in the liver. This microRNA has been observed in steatohepatitis and liver fibrosis, in addition to HCC[80,82]. Experimentally, the genetic deletion of miR-122 in mice has important effects on lipid metabolism, as well as on the progression of liver disease, from microsteatosis to HCC[80,82].
miR-33a/b regulates lipid metabolism through the ABCA1 cholesterol transporter. Its overexpression inhibits the oxidation of fatty acids in the liver cancer cell line HuH7, favoring the accumulation of triglycerides in larger lipid droplets. MiR-33 binding sites have been identified in the 3 'UTR of genes for mitochondrial proteins such as carnitine O-octaniltransferase (CROT) and carnitine palmitoyltransferase 1A (CPT1a). This miRNA inhibits also the insulin receptor substrate 2 (ISR2) and regulates insulin signaling[83,84]. There is very little information about the role of this miRNA in HCC, so we suggest that this could be studied more extensively to determine its importance in this pathology.
On the other hand, the role of mitochondrial genetic alterations has been investigated in HCC, and tumors contained significantly reduced mtDNA and TFAM overexpression, although neither condition correlated with the degree of cell differentiation; TFAM expression correlated with tumor size[85].
For all the above, the field of mitoepigenetics has attracted research with the aim of having more effective therapeutic targets in the treatment of HCC.
Mitoepigenetics in tumor-initiating stem-like cells in HCC
The liver cancer, as other epithelial cancers, has tumor-initiating stem-like cells (TICs) that are implicated in tumorigenesis and drug resistance[86]. TICs share some characteristics with embryonic stem cells (ESC) including expression of the pluripotency transcription factors, NANOG, OCT4, MYC, and SOX2. The expression of pluripotency transcription factors contributes to cancer progression by reprogramming mitochondrial metabolism[87,88].
NANOG represses OXPHOS genes, prevents mitochondrial ROS production, and activates fatty acid oxidation (FAO), contributing to TIC self-renewal and drug resistance[87]. Low levels of ROS are necessary to preserve stemness and self-renewal characteristics in TICs and protect them from drug-induced cell death. Elevated levels of ROS inhibit stemness genes by activation of the p38 MAPK pathway leading to polycomb suppressor protein complex I (BMI) protein degradation and FOXO3 activation. NANOG also acts synergistically with p53 inactivation and b-catenin activation to reprogram cellular metabolic pathways, since p53 promotes glycolysis and OXPHOS[88]. Other pluripotency transcription factors that are being studied that contribute to TICs metabolism reprogramming are MYC and OCT4. MYC regulates the glutaminolysis and glycolysis pathways and OCT4 also regulates OXPHOS[29,88].
MtDNA copy number has a very important role in tumorigenesis. Depending on the cancer types, the mtDNA copy number varies, an increase in prostate and endometrial cancer has been reported, whereas a decrease has been shown in HCC and gastric cancer[89]. Cancer stem like cells have a low mtDNA copy number that promotes their high proliferation rate and shifts their energy production by glycolysis. This low mtDNA copy number downregulates the expression of the catalytic subunit of the mitochondrial-specific polymerase POLGA by hypermethylation at exon 2[89]. The reduced expression of POLGA is necessary to maintain pluripotency of cancer stem like cells. Yamada et al.[90] reported a reduced copy number of mtDNA in patients with HCC, which correlated with malignant potential.
The removal of mtDNA from cells in culture induces alterations in nDNA methylation. For example, the content of 5mC in the genomic DNA of HCC (tumor tissue) is negatively correlated with the content of mtDNA[91]. These changes are reversible upon re-establishment of mtDNA[56]. One possible mechanism is that the expression of the enzyme DNMT1, crucial in DNA methylation, is dependent on the copy number of mtDNA[92].
miR122 is the most important miRNA in adult healthy liver and is associated with liver stem cells differentiation towards hepatocytes. In HCC, miR122 expression is lost. When miR122 expression is reestablished in a stem-like cell line derived from human HCC (BCLC9 cells), it decreases cell proliferation rate and reduces tumor size in vivo. This effect is achieved by down-regulating MYC, KLF4, FOXM1, AKT2, and AKT3 and up-regulating FOXO1 and FOXO3A gene expression[93].
Potential value of mitoepigenetics as biomarkers for cancer diagnosis and therapy
The main problem of HCC is the absence of early detection and effective therapies. The Asian Pacific Association for the Study of the Liver has recommended the use of alpha-fetoprotein (AFP) as a diagnostic biomarker for early detection of HCC complemented with orthodox imaging-based tools[94]. There are other candidates with clinical value for early HCC diagnosis; in this regard, glycoforms of AFP, des-γ-carboxyprothrombin, glypican-3, cytokeratin 19, annexin A2, and circulating miRNAs have been proposed, among others, to be used alone or in combination[95].
Cancer metabolic reprogramming regulated by mitochondrial enzymes is now one of the hallmarks of cancer. Tumor cells can acquire functional mtDNA from healthy cells to restore respiratory function and metabolic activity, which enabled them to proliferate[96].
The mtDNA acts as a critical message to travel and communicate between tumor cells and neighbor non-tumor cells. The outcome of mtDNA horizontal transfer could induce chemoresistance in the treatment[97].
In addition to the above mentioned, the mitochondrial cellular content and mutations have also been suggested as novel molecular markers[98]. Moreover, reduced expression of OXPHOS complexes has been associated with various form of cancer, including HCC. Carcinogenesis is a complex process that can be accompanied by epigenetic modifications. According to what has been described in this review, the epigenetic regulation of carcinogenesis can be used not only as a biomarker of cancer, but also to determine the stage of the carcinogenic process, because epigenetic patterns may be associated. Although there is very little information about mitoepigenetics in HCC, there are data that may be promising as a biomarker and even as a therapeutic target. The mtDNA heterogeneity epigenetics should be investigated by measuring single-cell DNA sequencing, comprehensive characterizations of mtDNA, and bidirectional effects between mtDNA and 3D genome, instability, and gene editing. It would be more helpful to combine the single-cell biology of CRIPRS to mtDNA function, given that copy number changes can also be regarded as biomarkers in cancer diagnosis and treatment[99-102].
Ye et al.[67] reported the methylation and hydroxymethylation profile of DNA in HCC related to HBV; they found some hypermethylated genes associated with metabolic pathways. Of these genes, the pc gene that codes for the enzyme pyruvate decarboxylase (PC) should be considered. PC is a nuclear-encoded mitochondrial enzyme involved in gluconeogenesis, it catalyzes the conversion of pyruvate to oxaloacetate in an adenosine triphosphate (ATP)-dependent form. Being a liver-specific enzyme, the hypermethylation of this gene could provide specificity as a biomarker of HCC.
Further studies are needed to find correlations between mtDNA methylation patterns and HCC in such a way as to get diagnostic tools through non-invasive techniques. Let us recall the work of Liu et al.[39], in which the detection of mtDNA methylation was below 2% in blood and saliva. However, there are other mitoepigenetic parameters in which significant correlations have been found with HCC and that point them out as potential biomarkers, such is the case of miR-122 that has been considered a molecule with great potential for diagnosis, prognosis of liver disease, and therapy. Studies demonstrated that miR-122 is reduced in rodent and human primary HCC[103]. Being it a miRNA that regulates hepatic homeostasis and having been found under-regulated in diseases. such as HCC, it becomes a possible biomarker and possible therapeutic target[104].
Final remarks
This review supports the suggestion that disrupted mitoepigenetics may contribute to tumorigenesis. The next generation experiments may elucidate the functional implications of mtDNA methylation and hydroxymethylation and could help clarify the role of these epigenetic markers.
The role of mtDNA is pluripotent, because it can affect processes like cellular differentiation, energy production, oxidative stress, metabolism, inflammation, and carcinogenesis. However, most of these emerging evidences could be modulated, at least in part, through changes in mitochondria, they could offer also a new opportunity to understand the causality of cancer [Figure 1].
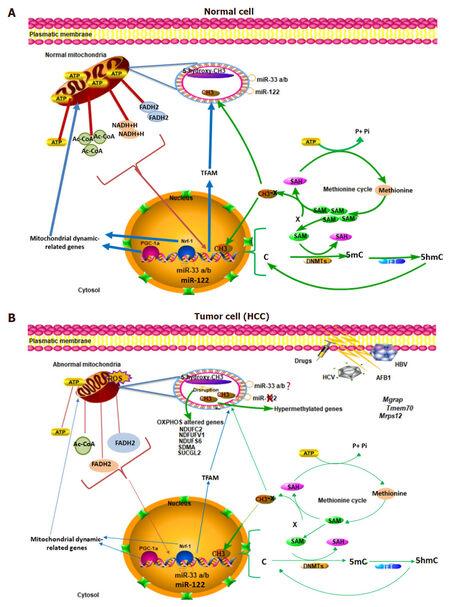
Figure 1. Epigenetic events in the expression of the mitochondrial genome and the nuclear-encoded mitochondrial genes. (A) Normal cell. There is an epigenetic flow of information between nucleus and mitochondria. The epigenetic events requires the production of both cytosolic and mitochondrial intermediates such as Ac-CoA, FADH2, NAD+/NADH and SAM generated by the TCA and methionine cycles, supporting the importance of the mitochondrial function. Methylation and hydroxymethylation reactions mediated by DNMTs and TET proteins can occur in both, mitochondria and nucleus, regulating the expression of the mitochondrial genome and the nuclear-encoded mitochondrial genes. In hepatic cells, miRNAS 122 and 33 a/b are also important to modulate the mitochondrial gene expression, modifying the metabolic state. The methylation in the PGC-1α and NRF-1 promoters regulates their expression; then, have an important effect on the mitochondrial dynamic; (B) Tumoral cell. There are dysfunctional mitochondria then, the production of mitochondrial intermediates are reduced and also an altered methionine cycle. In HCC, there are epigenetic changes, for example, global DNA hypomethylation occurs, and hypermethylation in some important genes of the mitochondrial genome. These changes promote the transcription of mitochondrial dynamic-related genes and the disruption of genes related to OXPHOS. Moreover, it has been shown that miR-122 is down-regulated in HCC, which has been considered as a tumor suppressor and a potential diagnostic marker tool. In HUH7 cell line, MiR-33a/b, a lipid metabolism regulator is overexpressed; however, its role in HCC needs to be elucidated
The studies of the role of mitoepigenetics modifications and the metabolic processes in the pathogenesis of HCC could be a relevant advancement in the diagnosis and future therapy for this and other types of cancer.
Given the impact of mitochondrial biology and genome, the mitoepigenetics field offers a new opportunity to understand mitochondrial diseases and others that are not known as mitochondrial diseases. In addition, the mitochondrial epigenome also provides new clues for possible therapeutic targets and favors the appearance of new pharmacological options.
Declarations
Authors’ contributionsAll authors contributed actively in the search of useful information and participated in the writing of this review.
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by Consejo Nacional de Ciencia y Tecnología (240315) and Dirección General de Asuntos del Personal Académico-Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (DGAPA-PAPIIT) (IN208915).
Conflicts of interestAll authors declare that they have no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Cardin R, Piciocchi M, Bortolami M, Kotsafti A, Barzon L, Lavezzo E, Sinigaglia A, Rodriguez-Castro KI, Rugge M, Farinati F. Oxidative damage in the progression of chronic liver disease to hepatocellular carcinoma: an intricate pathway. World J Gastroenterol 2014;20:3078-86.
2. Tu T, Budzinska MA, Maczurek AE, Cheng R, Di Bartolomeo A, Warner FJ, McCaughan GW, McLennan SV, Shackel NA. Novel aspects of the liver microenvironment in hepatocellular carcinoma pathogenesis and development. Int J Mol Sci 2014;15:9422-58.
4. Bellizzi D, D'Aquila P, Giordano M, Montesanto A, Passarino G. Global DNA methylation levels are modulated by mitochondrial DNA variants. Epigenomics 2012;4:17-27.
5. Barbosa IA, Machado NG, Skildum AJ, Scott PM, Oliveira PJ. Mitochondrial remodeling in cancer metabolism and survival: potential for new therapies. Biochim Biophys Acta 2012;1826:238-54.
6. Hernandez-Munoz R, Chagoya de Sanchez V. In vivo correlation between liver and blood energy status as evidenced by chronic treatment of carbon tetrachloride and adenosine to rats. Can J Physiol Pharmacol 1994;72:1252-6.
7. Hernandez-Munoz R, Glender W, Diaz-Munoz M, Suarez J, Lozano J, Chagoya de Sanchez V. Alterations of ATP levels and of energy parameters in the blood of alcoholic and nonalcoholic patients with liver damage. Alcohol Clin Exp Res 1991;15:500-3.
8. Chavez E, Lozano-Rosas MG, Dominguez-Lopez M, Velasco-Loyden G, Rodriguez-Aguilera JR, Jose-Nunez C, Tuena de Gomez-Puyou M, Chagoya de Sanchez V. Functional, metabolic, and dynamic mitochondrial changes in the rat cirrhosis-hepatocellular carcinoma model and the protective effect of IFC-305. J Pharmacol Exp Ther 2017;361:292-302.
10. Singh KK, Russell J, Sigala B, Zhang Y, Williams J, Keshav KF. Mitochondrial DNA determines the cellular response to cancer therapeutic agents. Oncogene 1999;18:6641-6.
11. Modica-Napolitano JS, Kulawiec M, Singh KK. Mitochondria and human cancer. Curr Mol Med 2007;7:121-31.
12. Modica-Napolitano JS, Singh KK. Mitochondrial dysfunction in cancer. Mitochondrion 2004;4:755-62.
13. Penta JS, Johnson FM, Wachsman JT, Copeland WC. Mitochondrial DNA in human malignancy. Mutat Res 2001;488:119-33.
14. Graier WF, Frieden M, Malli R. Mitochondria and Ca(2+) signaling: old guests, new functions. Pflugers Arch 2007;455:375-96.
15. Kasahara A, Scorrano L. Mitochondria: from cell death executioners to regulators of cell differentiation. Trends Cell Biol 2014;24:761-70.
16. Mposhi A, Van der Wijst MG, Faber KN, Rots MG. Regulation of mitochondrial gene expression, the epigenetic enigma. Front Biosci (Landmark Ed) 2017;22:1099-113.
17. Campbell A, Mrazek J, Karlin S. Genome signature comparisons among prokaryote, plasmid, and mitochondrial DNA. Proc Natl Acad Sci U S A 1999;96:9184-9.
18. Singh KK. Mitochondrial dysfunction is a common phenotype in aging and cancer. Ann N Y Acad Sci 2004;1019:260-4.
20. Wallace DC, Fan W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion 2010;10:12-31.
21. Wellen KE, Thompson CB. A two-way street: reciprocal regulation of metabolism and signalling. Nat Rev Mol Cell Biol 2012;13:270-6.
22. Lu SC, Mato JM. S-adenosylmethionine in liver health, injury, and cancer. Physiol Rev 2012;92:1515-42.
23. Agrimi G, Di Noia MA, Marobbio CM, Fiermonte G, Lasorsa FM, Palmieri F. Identification of the human mitochondrial S-adenosylmethionine transporter: bacterial expression, reconstitution, functional characterization and tissue distribution. Biochem J 2004;379:183-90.
24. Depeint F, Bruce WR, Shangari N, Mehta R, O'Brien PJ. Mitochondrial function and toxicity: role of B vitamins on the one-carbon transfer pathways. Chem Biol Interact 2006;163:113-32.
26. Chagoya de Sanchez V, Hernandez-Munoz R, Sanchez L, Vidrio S, Yanez L, Suarez J. Twenty-four-hour changes of S-adenosylmethionine, S-adenosylhomocysteine adenosine and their metabolizing enzymes in rat liver; possible physiological significance in phospholipid methylation. Int J Biochem 1991;23:1439-43.
27. van der Wijst MG, van Tilburg AY, Ruiters MH, Rots MG. Experimental mitochondria-targeted DNA methylation identifies GpC methylation, not CpG methylation, as potential regulator of mitochondrial gene expression. Sci Rep 2017;7:177.
28. Karpf AR, Matsui S. Genetic disruption of cytosine DNA methyltransferase enzymes induces chromosomal instability in human cancer cells. Cancer Res 2005;65:8635-9.
31. Manev H, Dzitoyeva S, Chen H. Mitochondrial DNA: a blind spot in neuroepigenetics. Biomol Concepts 2012;3:107-15.
32. Ferreira A, Serafim TL, Sardao VA, Cunha-Oliveira T. Role of mtDNA-related mitoepigenetic phenomena in cancer. Eur J Clin Invest 2015;45 Suppl 1:44-9.
33. Shock LS, Thakkar PV, Peterson EJ, Moran RG, Taylor SM. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc Natl Acad Sci U S A 2011;108:3630-5.
34. Rebelo AP, Williams SL, Moraes CT. In vivo methylation of mtDNA reveals the dynamics of protein-mtDNA interactions. Nucleic Acids Res 2009;37:6701-15.
35. Bellizzi D, D'Aquila P, Scafone T, Giordano M, Riso V, Riccio A, Passarino G. The control region of mitochondrial DNA shows an unusual CpG and non-CpG methylation pattern. DNA Res 2013;20:537-47.
36. Ghosh S, Sengupta S, Scaria V. Comparative analysis of human mitochondrial methylomes shows distinct patterns of epigenetic regulation in mitochondria. Mitochondrion 2014;18:58-62.
37. Metodiev MD, Lesko N, Park CB, Camara Y, Shi Y, Wibom R, Hultenby K, Gustafsson CM, Larsson NG. Methylation of 12S rRNA is necessary for in vivo stability of the small subunit of the mammalian mitochondrial ribosome. Cell Metab 2009;9:386-97.
38. Giordano M, Cristiani C, Crocco P, D'Aquila P, De Rango F, Pisani F, Scafone T, Tallaro F, Rose G, Passarino G, Beliizi D. Methylation of the human mitochondrial 12s rRNA gene is correlated with aging. Proceedings of 12th International Congress FISV Congress. Rome, Italy; 2012 Sep 24-27 .
39. Liu B, Du Q, Chen L, Fu G, Li S, Fu L, Zhang X, Ma C, Bin C. CpG methylation patterns of human mitochondrial DNA. Sci Rep 2016;6:23421.
40. Salminen A, Kauppinen A, Hiltunen M, Kaarniranta K. Krebs cycle intermediates regulate DNA and histone methylation: epigenetic impact on the aging process. Ageing Res Rev 2014;16:45-65.
41. Ghosh S, Sengupta S, Scaria V. Hydroxymethyl cytosine marks in the human mitochondrial genome are dynamic in nature. Mitochondrion 2016;27:25-31.
42. Iacobazzi V, Castegna A, Infantino V, Andria G. Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol Genet Metab 2013;110:25-34.
43. Tomasetti M, Santarelli L, Neuzil J, Dong L. MicroRNA regulation of cancer metabolism: role in tumour suppression. Mitochondrion 2014;19 Pt A:29-38.
44. Ameres SL, Zamore PD. Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol 2013;14:475-88.
45. Bienertova-Vasku J, Sana J, Slaby O. The role of microRNAs in mitochondria in cancer. Cancer Lett 2013;336:1-7.
46. Giuliani A, Prattichizzo F, Micolucci L, Ceriello A, Procopio AD, Rippo MR. Mitochondrial (Dys) function in inflammaging: do mitomiRs influence the energetic, oxidative, and inflammatory status of senescent cells? Mediators Inflamm 2017;2017:2309034.
47. Das S, Ferlito M, Kent OA, Fox-Talbot K, Wang R, Liu D, Raghavachari N, Yang Y, Wheelan SJ, Murphy E, Steenbergen C. Nuclear miRNA regulates the mitochondrial genome in the heart. Circ Res 2012;110:1596-603.
48. Wang JX, Jiao JQ, Li Q, Long B, Wang K, Liu JP, Li YR, Li PF. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat Med 2011;17:71-8.
49. Finley LW, Haigis MC. The coordination of nuclear and mitochondrial communication during aging and calorie restriction. Ageing Res Rev 2009;8:173-88.
50. Boland ML, Chourasia AH, Macleod KF. Mitochondrial dysfunction in cancer. Front Oncol 2013;3:292.
51. Chen J ZB, Wong N, Lo AW, To KF, Chan AW, Ng MH, Ho CY, Cheng SH, Lai PB, Yu J, Ng HK, Ling MT, Huang AL, Cai XF, Ko BC. Sirtuin 1 is upregulated in a subset of hepatocellular carcinomas where it is essential for telomere maintenance and tumor cell growth. Cancer Res 2012;72:4138-49.
52. Barres R, Osler ME, Yan J, Rune A, Fritz T, Caidahl K, Krook A, Zierath JR. Non-CpG methylation of the PGC-1alpha promoter through DNMT3B controls mitochondrial density. Cell Metab 2009;10:189-98.
53. Scarpulla RC. Nuclear control of respiratory gene expression in mammalian cells. J Cell Biochem 2006;97:673-83.
54. Hanaoka S, Ishida K, Tanaka S, Sakamoto S, Okuda K, Sanoh S, Ohta S, Kotake Y. Tributyltin induces epigenetic changes and decreases the expression of nuclear respiratory factor-1. Metallomics 2018;10:337-45.
56. Smiraglia DJ, Kulawiec M, Bistulfi GL, Gupta SG, Singh KK. A novel role for mitochondria in regulating epigenetic modification in the nucleus. Cancer Biol Ther 2008;7:1182-90.
57. Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 2005;39:359-407.
58. Chen C, Chen Y, Guan MX. A peep into mitochondrial disorder: multifaceted from mitochondrial DNA mutations to nuclear gene modulation. Protein Cell 2015;6:862-70.
59. Cassim S, Raymond VA, Dehbidi-Assadzadeh L, Lapierre P, Bilodeau M. Metabolic reprogramming enables hepatocarcinoma cells to efficiently adapt and survive to a nutrient-restricted microenvironment. Cell Cycle 2018; doi: 10.1080/15384101.2018.1460023.
60. Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, Walters H, Tantawy MN, Fu A, Manning HC, Horton JD, Hammer RE, McKnight SL, Tu BP. Acetate dependence of tumors. Cell 2014;159:1591-602.
61. Mato JM, Lu SC. Role of S-adenosyl-L-methionine in liver health and injury. Hepatology 2007;45:1306-12.
62. Mato JM, Martinez-Chantar ML, Lu SC. S-adenosylmethionine metabolism and liver disease. Ann Hepatol 2013;12:183-9.
63. Tohme S, Yazdani HO, Liu Y, Loughran P, van der Windt DJ, Huang H, Simmons RL, Shiva S, Tai S, Tsung A. Hypoxia mediates mitochondrial biogenesis in hepatocellular carcinoma to promote tumor growth through HMGB1 and TLR9 interaction. Hepatology 2017;66:182-97.
64. Zhang C, Li J, Huang T, Duan S, Dai D, Jiang D, Sui X, Li D, Chen Y, Ding F, Huang C, Chen G, Wang K. Meta-analysis of DNA methylation biomarkers in hepatocellular carcinoma. Oncotarget 2016;7:81255-67.
65. Malik AN, Czajka A. Is mitochondrial DNA content a potential biomarker of mitochondrial dysfunction? Mitochondrion 2013;13:481-92.
66. Singh KK, Kulawiec M, Still I, Desouki MM, Geradts J, Matsui S. Inter-genomic cross talk between mitochondria and the nucleus plays an important role in tumorigenesis. Gene 2005;354:140-6.
67. Ye C, Tao R, Cao Q, Zhu D, Wang Y, Wang J, Lu J, Chen E, Li L. Whole-genome DNA methylation and hydroxymethylation profiling for HBV-related hepatocellular carcinoma. Int J Oncol 2016;49:589-602.
68. Wu YL, Wang D, Peng XE, Chen YL, Zheng DL, Chen WN, Lin X. Epigenetic silencing of NAD(P)H:quinone oxidoreductase 1 by hepatitis B virus X protein increases mitochondrial injury and cellular susceptibility to oxidative stress in hepatoma cells. Free Radic Biol Med 2013;65:632-44.
69. Mizukami S, Yafune A, Watanabe Y, Nakajima K, Jin M, Yoshida T, Shibutani M. Identification of epigenetically downregulated Tmem70 and Ube2e2 in rat liver after 28-day treatment with hepatocarcinogenic thioacetamide showing gene product downregulation in hepatocellular preneoplastic and neoplastic lesions produced by tumor promotion. Toxicol Lett 2017;266:13-22.
70. Hejzlarova K, Tesarova M, Vrbacka-Cizkova A, Vrbacky M, Hartmannova H, Kaplanova V, Noskova L, Kratochvilova H, Buzkova J, Havlickova V, Zeman J, Kmoch S, Houstek J. Expression and processing of the TMEM70 protein. Biochim Biophys Acta 2011;1807:144-9.
71. Havlickova Karbanova V, Cizkova Vrbacka A, Hejzlarova K, Nuskova H, Stranecky V, Potocka A, Kmoch S, Houstek J. Compensatory upregulation of respiratory chain complexes III and IV in isolated deficiency of ATP synthase due to TMEM70 mutation. Biochim Biophys Acta 2012;1817:1037-43.
72. Shah ZH, Migliosi V, Miller SC, Wang A, Friedman TB, Jacobs HT. Chromosomal locations of three human nuclear genes (RPSM12, TUFM, and AFG3L1) specifying putative components of the mitochondrial gene expression apparatus. Genomics 1998;48:384-8.
73. Jin D, Li R, Mao D, Luo N, Wang Y, Chen S, Zhang S. Mitochondria-localized glutamic acid-rich protein (MGARP) gene transcription is regulated by Sp1. PLoS One 2012;7:e50053.
74. Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem 2007;76:75-100.
75. Bannister AJ, Schneider R, Kouzarides T. Histone methylation: dynamic or static? Cell 2002;109:801-6.
76. Minocherhomji S, Tollefsbol TO, Singh KK. Mitochondrial regulation of epigenetics and its role in human diseases. Epigenetics 2012;7:326-34.
77. Hayami S, Kelly JD, Cho HS, Yoshimatsu M, Unoki M, Tsunoda T, Field HI, Neal DE, Yamaue H, Ponder BA, Nakamura Y, Hamamoto R. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int J Cancer 2011;128:574-86.
78. Sakamoto A, Hino S, Nagaoka K, Anan K, Takase R, Matsumori H, Ojima H, Kanai Y, Arita K, Nakao M. Lysine demethylase LSD1 coordinates glycolytic and mitochondrial metabolism in hepatocellular carcinoma cells. Cancer Res 2015;75:1445-56.
79. Gailhouste L, Gomez-Santos L, Ochiya T. Potential applications of miRNAs as diagnostic and prognostic markers in liver cancer. Front Biosci (Landmark Ed) 2013;18:199-223.
80. Burchard J, Zhang C, Liu AM, Poon RT, Lee NP, Wong KF, Sham PC, Lam BY, Ferguson MD, Tokiwa G, Smith R, Leeson B, Beard R, Lamb JR, Lim L, Mao M, Dai H, Luk JM. microRNA-122 as a regulator of mitochondrial metabolic gene network in hepatocellular carcinoma. Mol Syst Biol 2010;6:402.
81. Bandiera S, Pfeffer S, Baumert TF, Zeisel MB. miR-122--a key factor and therapeutic target in liver disease. J Hepatol 2015;62:448-57.
82. Tsai WC, Hsu SD, Hsu CS, Lai TC, Chen SJ, Shen R, Huang Y, Chen HC, Lee CH, Tsai TF, Hsu MT, Wu JC, Huang HD, Shiao MS, Hsiao M, Tsou AP. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J Clin Invest 2012;122:2884-97.
83. Rayner KJ, Esau CC, Hussain FN, McDaniel AL, Marshall SM, van Gils JM, Ray TD, Sheedy FJ, Goedeke L, Liu X, Khatsenko OG, Kaimal V, Lees CJ, Fernandez-Hernando C, Fisher EA, Temel RE, Moore KJ. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature 2011;478:404-7.
84. Davalos A, Goedeke L, Smibert P, Ramirez CM, Warrier NP, Andreo U, Cirera-Salinas D, Rayner K, Suresh U, Pastor-Pareja JC, Esplugues E, Fisher EA, Penalva LO, Moore KJ, Suarez Y, Lai EC, Fernandez-Hernando C. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc Natl Acad Sci U S A 2011;108:9232-7.
85. Qiao L, Ru G, Mao Z, Wang C, Nie Z, Li Q, Huang-Yang Y, Zhu L, Liang X, Yu J, Jiang P. Mitochondrial DNA depletion, mitochondrial mutations and high TFAM expression in hepatocellular carcinoma. Oncotarget 2017;8:84373-83.
86. Machida K. Existence of cancer stem cells in hepatocellular carcinoma: myth or reality? Hepatol Int 2017;11:143-7.
87. Chen CL, Uthaya Kumar DB, Punj V, Xu J, Sher L, Tahara SM, Hess S, Machida K. NANOG metabolically reprograms tumor-initiating stem-like cells through tumorigenic changes in oxidative phosphorylation and fatty acid metabolism. Cell Metab 2016;23:206-19.
88. Machida K. Pluripotency transcription factors and metabolic reprogramming of mitochondria in tumor-initiating stem-like cells. Antioxid Redox Signal 2017; doi: 10.1089/ars.2017.7241.
89. Lee WT, St John J. The control of mitochondrial DNA replication during development and tumorigenesis. Ann N Y Acad Sci 2015;1350:95-106.
90. Yamada S, Nomoto S, Fujii T, Kaneko T, Takeda S, Inoue S, Kanazumi N, Nakao A. Correlation between copy number of mitochondrial DNA and clinico-pathologic parameters of hepatocellular carcinoma. Eur J Surg Oncol 2006;32:303-7.
91. Shen F, Huang W, Qi JH, Yuan BF, Huang JT, Zhou X, Feng YQ, Liu YJ, Liu SM. Association of 5-methylcytosine and 5-hydroxymethylcytosine with mitochondrial DNA content and clinical and biochemical parameters in hepatocellular carcinoma. PLoS One 2013;8:e76967.
92. Xie CH, Naito A, Mizumachi T, Evans TT, Douglas MG, Cooney CA, Fan CY, Higuchi M. Mitochondrial regulation of cancer associated nuclear DNA methylation. Biochem Biophys Res Commun 2007;364:656-61.
93. Boix L, Lopez-Oliva JM, Rhodes AC, Bruix J. Restoring miR122 in human stem-like hepatocarcinoma cells, prompts tumor dormancy through Smad-independent TGF-beta pathway. Oncotarget 2016;7:71309-29.
94. Omata M, Lesmana LA, Tateishi R, Chen PJ, Lin SM, Yoshida H, Kudo M, Lee JM, Choi BI, Poon RT, Shiina S, Cheng AL, Jia JD, Obi S, Han KH, Jafri W, Chow P, Lim SG, Chawla YK, Budihusodo U, Gani RA, Lesmana CR, Putranto TA, Liaw YF, Sarin SK. Asian Pacific Association for the Study of the Liver consensus recommendations on hepatocellular carcinoma. Hepatol Int 2010;4:439-74.
95. Lou J, Zhang L, Lv S, Zhang C, Jiang S. Biomarkers for hepatocellular carcinoma. Biomark Cancer 2017;9:1-9.
96. Berridge MV, Dong L, Neuzil J. Mitochondrial DNA in tumor initiation, progression, and metastasis: role of horizontal mtDNA transfer. Cancer Res 2015;75:3203-8.
97. Pasquier J, Guerrouahen BS, Al Thawadi H, Ghiabi P, Maleki M, Abu-Kaoud N, Jacob A, Mirshahi M, Galas L, Rafii S, Le Foll F, Rafii A. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J Transl Med 2013;11:94.
98. Chatterjee A, Dasgupta S, Sidransky D. Mitochondrial subversion in cancer. Cancer Prev Res (Phila) 2011;4:638-54.
99. Chu MP, Kriangkum J, Venner CP, Sandhu I, Hewitt J, Belch AR, Pilarski LM. Addressing heterogeneity of individual blood cancers: the need for single cell analysis. Cell Biol Toxicol 2017;33:83-97.
100. Wang W, Gao D, Wang X. Can single-cell RNA sequencing crack the mystery of cells? Cell Biol Toxicol 2018;34:1-6.
101. Wang W, Wang X. Single-cell CRISPR screening in drug resistance. Cell Biol Toxicol 2017;33:207-10.
102. Qian M, Spada C, Wang X. Approach, application, and bioethics of mtDNA sequencing in cancer. Adv Exp Med Biol 2017;1038:23-38.
103. Kutay H, Bai S, Datta J, Motiwala T, Pogribny I, Frankel W, Jacob ST, Ghoshal K. Downregulation of miR-122 in the rodent and human hepatocellular carcinomas. J Cell Biochem 2006;99:671-8.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Lozano-Rosas MG, Chávez E, Aparicio-Cadena AR, Velasco-Loyden G, Sánchez VC. Mitoepigenetics and hepatocellular carcinoma. Hepatoma Res 2018;4:19. http://dx.doi.org/10.20517/2394-5079.2018.48
AMA Style
Lozano-Rosas MG, Chávez E, Aparicio-Cadena AR, Velasco-Loyden G, Sánchez VC. Mitoepigenetics and hepatocellular carcinoma. Hepatoma Research. 2018; 4: 19. http://dx.doi.org/10.20517/2394-5079.2018.48
Chicago/Turabian Style
Lozano-Rosas, María Guadalupe, Enrique Chávez, Alejandro Rusbel Aparicio-Cadena, Gabriela Velasco-Loyden, Victoria Chagoya de Sánchez. 2018. "Mitoepigenetics and hepatocellular carcinoma" Hepatoma Research. 4: 19. http://dx.doi.org/10.20517/2394-5079.2018.48
ACS Style
Lozano-Rosas, MG.; Chávez E.; Aparicio-Cadena AR.; Velasco-Loyden G.; Sánchez VC. Mitoepigenetics and hepatocellular carcinoma. Hepatoma. Res. 2018, 4, 19. http://dx.doi.org/10.20517/2394-5079.2018.48
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 11 clicks
Cite This Article 11 clicks

 Like This Article 31
likes
Like This Article 31
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.