
Comparison of p53 and prohibitin expression in the spectrum of hepatitis, cirrhosis, and hepatocellular carcinoma


Abstract
Aim: To investigate the correlation of p53 and prohibitin (PHB) expression in the spectrum of hepatitis, cirrhosis, and human hepatocellular carcinoma (HCC).
Methods: Hepatic biopsies from patients with HCC (n = 60), cirrhosis (CIR, n = 30), hepatitis C virus (HCV, n = 30), and normal livers (NL, n = 20) were examined for immunohistochemical expression of RelA/p65, tumor necrosis factor receptor-1 (TNFR1), TNF-related apoptosis-inducing ligand (TRAIL), p53, and PHB. The samples were also analysed by nuclear factor kappa B (NFκB) Southwestern histochemistry and a transferase-mediated dUTP-biotin nick-end labelling assay.
Results: Expression of NFκB and RelA/p65 was detected increasingly from NL to CIR, but had a diminished labelling in the HCC cases (P < 0.05). Expression levels of TNFR1 and TRAIL followed the same pattern (P < 0.05). Apoptosis was increased in HCC, but was progressively reduced from CIR to NL (P < 0.05). p53 and PHB nuclear expressions were amplified in cases of HCC, but diminished in NL, HCV, and CIR (P < 0.05).
Conclusion: These results suggest that in addition to well-understood sequences of proinflammatory events such as TNF-induced NFκB activation and NFκB/TRAIL pathway-mediated apoptosis, development of HCC is also influenced by regulation of p53 and PHB tumour suppressor function. Additional studies are necessary to explain the contradictory mechanisms of the tumour microenvironment observed in the sequence of HCV, CIR, and HCC.
Keywords
Introduction
Hepatocellular carcinoma (HCC), the most frequent primary hepatic cancer, is the third highest cause of cancer-related death worldwide.[1] Accumulation of genetic and epigenetic alterations results in the development of HCC.[2] Therefore, the molecular pathways involved in hepatic cancer have been the focus of numerous studies.
Activation of the transcriptional factor nuclear factor kappa B (NFκB), an important modulator of inflammatory and cell survival responses,[3] has been associated with hepatic carcinogenesis.[4] NFκB may be activated in hepatocytes as a consequence of chronic inflammation, which occurs in viral hepatitis.[5] However, little is known about the interaction of NFκB activation and tumour microenvironment in the sequence of hepatitis C virus (HCV), cirrhosis (CIR), and HCC.
The inflammatory cytokine tumor necrosis factor-α (TNFα) participates in the control of cellular proliferation and differentiation and cell death.[6] Although TNFα was initially identified as inducing cell death in some tumours, an association between activation of the TNFα/NFκB pathway by inflammation and hepatocarcinogenesis has also been reported.[7] Binding of TNFα to TNF receptor-1 (TNFR1) results in NFκB activation and may induce hepatocyte survival and proliferation.[8] Additionally, activated NFκB can be considered as a protector of TNFα-induced apoptosis.[9]
TNF-related apoptosis-inducing ligand (TRAIL) has been demonstrated as a proapoptotic mediator of various tumour cells.[10] Similarly, TRAIL caused cytotoxic effects in transformed hepatocytes of HCC, perhaps related to inhibition of NFκB survival signalling.[11] In contrast, participation of TRAIL in hepatocyte’s apoptosis during chronic hepatic diseases remains controversial.
The p53 gene is a classical tumor suppressor gene. p53 mutations occur in diverse human cancers, including HCC. p53 tumor suppressor function involves cell cycle control, transcriptional regulation, and apoptosis.[2] In spite of the p53 mutation being rare in HCC not induced by aflatoxin, the high incidence of HCV-related HCC justifies novel studies even in this context. NFκB linked to HCV non-structural 5A (NS5A) protein inhibits the p53 tumor suppressor role and leads to cell survival and hepatocarcinogenesis.[12]
Human prohibitin (PHB), a pleiotropic protein, was identified first as a potential tumour suppressor[13] and has been implicated in cellular differentiation, antiproliferation, and morphogenesis.[14] Although a liver-specific deletion of PHB has been observed in a wide spectrum of liver injury types, fibrosis, and hepatocarcinogenesis in mice,[15] PHB has also been reported to be overexpressed in cases of human HCV.[16] On the other handit has been suggested that PHB may have a pivotal role in cellular proliferation and malignant transformation.[17] Thus, the role of PHB in development of human hepatic cancer remains controversial.
Our study aimed to compare the importance of the p53 and PHB pathway through the NFκB signalling in the spectrum of HCV, CIR, and HCC, considering the potential differences in the pathogenesis of human hepatic cancer.
Methods
Tissue specimens
Primary liver carcinoma samples were obtained from the Department of Pathology at the University Hospital of the Faculty of Medicine of Ribeirão Preto, University of São Paulo, Ribeirão Preto, São Paulo, Brazil. Twenty samples of normal liver from autopsies with causes other than liver disease (NL), 30 cases of HCV, 30 cases of CIR associated with HCV and 60 cases of HCC related to HCV-CIR were selected for this study [Table 1]. Patients with evidence of any other cause of liver disease were excluded. The study was approved by the local Ethics Committee (number 1611/2011).
Clinicopathologic variables of NL, CIR, HCV, and HCC
| Clinicopathologic variables | Etiology (% cases) | P value | |||
|---|---|---|---|---|---|
| NL (n = 20) | HCV (n = 30) | CIR (n = 30) | HCC (n = 60) | ||
| Age (years) | 0.5530 | ||||
| ≤ 50 | 0 | 40 | 20 | 35 | |
| > 50 | 100 | 60 | 80 | 65 | |
| Gender | 0.4116 | ||||
| Male | 60 | 80 | 70 | 80 | |
| Female | 40 | 20 | 30 | 20 | |
| Cirrhosis | 0.0001 | ||||
| Absence | 100 | 100 | 0 | 3 | |
| Presence | 0 | 0 | 100 | 97 | |
| Serum AFP (μg/L) | 0.0001 | ||||
| ≤ 20 | 100 | 100 | 100 | 40 | |
| > 20 | 0 | 0 | 0 | 60 | |
Immunohistochemistry
Liver preparations were submitted for immuno-histochemical analysis. Sections were incubated with monoclonal primary antibodies specific for TNFR1, TRAIL, RelA/p65, and p53 (Santa Cruz Biotechnology, Santa Cruz, CA, USA, dilution 1:100) and PHB (Thermo Fisher Scientific Waltham, MA, USA, dilution 1:100). Following this, a secondary antibody (Vectastain Elite ABC Kit, Universal, Vector Laboratories Inc.) was applied. Next, the slides were incubated with avidin-biotin-peroxidase complex (Vectastain Elite ABC Kit) and developed with Vector NovaRED kit (Vector Laboratories Inc.) for 5 min. The slides were counterstained with Harris haematoxylin and mounted with Permount (Biomeda, Foster City, CA, USA). Percentages of nuclear RelA/p65 and p53, cytoplasmic TNFR1 and TRAIL, and nuclear/cytoplasmic PHB-positive cells were obtained blindly for each case at least 10 representative high-power fields (40×) by two of the authors (LDP and LNR). For statistical purposes, the samples were scored as follows: -, no stained cells; +, weak or moderate staining in less than 25% positive cells; ++, moderate to strong staining in 25-50% positive cells; and +++, strong staining in more than 50% positive cells.[18]
Southwestern histochemistry analysis
Non-radioactive in situ detection of NFκB in paraffin-embedded liver tissue preparations was performed using the Southwestern histochemistry method, with digoxigenin labelling and detection kits (Roche Applied Science, Indianapolis, USA). Briefly, synthetic sense DNAs (Imprint Genetics Corporation, Hialeah, USA), which contain sequences of NFκB, were used as probes. After annealing with the complementary sequence, the DNA probe was labelled with digoxigenin. The percentage of NFκB-positive cells was obtained blindly for each case, at least 10 representative high-power fields (40×) by two of the authors (LDP and LNR). For statistical purposes, the samples were scored similarly as was presented in the immunohistochemistry section.[18]
TUNEL assay
Paraffin-embedded liver tissue sections were deparaffinised and incubated with 20 µg/mL proteinase K (Promega Corporation, Madison, USA). A DeadEnd peroxidase in situ apoptosis detection kit (DeadEnd TUNEL; Promega Corporation) was used for transferase-mediated dUTP-biotin nick-end labelling (TUNEL) staining. The percentage of TUNEL-positive cells was obtained blindly, for each case at least 10 representative high-power fields (40×) by two of the authors (LDP and LNR). For statistical purposes, the samples were scored similarly as was presented in the immunohistochemistry section.[18]
Statistical evaluation
Statistical analysis was performed using GraphPad Prism v4.0 software (GraphPad Software, Inc., San Diego, CA). Association between the expression of TNFR1, TRAIL, NFκB, RelA/p65, p53, PHB, and TUNEL and histologic variables was determined by Fisher’s exact test (2 groups) or a chi-square test (3 or more groups). One-way analysis of variance followed by Dunn’s post-test was also performed. All tests were two-tailed, and P < 0.05 was considered significant.
Results
Study tissue specimens
The most relevant data concerning clinic and pathological variables was the occurrence of cirrhosis or viral hepatitis in almost all HCC cases, as well as absence of cirrhosis or other inflammatory conditions in the normal liver (P = 0.0001). Additionally, the level serum α-fetoprotein was > 20 μg/L in the majority of HCC cases, but ≤ 20 μg/L in the other types (P = 0.0001) [Table 1].
Expression and clinicopathological features
Table 2 summarizes the comparative TNFR1, TRAIL, NFκB, nuclear RelA/p65, p53, nuclear PHB and cytoplasmic PHB and TUNEL expression by HCC, CIR, HCV, and NL.
Comparative TNFR1, TRAIL, NFkB, nuclear RelA/p65, p53, nuclear PHB, cytoplasmic PHB and TUNEL expression by NL, CIR, HCV, and HCC
| Markers | Expression | NL | HCV | CIR | HCC | P < 0.05 |
|---|---|---|---|---|---|---|
| (n = 20) | (n = 30) | (n = 30) | (n = 60) | |||
| TNFR1 | Negative | 18 | 3 | 3 | 32 | CIR vs. HCC CIR vs. HCV HCV vs. NL |
| + | 2 | 24 | 9 | 13 | ||
| ++ | 0 | 3 | 15 | 12 | ||
| +++ | 0 | 0 | 3 | 3 | ||
| TRAIL | Negative | 18 | 12 | 9 | 14 | HCC vs. CIR HCC vs. HCV HCV vs. NL |
| + | 2 | 18 | 21 | 18 | ||
| ++ | 0 | 0 | 0 | 18 | ||
| +++ | 0 | 0 | 0 | 10 | ||
| RelA/p65 | Negative | 16 | 3 | 0 | 21 | CIR vs. HCC CIR vs. HCV HCV vs. NL |
| + | 4 | 18 | 9 | 19 | ||
| ++ | 0 | 9 | 15 | 13 | ||
| +++ | 0 | 0 | 6 | 7 | ||
| NFκB | Negative | 18 | 0 | 3 | 26 | CIR vs. HCC CIR vs. HCV HCV vs. NL |
| + | 2 | 6 | 18 | 22 | ||
| ++ | 0 | 15 | 6 | 10 | ||
| +++ | 0 | 9 | 3 | 2 | ||
| p53 | Negative | 17 | 27 | 27 | 20 | HCC vs. CIR HCC vs. HCV HCC vs. NL |
| + | 3 | 3 | 3 | 26 | ||
| ++ | 0 | 0 | 0 | 12 | ||
| +++ | 0 | 0 | 0 | 2 | ||
| Cytoplasmic PHB | Negative | 16 | 3 | 3 | 35 | CIR vs. HCC CIR vs. HCV HCV vs. NL |
| + | 4 | 5 | 7 | 23 | ||
| ++ | 0 | 10 | 11 | 2 | ||
| +++ | 0 | 12 | 9 | 0 | ||
| Nuclear PHB | Negative | 18 | 27 | 24 | 19 | HCC vs. CIR HCC vs. HCV HCC vs. NL |
| + | 2 | 3 | 6 | 24 | ||
| ++ | 0 | 0 | 0 | 13 | ||
| +++ | 0 | 0 | 0 | 4 | ||
| TUNEL | Negative | 17 | 3 | 2 | 4 | HCC vs. CIR HCC vs. HCV HCC vs. NL |
| + | 3 | 16 | 18 | 15 | ||
| ++ | 0 | 11 | 10 | 32 | ||
| +++ | 0 | 0 | 0 | 9 |
TNFR1 expression was higher in CIR [Figure 1C] in comparison to HCC [Figure 1D] and HCV [Figure 1B] (P = 0.0294 and P = 0.0037, respectively), as well as in HCV compared to NL [Figure 1A] (P = 0.0313) [Figure 1Q]. TRAIL expression was amplified in HCC [Figure 1H] compared to CIR [Figure 1G] and HCV [Figure 1F] (P = 0.0377 and P = 0.0371, respectively), as well as in HCV in relation to NL [Figure 1E] (P = 0.0462) [Figure 1R].
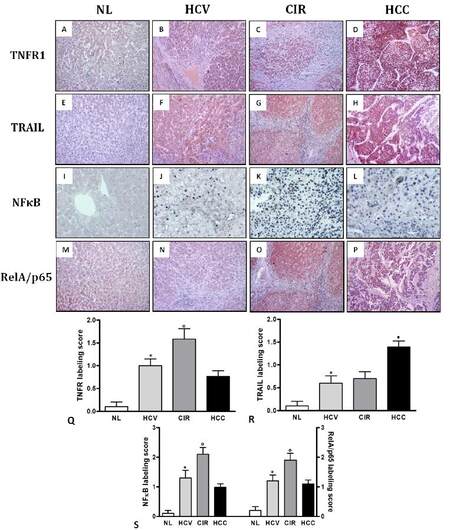
Figure 1. Comparative expression of TNFR1 (immunohistochemistry, ×200), TRAIL (immunohistochemistry, ×200), NFkB (Southwestern histochemistry, ×200) and RelA/p65 (immunohistochemistry, ×200) by NL, HCV, CIR and HCC. A: low expression of TNFR1 in NL; B: increased expression of TNFR1 in HCV; C: high expression of TNFR1 in CIR; D: strong expression of TNFR1 in HCC; E: minimal expression of TRAIL in NL; F: increased expression of TRAIL in HCV; G: augmented expression of TRAIL in CIR; H: high expression of TRAIL in HCC; I: low expression of NFkB in NL; J: high expression of NFkB in HCV; K: marked expression of NFkB in CIR; L: increased expression of NFkB in HCC; M: minor expression of RelA/p65 in NL; N: increased expression of RelA/p65 in HCV; O: evident expression of RelA/p65 in CIR; P: high expression of RelA/p65 in HCC; (Q, R, S) comparison of the TNFR1 (Q), TRAIL (R), NFkB and RelA/p65 (S) labeling scores by NL, HCV, CIR and HCC. *P < 0.05 vs. NL; 0P < 0.05 vs. HCV; •P < 0.05 vs. CIR. TNFR1: tumor necrosis factor receptor-1; TRAIL: TNF-related apoptosis-inducing ligand; NFkB: nuclear factor kappa B; NL: normal liver; CIR: cirrhosis; HCV: hepatitis C virus; HCC: hepatocellular carcinoma
NFκB expression was increased in CIR [Figure 1K] in comparison to HCC [Figure 1L] (P = 0.0464). NFκB expression was also higher in CIR compared to HCV [Figure 1J] (P = 0.0031), as well as in HCV [Figure 1D] in relation to NL [Figure 1I] (P = 0.0477) [Figure 1S]. In a pattern similar to that of NFκB, nuclear RelA/p65 expression was increased in CIR [Figure 1O] compared to HCC [Figure 1P] and HCV [Figure 1N] (P = 0.0228 and P = 0.0426, respectively), as well as in HCV relative to NL [Figure 1M] (P = 0.0288) [Figure 1S].
Since p53 expression was almost solely found in HCC [Figure 2D], this group presented a higher p53 expression relative to NL [Figure 2A], HCV [Figure 2B], and CIR [Figure 2C] (P = 0.011, P = 0.014 and P = 0.0013, respectively). However, no difference in p53 expression was observed between CIR and HCV (P = 0.9421), as well as in HCV in relation to NL (P = 0.9421) [Figure 2M].
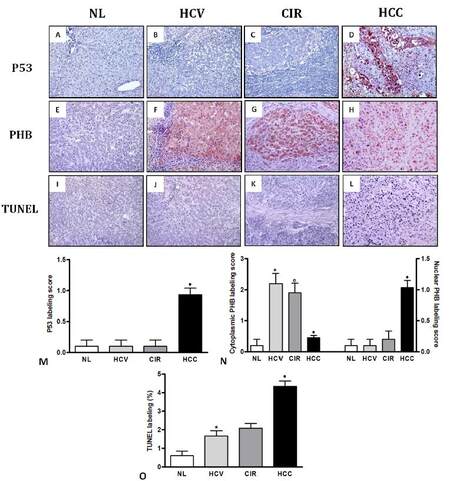
Figure 2. Comparative expression of p53 (immunohistochemistry, ×200), PHB (immunohistochemistry, ×400) and TUNEL (immunoassay, ×200) by NL, HCV, CIR and HCC. A: absence of p53 expression in NL; B: low expression of p53 in HCV; C: minor expression of p53 in CIR; D: high nuclear and cytoplasmic expression of p53 in HCC; E: slight cytoplasmic expression of PHB in NL; F: augmented cytoplasmic expression of PHB in HCV; G: high cytoplasmic expression of PHB in CIR; H: strong nuclear and cytoplasmic expression of PHB in HCC; I: slight expression of apoptosis in NL; J: increased expression of apoptosis in HCV; K: augmented expression of apoptosis in CIR; L: marked expression of apoptosis in HCC; (M, N, O) comparison of the p53 (M), cytoplasmic and nuclear PHB (N) and TUNEL (O) labeling scores by NL, HCV, CIR and HCC. *P < 0.05 vs. NL; 0P < 0.05 vs. HCV; •P < 0.05 vs. CIR. PHB: prohibitin; TUNEL: transferase-mediated dUTP-biotin nick-end labelling; NL: normal liver; CIR: cirrhosis; HCV: hepatitis C virus; HCC: hepatocellular carcinoma
Cytoplasmic PHB expression was augmented in CIR [Figure 2G] in relation to HCC [Figure 2H] (P = 0.0001), whereas PHB cytoplasmic expression was similar between CIR and HCV [Figure 2F] (P = 0.4468). PHB cytoplasmic expression was increased in HCV, CIR, and HCC in comparison to NL [Figure 2E] (P = 0.0088). Because PHB nuclear expression was almost exclusively observed in HCC, this group presented a higher PHB nuclear expression in contrast to CIR, HCV, and NL (P = 0.0041, P = 0.0011 and P = 0.0011, respectively) [Figure 2N].
The percentage of apoptosis was increased in HCC [Figure 2L] in contrast to CIR [Figure 2K] and HCV [Figure 2J] (P = 0.0054 and P = 0.0217, respectively). Moreover, the percentage of apoptosis was increased in HCV in relation to NL [Figure 2I] (P = 0.0161) [Figure 2O].
Discussion
The findings of the present study reveal the importance of p53 and PHB participation in human hepatic cancer development, which is dependent on different pathogenic mechanisms of the tumour microenvironment.
TNFα cytokine is increased in patients with chronic HCV infection.[19] TNFα and TNF-receptors levels are augmented during chronic HCV infection and result in disease progression.[20] Similarly, we found a progressive increase of TNFR1 expression in cases of HCV and CIR in comparison to NL. Indeed, TNFα proinflammatory stimuli can be responsible for NFκB activation, which results in protection of hepatocytes from apoptosis.[9] Moreover, the HCV core protein potentiates NFκB activation and chronically activated NFκB leads to infected hepatocytes survival and consequent HCV infection persistence.[21]
In the present study, RelA/p65 nuclear labelling followed the same profile of activated NFκB expression by Southwestern histochemistry, as previously reported.[22] Thus, these data were discoursed together as activated NFκB expression. We observed a crescent augmentation of activated NFκB expression in HCV and CIR in relation to NL. NFκB activation may be induced by TNFα secondarily to the increase of proinflammatory status in HCV and CIR. Since TNFα-induced NFκB activation acts as an important survival factor for hepatocytes,[9] higher activated NFκB expression could explain the slight number of apoptotic hepatocytes in HCV and CIR samples in comparison to HCC. In addition, constant activation of NFκB during chronic liver disease performs as an early molecular change during HCC progression.[11] Nevertheless, in the later stages of hepatocarcinogenesis, NFκB inhibition can accelerate development of HCC by cellular proliferation.[23] Thus, in spite of the reduction of activated NFκB expression in HCC patients which may be related to the significant increase of apoptosis, other mechanisms could be associated with this phenomenon.
TRAIL expression was increased and accompanied by decreased NFκB activation and frequent apoptosis in the cases of HCC when compared to HCV and CIR. In addition to inducing apoptosis in normal hepatocytes,[24] TRAIL can mediate NFκB inhibition with consequent apoptosis in transformed hepatocytes.[11] This finding may be related to the attempt of preventing tumorigenesis during chronic inflammatory disease. Moreover, increase of TRAIL expression in HCC with consequent reduction of NFκB activation and augment of hepatocytes apoptosis may occur by a p53-independent pathway.[25]
p53 has been widely considered as a tumor suppressor gene associated with induction of apoptosis in different types of neoplasms, including HCC.[26] Furthermore, p53 is usually less expressed during HCV than in HCC.[27] Accordingly, we found p53 expression almost solely in HCC, without significant changes during chronic liver inflammation. Increased p53 expression was also followed by augmentation of apoptosis in HCC. Moreover, the RelA/p65 subunit can inhibit p53 activation, as well as p53 also suppresses NFκB transcriptional effects.[28] Perhaps during HCV and CIR, the inflammatory stimulus induces an increase of TNFα with consequent NFκB activation and p53 inhibition, which leaked from hepatocytes from apoptosis. Concerning HCC, which present a less evident inflammatory process, TNFα-induced NFκB activation is discrete and, in addition to inhibition of NFκB activation by p53, augments apoptosis. Furthermore, another possible suppressor tumor gene, PHB, has been associated with an increase of p53 activity in the induction of apoptosis in cancer.[29]
The role of PHB, in particular, is not fully understood. There are controversies because both antitumorigenic and protumorigenic functions have been reported for PHB, depending on its subcellular localization.[30] In addition to regulation of various cellular functions, the mitochondrial location of PHB is mainly engaged in reducing damage caused by oxidative stress in the chronic phase of various diseases.[31] In our HCV and CIR samples, we detected a high expression of cytoplasmic PHB, likely due to chronic inflammation associated with these diseases. Moreover, in spite of PHB reducing NFκB activation mediated by TNFα, which permits occurrence of apoptosis, high levels of TNFα can also inhibit tumour suppressor PHB activity during chronic inflammation.[32] This regulatory mechanism may explain increased NFκB activation and higher hepatocytes survival in HCV and CIR in comparison with HCC, despite high cytoplasmic PHB expression in HCV and CIR. In addition, the failure of apoptosis of mutated hepatocytes may represent a primary event in the hepatocarcinogenesis associated with chronic liver disease.
Our data show that PHB expression was exclusively identified as nuclear labelling in HCC. It has been reported that nuclear PHB appears to be essential for regulation of cellular processes such as apoptosis, proliferation, and gene transcription.[30] PHB was also detected in the nucleus associated with retinoblastoma and p53, inducing changes in transcription factors, resulting in cell cycle inhibition and induction of apoptosis.[33] The tumour suppressor action of PHB was associated with nuclear expression in cells of several neoplasms.[29,34] In agreement, the HCC group presented exclusively a PHB nuclear expression in contrast to CIR and HCV. Because NFκB may be acting as an antiapoptotic mediator, and once inhibited by the action of PHB in HCC, it may increase the rate of apoptosis in these cases.
In conclusion, our results suggest that, in addition to well-understood sequences of proinflammatory events such as TNF-induced NFκB activation and NFκB/TRAIL pathway-mediated apoptosis, development of human hepatic cancer may be influenced by regulation of p53 and PHB tumour suppressor function. Another possibility would be that expression of p53 and PHB may have been altered as a consequence of an already-established hepatocellular carcinoma. Additional studies are necessary to explain the contradictory mechanisms of the tumour microenvironment observed in the sequence of HCV, CIR, and HCC.
Authors’ contributions
Casuistic retrieval and samples analysis: L.M. Della Porta
Statistical analysis and writing: F.S. Ramalho
Samples analysis and discussion: C.A. Oliveira
Histological and immunohistochemical preparations and analysis: D.M. Silva
Southwestern histochemistry and analysis: M.J. Augusto
Paper scientific design and writing: L.N.Z. Ramalho
Financial support and sponsorship
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP)-nº2010/20895-4, Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq)-nº302547/2012-6 and Fundação de Amparo à Fundação de Apoio ao Ensino, Pesquisa e Assistência (FAEPA).
Conflict of interests
There are no conflicts of interests.
Patient consent
Each patient was informed of the study and gave their consent.
Ethics approval
The study was approved by the local Ethics Committee (nº1611/2011).
REFERENCES
2. Shiraha H, Yamamoto K, Namba M. Human hepatocyte carcinogenesis (review). Int J Oncol 2013;42:1133-8.
5. Guan YS, He Q, Wang MQ, Li P. Nuclear factor kappa B and hepatitis viruses. Expert Opin Ther Targets 2008;12:265-80.
6. Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol 2013;59:583-94.
7. Liedtke C, Trautwein C. The role of TNF and Fas dependent signaling in animal models of inflammatory liver injury and liver cancer. Eur J Cell Biol 2012;91:582-9.
8. Papa S, Bubici C, Zazzeroni F, Franzoso G. Mechanisms of liver disease: cross-talk between the NF-kappaB and JNK pathways. Biol Chem 2009;390:965-76.
9. Schwabe RF, Brenner DA. Mechanisms of liver injury. I. TNF-alpha-induced liver injury: role of IKK, JNK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol 2006;290:G583-9.
10. Daniels RA, Turley H, Kimberley FC, Liu XS, Mongkolsapaya J, Ch'En P, Xu XN, Jin BQ, Pezzella F, Screaton GR. Expression of TRAIL and TRAIL receptors in normal and malignant tissues. Cell Res 2005;15:430-8.
11. Chu L, Gu J, He Z, Xiao T, Liu X. Adenoviral vector expressing CYLD augments antitumor activity of TRAIL by suppression of NF-kappaB survival signaling in hepatocellular carcinoma. Cancer Biol Ther 2006;5:615-22.
12. Jiang YF, He B, Li NP, Ma J, Gong GZ, Zhang M. The oncogenic role of NS5A of hepatitis C virus is mediated by up-regulation of survivin gene expression in the hepatocellular cell through p53 and NF-κB pathways. Cell Biol Int 2011;35:1225-32.
13. Nuell MJ, Stewart DA, Walker L, Friedman V, Wood CM, Owens GA, Smith JR, Schneider EL, Dell' Orco R, Lumpkin CK, Danner DB, McClung JK. Prohibitin, an evolutionarily conserved intracellular protein that blocks DNA synthesis in normal fibroblasts and HeLa cells. Mol Cell Biol 1991;11:1372-81.
14. Mishra S, Murphy LC, Murphy LJ. The prohibitins: emerging roles in diverse functions. J Cell Mol Med 2006;10:353-63.
15. Ko KS, Tomasi ML, Iglesias-Ara A, French BA, French SW, Ramani K, Lozano JJ, Oh P, He L, Stiles BL, Li TW, Yang H, Martínez-Chantar ML, Mato JM, Lu SC. Liver-specific deletion of prohibitin 1 results in spontaneous liver injury, fibrosis, and hepatocellular carcinoma in mice. Hepatology 2010;52:2096-108.
16. Tsutsumi T, Matsuda M, Aizaki H, Moriya K, Miyoshi H, Fujie H, Shintani Y, Yotsuyanagi H, Miyamura T, Suzuki T, Koike K. Proteomics analysis of mitochondrial proteins reveals overexpression of a mitochondrial protein chaperon, prohibitin, in cells expressing hepatitis C virus core protein. Hepatology 2009;50:378-86.
18. Cong X, Lu C, Huang X, Yang D, Cui X, Cai J, Lv L, He S, Zhang Y, Ni R. Increased expression of glycinamide ribonucleotide transformylase is associated with a poor prognosis in hepatocellular carcinoma, and it promotes liver cancer cell proliferation. Hum Pathol 2014;45:1370-8.
19. Nelson DR, Lim HL, Marousis CG, Fang JW, Davis GL, Shen L, Urdea MS, Kolberg JA, Lau JY. Activation of tumor necrosis factor-alpha system in chronic hepatitis C virus infection. Dig Dis Sci 1997;42:2487-94.
20. Kallinowski B, Haseroth K, Marinos G, Hanck C, Stremmel W, Theilmann L, Singer MV, Rossol S. Induction of tumour necrosis factor (TNF) receptor type p55 and p75 in patients with chronic hepatitis C virus (HCV) infection. Clin Exp Immunol 1998;111:269-77.
21. You LR, Chen CM, Lee YH. Hepatitis C virus core protein enhances NF-kappaB signal pathway triggering by lymphotoxin-beta receptor ligand and tumor necrosis factor alpha. J Virol 1999;73:1672-81.
22. Braz MM, Ramalho FS, Cardoso RL, Zucoloto S, Costa RS, Ramalho LN. Slight activation of nuclear factor kappa-B is associated with increased hepatic stellate cell apoptosis in human schistosomal fibrosis. Acta Trop 2010;113:66-71.
23. Luedde T, Schwabe RF. NF-κB in the liver-linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 2011;8:108-18.
24. Jo M, Kim TH, Seol DW, Esplen JE, Dorko K, Billiar TR, Strom SC. Apoptosis induced in normal human hepatocytes by tumor necrosis factor-related apoptosis-inducing ligand. Nat Med 2000;6:564-7.
25. Yeh CH, Yang YY, Huang YF, Chow KC, Chen MF. Induction of apoptosis in human Hep3B hepatoma cells by norcantharidin through a p53 independent pathway via TRAIL/DR5 signal transduction. Chin J Integr Med 2012;18:676-82.
26. Martin J, Dufour JF. Tumor suppressor and hepatocellular carcinoma. World J Gastroenterol 2008;14:1720-33.
27. Kasprzak A, Adamek A, Przybyszewska W, Czajka A, Olejniczak K, Juszczyk J, Biczysko W, Zabel M. p53 immunocytochemistry and TP53 gene mutations in patients with chronic hepatitis C virus (HCV) infection. Folia Histochem Cytobiol 2009;47:35-42.
28. Webster GA, Perkins ND. Transcriptional cross talk between NF-kappaB and p53. Mol Cell Biol 1999;19:3485-95.
29. Fusaro G, Dasgupta P, Rastogi S, Joshi B, Chellappan S. Prohibitin induces the transcriptional activity of p53 and is exported from the nucleus upon apoptotic signaling. J Biol Chem 2003;278:47853-61.
30. Theiss AL, Sitaraman SV. The role and therapeutic potential of prohibitin in disease. Biochim Biophys Acta 2011;1813:1137-43.
31. Theiss AL, Vijay-Kumar M, Obertone TS, Jones DP, Hansen JM, Gewirtz AT, Merlin D, Sitaraman SV. Prohibitin is a novel regulator of antioxidant response that attenuates colonic inflammation in mice. Gastroenterology 2009;137:199-208.
32. Theiss AL, Jenkins AK, Okoro NI, Klapproth JM, Merlin D, Sitaraman SV. Prohibitin inhibits tumor necrosis factor alpha-induced nuclear factor-kappa B nuclear translocation via the novel mechanism of decreasing importin alpha3 expression. Mol Biol Cell 2009;20:4412-23.
33. Sánchez-Quiles V, Santamaría E, Segura V, Sesma L, Prieto J, Corrales FJ. Prohibitin deficiency blocks proliferation and induces apoptosis in human hepatoma cells: molecular mechanisms and functional implications. Proteomics 2010;10:1609-20.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Porta LMD, Ramalho FS, Oliveira CA, Silva DM, Augusto MJ, Ramalho LNZ. Comparison of p53 and prohibitin expression in the spectrum of hepatitis, cirrhosis, and hepatocellular carcinoma. Hepatoma Res 2017;3:34-42. http://dx.doi.org/10.20517/2394-5079.2016.39
AMA Style
Porta LMD, Ramalho FS, Oliveira CA, Silva DM, Augusto MJ, Ramalho LNZ. Comparison of p53 and prohibitin expression in the spectrum of hepatitis, cirrhosis, and hepatocellular carcinoma. Hepatoma Research. 2017; 3: 34-42. http://dx.doi.org/10.20517/2394-5079.2016.39
Chicago/Turabian Style
Porta, Lívia Maria Della, Fernando Silva Ramalho, Carlos Augusto Oliveira, Deisy Mara Silva, Marlei Josiele Augusto, Leandra Náira Zambelli Ramalho. 2017. "Comparison of p53 and prohibitin expression in the spectrum of hepatitis, cirrhosis, and hepatocellular carcinoma" Hepatoma Research. 3: 34-42. http://dx.doi.org/10.20517/2394-5079.2016.39
ACS Style
Porta, LMD.; Ramalho FS.; Oliveira CA.; Silva DM.; Augusto MJ.; Ramalho LNZ. Comparison of p53 and prohibitin expression in the spectrum of hepatitis, cirrhosis, and hepatocellular carcinoma. Hepatoma. Res. 2017, 3, 34-42. http://dx.doi.org/10.20517/2394-5079.2016.39
About This Article
Copyright

Data & Comments
Data


 Cite This Article 1 clicks
Cite This Article 1 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.