
Identifying microRNA panels specifically associated with hepatocellular carcinoma and its different etiologies


Abstract
Aim: Deregulation of microRNAs (miRNAs) expression has been identified in hepatocellular carcinoma (HCC), but few results are consistent. The objective of this study is to investigate “HCC tumor type specific” and “tumor common” miRNA panels.
Methods: The authors integrate and analyze clinical, etiologic and miRNA profiles data from 9 types of solid tumors in The Cancer Genome Atlas (TCGA) and HCC data from Columbia University Medical Center (CUMC).
Results: Levels of 33 miRNAs were significant different between HCC tumor and paired non-tumor tissues (over 2-fold changes) after Bonferroni correction for multiple comparisons, and most (28 miRNAs) were down-regulated in HCC tumors. Using this panel, the authors well classified HCC tumor tissues with 4 misclassifications among 48 paired tissues. Validating this panel in an additional 302 HCC tumor tissues, the authors almost perfectly distinguished tumor from non-tumor tissues with only two misclassifications (99% of HCC tissues correctly classified). Evaluating miRNA profiles in 32 independent HCC paired tissues from CUMC, the authors observed 40 miRNAs significantly deregulated in HCC with over 2-fold changes; 14 overlapped with those identified in TCGA. Subgroup analyses by HCC etiology found that 4 upregulated and 8 downregulated miRNAs were significantly associated with alcohol-related HCC. There were 7 and 4 miRNAs significantly associated with hepatitis B virus- and hepatitis C virus-related HCC, respectively. Data for the first time revealed that miR-24-1, miR-130a and miR-505 were significantly down-regulated only in HCC tumors; miR-142 and miR-455 were significantly down-regulated in HCC, but up-regulated in 5 other solid tumors; suggesting their HCC “tumor type specific” characteristics. A panel of 8 miRNAs was significant in at least 5 tumor types, including HCC, and was identified as “tumor common” marker.
Conclusion: The authors concluded that aberrant miRNA panels have HCC “tumor type specificity” and may be affected by etiologic factors.
Keywords
Introduction
MicroRNAs (miRNA) are important biological regulators and play a critical role in controlling protein-coding genes’ expression at the post-transcriptional level. It is estimated that one third of human genes are directly or indirectly governed by miRNAs and they impact multiple cellular pathways involved in tumorigenesis.[1,2] Anomalous expression of miRNAs have been implicated in a wide variety of cancers, including hepatocellular carcinoma (HCC), one of the most common cancers and the third leading cause of cancer death worldwide. The incidence of HCC has tripled over the past 30 years in the United States,[3,4] which may be attributed to increased hepatitis C virus (HCV) infection and obesity-related nonalcoholic fatty liver disease (NAFLD).[4,5] Other established etiologies of HCC are hepatitis B virus (HBV) infection, alcohol abuse and aflatoxin B1 (AFB1) exposure.[6,7] Most previous studies examining miRNA profiles in HCC tumor tissue or blood focused on investigation of the main effects of aberrant miRNAs associated with cancer status without consideration of the potential influence of etiologic risk factors on miRNA levels that may bias the miRNA patterns observed in HCC tumors with heterogeneous etiologies. That may be one reason for the discrepant results of previous miRNA marker studies of HCC. Although some studies do examine miRNA profiles in HCC patients carrying specific etiologies,[8-10] it is still unclear whether the identified miRNAs are etiology-specific due to lack of transverse comparisons with HCC patients carrying other etiologic factors. Another challenge is the lack of a comparison of miRNA panels between HCC and other solid tumor types, and no evidence to indicate HCC tumor type specific miRNA alterations that may limit future clinical application. In the current study, we integrate HCC etiologies and miRNA sequencing data from HCC and 8 other types of solid tumors in The Cancer Genome Atlas (TCGA) resource, and investigate whether miRNA panels identified in HCC tumor are organ specific and affected by important etiologic factors. These results can be used for more precise clinical early diagnosis of HCC subtypes and screening of high risk populations with specific HCC etiologies.
Methods
Demographic, etiologies, clinical and miRNA data in HCC patients from TCGA dataset
TCGA is a comprehensive and coordinated project supported by the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI) to characterize the genomic data of more than 30 different types of cancers, and accelerate understanding of the molecular basis of cancer. Currently, there are 366 cancer patients in the cancer type of liver hepatocellular carcinoma, who provided demographic, etiologic and clinical data, as well as tissue samples for TCGA study. The miRNA expression and corresponding etiologies and clinical data were downloaded (up to June 16, 2015) from TCGA data portal (https://tcga-data.nci.nih.gov/tcga/tcgaHome2.jsp), and a total of 414 samples from 366 histologically confirmed liver cancer patients have completed miRNAs, etiologic and clinical data. After checking histologic diagnosis and tissue types, we excluded 10 non-HCC cases (either mixed hepatocholangiocarcinoma or fibrolamellar carcinoma); 5 recurrent HCC tumors and 1 HCC non-tumor tissue without relevant paired tumor tissue. Finally, data from 48 HCC patients with paired tumor and non-tumor tissues and 302 HCC patients with tumor tissues alone were analyzed in the current study.
Demographic, etiologic and clinical data include co-variates of age, gender, race/ethnicity, height (m) and weight (kg) at cancer diagnosis, body mass index (BMI, kg/m2), HCC risk factors (alcohol consumption, HBV, HCV, NAFLD, mixed and none), tumor status (free vs. not free), family history of cancer (no vs. yes), alpha fetoprotein (AFP, ng/mL), histologic tumor grade (G1-G2 vs. G3-G4), the American Joint Committee on Cancer (AJCC)[11] tumor-node-metastasis (TNM) (T0-T2 vs. T3-Tx), lymph node involvement (N0-N1 vs. Nx), pathological stage (stage I-II vs. stage III-IV), metastasis status (M0-M1 vs. Mx), vital status (alive vs. dead), survival days (either days to last follow-up or days to death). Other clinical variables (Child-Pugh classification, vascular tumor invasion, adjuvant treatment, surgical types and new tumor event after initial treatment) were not analyzed in the current study due to either a large amount of missing data or small sample sizes in subgroups.
The level 3 (archive type) miRNA expression data were generated from the Illumina HiSeq 2000 platform (Illumina Inc., San Diego, CA) and annotated to reference miRBase v16 of UCSC hg19 alignments.[12] A total of 1,046 unique mature miRNAs were obtained. The sequencing data are presented as raw read counts and reads per million (RPM) mapped miRNAs reads. The RPM indicates the expression level of miRNA and is calculated according to the formula: RPM = (NmiR/Nall) × 106, NmiR: number of reads mapped to the specific miRNA reference; Nall: total number of reads mapped in the sample. Because all demographic, clinical and miRNA data are derived from the de-identified publically available TCGA dataset, it is not possible to link to any individual. Therefore, no Institutional Review Board (IRB) approval was required.
MiRNA sequencing data from 8 other solid tumors in TCGA dataset
MiRNA sequencing and clinical data from other solid cancers were also downloaded from TCGA data portal. Eight solid tumors with available miRNA and clinical data in over 40 paired tumor and non-tumor tissues were considered in the final statistical analyses, including female breast invasive carcinoma (BRCA), head and neck squamous cell carcinoma (HNSC), kidney renal cell carcinoma (KIRC), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), prostate adenocarcinoma (PRAD), stomach adenocarcinoma (STAD), and thyroid carcinoma (THCA). The samples sizes (pairs) were 102 for BRCA, 71 for KIRC, 59 for THCA, 52 for PRAD, 46 for LUAD, 43 for HNSC, and 41 for both STAD and LUSC.
HCC patients and miRNA data used as the validation set
For the first set of validation, we used 32 HCC frozen tumor and adjacent non-tumor tissues (16 pairs) that were collected by the Center for Liver Disease and Transplantation, and stored in the Molecular Pathology Shared Resource of the Herbert Irving Comprehensive Cancer Center, Columbia University Medical Center (CUMC). This study has been approved by the IRB of CUMC. Total RNA, including miRNAs was isolated from HCC tissues by RNeasy Microarray Tissue Mini Kits (Qiagen, Frederick, MA) according to the manufacturer’s protocol. TaqMan Low Density Arrays (TLDA, Applied Biosystems, Foster City, CA), covering 733 miRNAs (670 unique human mature miRNAs), were used to generate miRNA profiles thatwere deposited in NCBI’s Gene Expression Omnibus database (accession number GSE54751).[13] TaqMan MicroRNA assays were used to further evaluate the consistence of candidate miRNA expression patterns in 66 paired HCC tumor and non-tumor tissues from CUMC. U6 snRNA stable in liver tumor/adjacent tissues (Ct: 21.19 vs. 21.08, P = 0.398) was used as an endogenous control to normalize the expression of miRNAs using the 2(-ΔΔCt) approach.[14]
Statistical analysis
We applied stringent criteria to filter available miRNA sequencing data before performing any statistical analysis to ensure the reliability and abundance of candidate miRNAs in the target tissues. MiRNAs were excluded from further data analyses if the RPM was less than 10 counts and missing data exceeded 10% of all subjects. MiRNAs with less than 10 counts per million may be due to sequencing errors.[15] A low missing value (< 10%) provides the most reliable and consistent result without the need for further normalization.[16] A total of 153 miRNAs passed the filtering criteria and data were log2-transformed for final statistical analysis in HCC.
Paired t-test with Bonferroni correction for multiple comparisons was used to identify miRNAs that were significant different (P < 0.0001) with at least a 2-fold expression change between the 48 paired HCC tumor and adjacent non-tumor tissues. The volcano plot and hierarchical clustering were performed using the panel of significant miRNAs to describe the distribution of miRNAs and tumor classification, respectively. The same miRNA panel was used to construct a heat-map and classify the 302 unpaired tumor tissues. The general linear model was used to compare miRNAs expression levels between unpaired HCC tumor and non-tumor tissues adjusted for covariates significantly different between groups. Prediction analysis of microarrays using the nearest shrunken centroid methodology was used to separately evaluate the classification of tissues (tumor vs. non-tumor) for paired and unpaired tumors by those significantly altered miRNAs, and estimate prediction error, sensitivity, specificity, positive predictive value and negative predictive value via cross-validation.[17] Two-sample t-tests were applied to identify significant miRNAs (P < 0.0001) with over 2-fold changes by age group (< 60 vs. ≥ 60 years), gender (male vs. female), BMI (≥ 25 vs. < 25), etiologies [alcohol vs. hepatitis B surface antigen (HBsAg) positive vs. anti-HCV positive], AFP (≥ 400 vs. < 400 ng/mL), and other clinicopathological covariates described above. Subgroups analyses were further conducted among HCC tumor and non-tumor tissues carrying one specific risk factor (alcohol, HBsAg or anti-HCV) to identify etiologic-specific miRNA panels.
Similar stringent filtering criteria and statistical analysis strategies were used to identify aberrantly expressed miRNA profiles from the other 8 different solid tumors. The identified miRNA panels from different tumors were compared to each other to discover “tumor type specific” or “tumor common” miRNA panels. We define “tumor common” miRNAs as those significant for at least 5 tumor types, including HCC, and with fold-changes in the same direction. “Tumor type specific” miRNAs are defined as only significant for one type of tumor among the 9 investigated tumors. If miRNAs are significant for several different tumor types, but the direction in one type of tumor is opposite to all others, we also define them as “tumor type specific” miRNAs. The most commonly or uniquely expressed miRNAs were selected as “tumor common” or “tumor type specific” markers, respectively, for further bioinformatics validation.
All statistical data analyses were performed using BRB-ArrayTools (version 4.4) developed by Dr. Richard Simon and the BRB-ArrayTools Development Team (http://linus.nci.nih.gov/BRB-ArrayTools.html)[18] and Statistical Analysis System 9.0 (SAS Institute). TCGA data used in this study meet the publication guidelines provided by TCGA (http://cancergenome.nih.gov/publications/publicationguidelines).
Bioinformatics analyses of miRNA targets and pathways enrichment
The targets of the miRNAs were predicted by mirsystem, which integrates seven well known miRNA target gene prediction programs (http://mirsystem.cgm.ntu.edu.tw/index.php),[19] as well as the experimentally validated miRNA-target data from miRecords (http://c1.accurascience.com/miRecords/) and TarBase (http://www.hsls.pitt.edu/obrc/index.php?page=URL1237572545). The seven predictive tools include DIANA (http://diana.imis.athena-innovation.gr/DianaTools/index.php), miRanda (http://www.microrna.org/microrna/home.do), mirBridge (http://mirbridge.org/), PicTar (http://pictar.mdc-berlin.de/), PITA (http://genie.weizmann.ac.il/pubs/mir07/mir07_data.html), RNA22 (https://cm.jefferson.edu/rna22v2/), and TargetScan v6.2 (http://www.targetscan.org/). The concordant targets in the current study were defined as genes predicted by at least 5 out of 7 algorithms or validated by functional experiment. These genes were the most likely miRNA targets that were further evaluated by ToppGene (https://toppgene.cchmc.org/prioritization.jsp)[20] to identify significant biological processes, pathways, molecular functions and cellular components after Bonferroni correction P < 0.05.
Results
Demographic and clinical characteristics of HCC patients
We compared the demographic and clinical characteristics between 48 HCC patients with paired tumor and non-tumor tissues, and 302 patients with tumor tissue alone [Supplementary Table 1]. There were no significant differences for the co-variates of age (means of 61.1 vs. 59.5 years), gender, etiology, BMI, AFP level, tumor grade, lymph node involvement, metastasis status, AJCC pathological stage and TNM. The 48 HCC patients were more often white, had less surgical remove of their tumors, more frequently had a cancer family history and had a longer survival time compared to the 302 patients.
MiRNA abundance and classification of tumor tissues
A pie graph shows the distribution of the most abundant miRNAs (top 20) in HCC tumor and non-tumor tissues [Supplementary Figure 1]. The most abundant 5 miRNAs are miR-21, miR-22, miR-143, miR-148a and miR-192; they account for more than 58% of all detectable miRNAs. Paired t-test analysis revealed that 33 miRNAs were significantly differentially expressed between the 48 paired HCC tumor and non-tumor tissues with over 2-fold changes at the significance level of P < 0.0001 [Table 1, Supplementary Figure 2]. However, only 5 overlap with the top 20 most abundant miRNAs, suggesting the most significant miRNAs are infrequently expressed in liver tissues a more sensitive approach for their detection is needed. Five miRNAs (miR-10b, miR-182, miR-183, miR-21 and miR-452) were significantly up-regulated in HCC tumor tissue with fold changes ranging from 11.13 to 2.25, while 28 miRNAs showed significant down-regulation in tumor tissue (fold-change from -5.26 to -2.08). The same expression patterns for the 33 significant miRNAs were also observed in additional 302 HCC tissues comparing with unpaired 48 non-tumor tissues [Supplementary Table 2]. After adjusting for covariates of race, survival time, tumor status and family history of cancer, these miRNAs still kept significance with over 2-fold changes, indicating the aberrant miRNAs mainly caused by tumor itself. Using the 33 significant miRNAs as a panel to generate a hierarchical heat map, only 4 tumor/non-tumor tissues were misclassified among the 48 paired tissues [Figure 1]. The same panel of miRNAs was used to classify the additional 302 HCC patients with tumor tissues alone, and excellent clustering was observed with only two misclassifications [Supplementary Figure 3]. Percentages of correctly classified HCC tissues were 96% and 99% for the 48 paired and 302 unpaired tumor tissues respectively [Table 2], suggesting the promise of aberrantly expressed miRNAs as HCC biomarkers.
Differentially expressed miRNAs between 48 paired hepatocellular carcinoma tumor and non-tumor tissues
| miRNAs | Geometric mean of RPM in tumor tissue | Geometric mean of RPM in non-tumor tissue | Fold-change | P-value | FDR |
|---|---|---|---|---|---|
| Upregulated | |||||
| miR-10b | 15,976.87 | 1,435.06 | 11.13 | < 1e-07 | < 1e-07 |
| miR-183 | 1,451.95 | 261.13 | 5.56 | < 1e-07 | < 1e-07 |
| miR-182 | 4,034.39 | 971.8 | 4.15 | 1.00E-07 | 4.50E-07 |
| miR-452 | 161.94 | 52.46 | 3.09 | 6.00E-07 | 2.30E-06 |
| miR-21 | 165,875.22 | 73,641.4 | 2.25 | < 1e-07 | < 1e-07 |
| Downregulated | |||||
| miR-199a-1 | 242.98 | 1,308.64 | -5.26 | < 1e-07 | < 1e-07 |
| miR-199a-2 | 404.21 | 2,176.44 | -5.26 | < 1e-07 | < 1e-07 |
| miR-199b | 490.46 | 2,611.64 | -5.26 | < 1e-07 | < 1e-07 |
| miR-139 | 101.67 | 426.13 | -4.17 | < 1e-07 | < 1e-07 |
| miR-375 | 1,338.06 | 5,364.67 | -4.00 | 1.87E-05 | 5.50E-05 |
| miR-424 | 133.58 | 537.65 | -4.00 | < 1e-07 | < 1e-07 |
| miR-130a | 37.45 | 140.38 | -3.70 | < 1e-07 | < 1e-07 |
| miR-451 | 382.94 | 1,433.49 | -3.70 | < 1e-07 | < 1e-07 |
| miR-144 | 94.72 | 315.05 | -3.33 | < 1e-07 | < 1e-07 |
| miR-142 | 996.3 | 2,968.42 | -2.94 | < 1e-07 | < 1e-07 |
| miR-486 | 94.12 | 279.26 | -2.94 | < 1e-07 | < 1e-07 |
| miR-99a | 535.01 | 1,568.16 | -2.94 | < 1e-07 | < 1e-07 |
| let-7c | 1,921.73 | 5,196.5 | -2.70 | < 1e-07 | < 1e-07 |
| miR-101-2 | 67.78 | 176.16 | -2.63 | < 1e-07 | < 1e-07 |
| miR-145 | 830.74 | 2,177.06 | -2.63 | < 1e-07 | < 1e-07 |
| miR-379 | 470.22 | 1,209.95 | -2.56 | 2.00E-07 | 8.27E-07 |
| miR-150 | 218.83 | 546.98 | -2.50 | 5.20E-06 | 1.69E-05 |
| miR-223 | 90.56 | 228.55 | -2.50 | < 1e-07 | < 1e-07 |
| miR-24-1 | 34.72 | 85.92 | -2.50 | < 1e-07 | < 1e-07 |
| miR-125b-1 | 445.23 | 1,078.99 | -2.44 | < 1e-07 | < 1e-07 |
| miR-542 | 195.84 | 462.64 | -2.38 | < 1e-07 | < 1e-07 |
| miR-101-1 | 18,399.45 | 42,453.11 | -2.33 | < 1e-07 | < 1e-07 |
| miR-10a | 10,426.51 | 23,842.5 | -2.27 | 6.00E-07 | 2.30E-06 |
| miR-134 | 138.39 | 318.13 | -2.27 | 9.10E-06 | 2.90E-05 |
| miR-378 | 767.99 | 1,756.29 | -2.27 | < 1e-07 | < 1e-07 |
| miR-455 | 681.31 | 1,547.61 | -2.27 | 3.40E-06 | 1.13E-05 |
| miR-505 | 66.11 | 149.37 | -2.27 | < 1e-07 | < 1e-07 |
| miR-29c | 2,061 | 4,281.77 | -2.08 | < 1e-07 | < 1e-07 |
Accuracy of hepatocellular carcinoma tumor tissues classification by 33 significant miRNAs panel
| Classification | Sensitivity | Specificity | PPV | NPV | Correct classification (%) | Misclassification (%) |
|---|---|---|---|---|---|---|
| 48 paired tumors | 0.917 | 1.000 | 1.000 | 0.923 | 95.9 | 4.1 |
| 302 unpaired tumors | 0.990 | 0.979 | 0.997 | 0.940 | 98.5 | 1.5 |
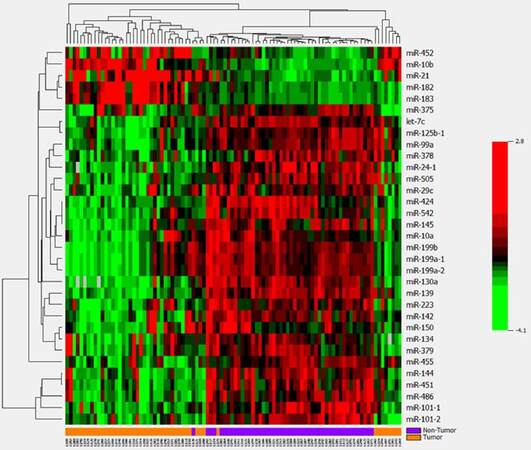
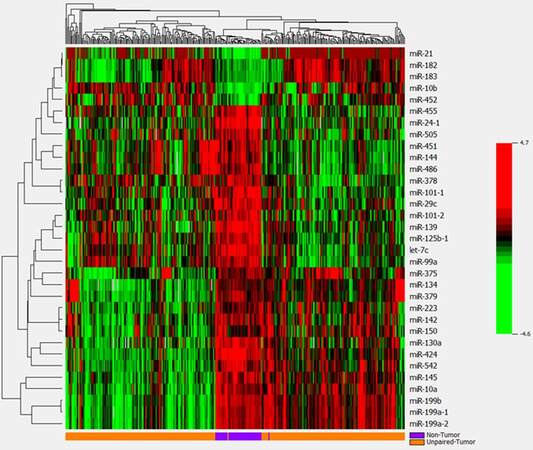
Figure 1. Hierarchical cluster analysis of 33 significantly differentially expressed miRNAs between 48 paired tumor and adjacent non-tumor tissues. Most miRNAs are down-regulated (green), while a few are up-regulated (red) in tumor tissues. The panel of miRNAs can well classify tissue types with 4 misclassified tumor and non-tumor tissues
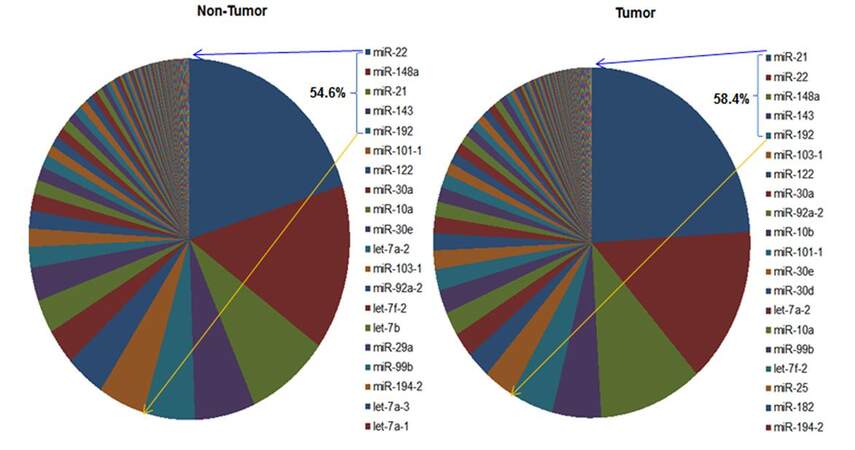
Supplementary Figure 1. Pie graph showing the distribution of the most abundant miRNAs (top 20) in HCC tumor and non-tumor tissues. Of these, 16 abundant miRNAs were found in both tumor and non-tumor tissues. The top 5 abundant miRNAs account for more than 58% of all detectable miRNAs. MiR-122, the most abundant miRNA previously reported in liver tissue is the 7th most common in both tumor and non-tumor tissues
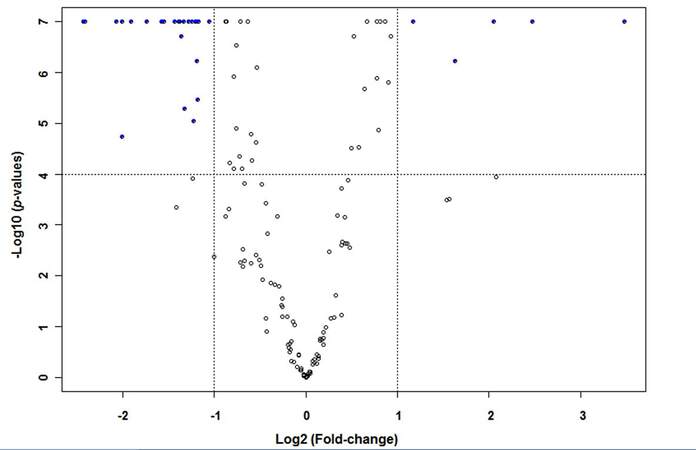
Supplementary Figure 2. Volcano plot of 153 tested miRNAs in 48 paired HCC tissues. Solid blue dots indicated that 5 miRNAs were significantly up-regulated, while 28 miRNAs showed significant down-regulation in tumor tissues with over 2-fold changes after Bonferroni correction for multiple comparison (P < 0.0001)
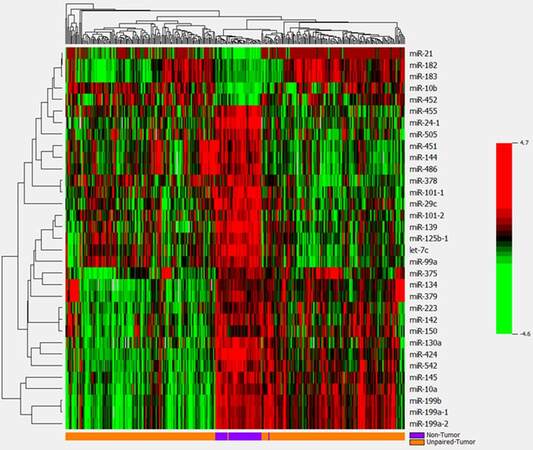
Supplementary Figure 3. Hierarchical cluster analysis of the same 33 significant miRNAs in differentiating 302 unpaired tumors from 48 non-tumor tissues. The panel of miRNAs can also well cluster tumor tissues with two misclassifications
We validated the findings from TCGA data by measuring miRNA profiles in 32 paired HCC tissues from CUMC. We observed 40 miRNAs significantly deregulated in HCC tumors (P < 0.05) with over 2-fold changes [Supplementary Figure 4], and 14 (let-7c, miR-21, miR-99a, miR-125b, miR-130a, miR-139, miR-144, miR-145, miR-150, miR-199a, miR-223, miR-378, miR-455 and miR-486) overlap with those identified in TCGA data. Eight miRNAs (miR-122, miR-1180, miR-199a, miR-182, miR-152, miR-125b, miR-18a and miR-10a) with various expression levels in TCGA data were randomly selected and evaluated by TaqMan quantitative reverse transcription polymerase chain reaction (RT-PCR) in 66 paired HCC tissues from CUMC. Seven out of 8 miRNAs had consistent fold-changes as in TCGA data [Supplementary Table 3]. Only miR-152 showed an inconsistent fold-change (1.01 in RT-PCR and -1.03 in TCGA). The raw expression data of 8 miRNAs were showed in [Supplementary Table 4].
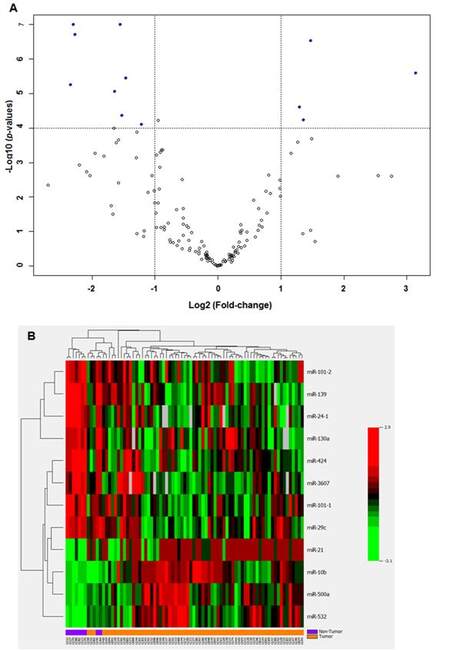
Supplementary Figure 4. Volcano plot of 40 significant miRNAs (P < 0.05) with over 2-fold expression changes validated in 32 CUMC HCC patients
Aberrant miRNAs panels associated with etiology-specific HCC
Subgroup analyses for three HCC-specific major etiologic factors (alcohol abuse, HBV and HCV infection) by two-sample t-tests, we identified 4 upregulated (miR-10b, miR-21, miR-500a and miR-532) and 8 downregulated miRNAs panel significantly associated with alcohol-related HCC [Table 3, Supplementary Figure 5A]. The 12-miRNAs panel can distinguish alcohol-related HCC tumor from non-tumor with 3 misclassifications [Supplementary Figure 5B]. There were panels of 7 and 4 significant aberrantly expressed miRNAs observed in HBV- or HCV-related HCC, respectively, with over 2-fold expression changes [Table 3, Supplementary Figure 6]. These miRNA panels can also correctly classify HBV- or HCV-infected tumors with 1-2 misclassifications [Supplementary Figure 7]. Comparison of significant miRNAs for HCCs with different etiologies and overall HCCs, only miR-424 was consistently down-regulated among all HCC groups; miR-6b was only significantly repressed in HBV-related HCC; and up-regulated miR-93 was identified in both HBV and HCV related HCC [Supplementary Figure 8A]. Several etiology-specific miRNAs were identified that do not overlap with those found in overall HCC [Supplementary Figure 8B], but most significant miRNAs identified in etiology-specific HCC were also consistently observed in overall HCC, which indicates that the fundamental mechanisms may be similar for hepatocarcinogenesis regardless of etiology.However, the results should be explainedwith caution because of the small sample sizes in subgroup analyses.
Aberrant miRNAs associated with etiologic specific HCC tumors
| Etiologies | miRNAs | Geometric mean of RPM in tumor tissue | Geometric mean of RPM in non-tumor tissue | Fold-change | P-value | FDR |
|---|---|---|---|---|---|---|
| Alcoholic HCC | ||||||
| (n = 79) | miR-10b | 12,283.98 | 1,395.27 | 8.80 | 2.60E-06 | 7.96E-05 |
| miR-21 | 189,758.19 | 68,433.86 | 2.77 | 3.00E-07 | 1.15E-05 | |
| miR-500a | 423.56 | 165.36 | 2.56 | 5.73E-05 | 7.57E-04 | |
| miR-532 | 1491.55 | 607.79 | 2.45 | 2.47E-05 | 4.20E-04 | |
| miR-424 | 98.38 | 483.13 | -5.00 | 1.00E-07 | 7.65E-06 | |
| miR-3607 | 43.77 | 221.60 | -5.00 | 5.60E-06 | 1.22E-04 | |
| miR-139 | 101.16 | 487.34 | -4.76 | 2.00E-07 | 1.02E-05 | |
| miR-130a | 48.83 | 152.77 | -3.13 | 8.60E-06 | 1.64E-04 | |
| miR-24-1 | 32.81 | 96.18 | -2.94 | < 1e-07 | < 1e-07 | |
| miR-29c | 1,510.76 | 4,355.13 | -2.86 | 4.31E-05 | 6.59E-04 | |
| miR-101-1 | 16,403.75 | 45,299.71 | -2.78 | 3.60E-06 | 9.18E-05 | |
| miR-101-2 | 81.66 | 189.96 | -2.33 | 7.73E-05 | 9.10E-04 | |
| HBV-related HCC | ||||||
| (n = 79) | miR-532 | 1,665.49 | 605.92 | 2.75 | 6.40E-05 | 1.40E-03 |
| miR-93 | 5448.5 | 2,193.92 | 2.48 | 5.11E-05 | 1.30E-03 | |
| miR-21 | 205,313.3 | 84,355.47 | 2.43 | 1.60E-06 | 8.16E-05 | |
| miR-424 | 85.19 | 535.38 | -6.25 | < 1e-07 | < 1e-07 | |
| miR-139 | 104.28 | 383.68 | -3.70 | 2.22E-05 | 6.79E-04 | |
| miR-24-1 | 28.91 | 73.80 | -2.56 | 1.50E-06 | 8.16E-05 | |
| miR-26b | 865.69 | 2,027.85 | -2.33 | 1.04E-05 | 3.98E-04 | |
| HCV-related HCC | ||||||
| (n = 31) | miR-93 | 5,978.72 | 1,423.17 | 4.20 | 1.99E-05 | 1.60E-03 |
| miR-500a | 457.17 | 125.83 | 3.63 | 9.53E-05 | 3.65E-03 | |
| miR-424 | 91.62 | 611.42 | -6.67 | 3.13E-05 | 1.60E-03 | |
| miR-3607 | 46.28 | 249.57 | -5.26 | 2.90E-05 | 1.60E-03 | |
Supplementary Figure 5. Volcano plot (A) and heat-map (B) of 12 significant miRNAs (P < 0.0001) associated with alcoholic related HCC with over 2-fold expression changes
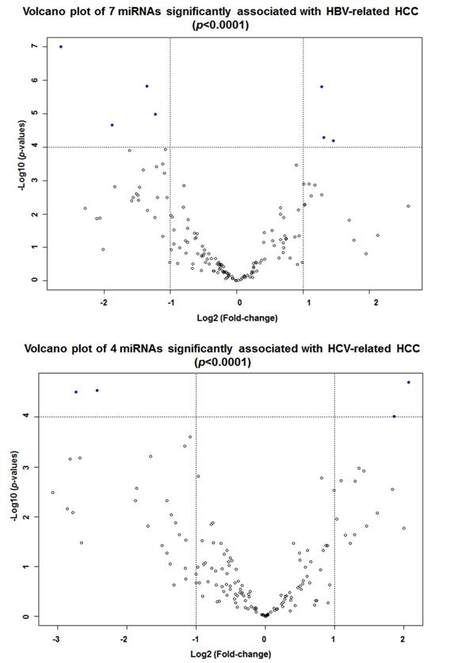
Supplementary Figure 6. Volcano plots to display significant miRNAs associated with HBV- or HCV-related HCC. Seven miRNAs showed significant associations with HBV-related HCC (P < 0.0001) having over 2-fold expression changes, and four miRNAs revealed significant associations with HCV-related HCC (P < 0.0001) having over 2-fold expression changes
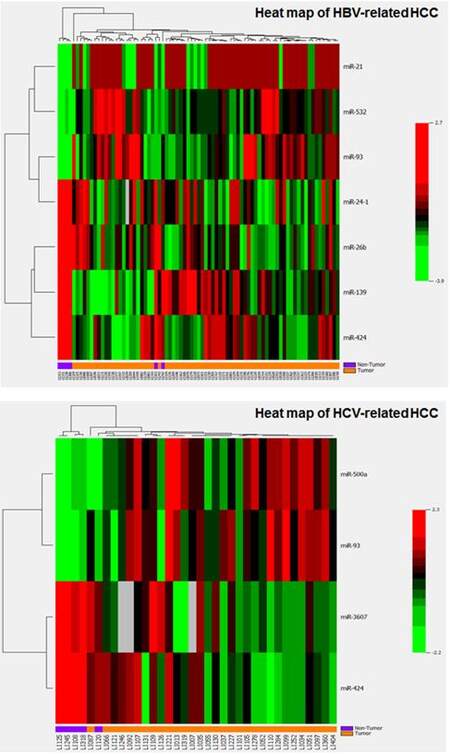
Supplementary Figure 7. Heat maps of hierarchical cluster analysis for significant miRNAs in classification of HBV- or HCV-related HCC. Both panels of miRNAs can correctly classify HBV- or HCV-infected tumors with only 1-2 misclassifications
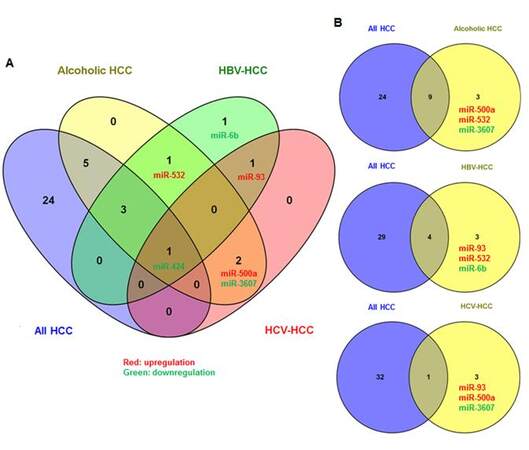
Supplementary Figure 8. Venn diagrams displaying the overlapping and unique miRNAs between overall and etiology-specific HCC. Only miR-424 was consistently down-regulated among all groups, and miR-6b was only significantly repressed in HBV-related HCC (A). Most etiology-related significant miRNAs were consistently observed in overall HCC, and only a few miRNAs were specifically identified in HCCs of individual etiology (B)
Exploring HCC “tumor type specific” and “common tumor” miRNAs panels
Using the same filtering criteria and statistical analysis strategies as for HCC, we examined miRNA profiles in an additional 8 solid tumor types with TCGA data available on at least 40 paired tumor and adjacent non-tumor tissues. Different panels consisting of 15 to 52 significant miRNAs (P < 0.0001) with over 2-fold changes were obtained for these tumors [Supplementary Figure 9]. Many more up-regulated miRNAs were found for LUAD, LUSC, STAD and PRAD compared to HCC which had more down-regulated miRNAs [Supplementary Figures 2 and 4]. Other tumors have similar numbers of up- or down-regulated miRNAs. Our data for the first time revealed that certain miRNAs have “tumor type specificity” and are only significant in one type of tumor, but not others, or aberrant expression in one tumor type is in the opposite direction compared with all others. Significantly down-regulated miR-24-1, miR-130a and miR-505 were only observed in HCC tumors with fold-changes ranging from -2.27 to -3.7 [Figure 2]. Similar down-regulation was confirmed by TaqMan arrayin the validation set of HCC patients from CUMC although only miR-24 and miR-130a achieved statistical significance [Supplementary Table 5]. The expression pattern of the 3 miRNAs was consistently repressed in KIRC and PRAD, but was not statistically significant. An up-regulated expression pattern was observed for 3 miRNAs in HNSC, LUAD and STAD, but none was significant. Inconsistent regulation patterns (up- or down-) for the 3 miRNAs were obtained in BRCA, LUSC and THCA, suggesting HCC tumor specificity for these miRNAs. We also identified 2 “tumor specific” miRNAs for LUAD, 3 for PRAD, 4 for HNSC, 5 for BRCA, 6 for KIRC, and 8 for LUSA, STAD and THCA (data not shown). These data provide promising evidence to justify further investigation of “tumor type specific” miRNAs in other types of tumors.
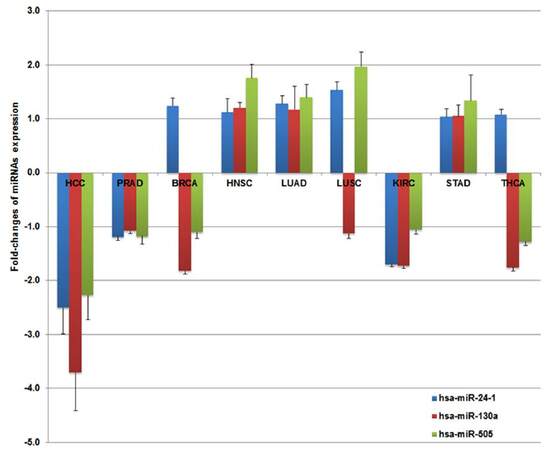
Figure 2. Hepatocellular carcinoma (HCC) "tumor specific" miRNA expression patterns (fold-changes and standard errors) compared to 8 other types of solid tumors. Three miRNAs (miR-24-1, miR-130a and miR-505) were significantly down-regulated in HCC with over 2-fold changes. Although the expression pattern of 3 miRNAs was consistently repressed in kidney renal cell carcinoma (KIRC) and prostate adenocarcinoma (PRAD), none were statistically significant. An up-regulated expression pattern was observed for the 3 miRNAs in head and neck squamous cell carcinoma (HNSC), lung adenocarcinoma (LUAD) and stomach adenocarcinoma (STAD), but also no significant difference. Both up- and down-regulation patterns were obtained for the 3 miRNAs in female breast invasive carcinoma (BRCA), lung squamous cell carcinoma (LUSC) and thyroid carcinoma (THCA), suggesting their tumor specificity
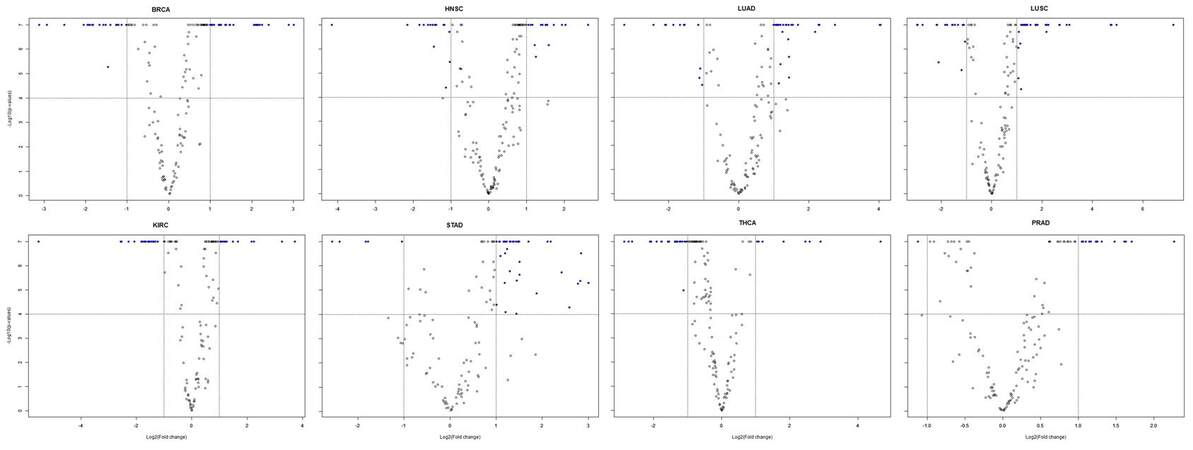
Supplementary Figure 9. Volcano plots displaying significant miRNA panels associated with 8 other types of solid tumors, including female breast invasive carcinoma (BRCA), head and neck squamous cell carcinoma (HNSC), kidney renal cell carcinoma (KIRC), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), prostate adenocarcinoma (PRAD), stomach adenocarcinoma (STAD), and thyroid carcinoma (THCA) using the same criteria as for HCC. More up-regulated miRNAs were found for LUAD, LUSC, STAD and PRAD compared to HCC
We also identified 8 “tumor common” miRNAs fitting into the above definition including up-regulated miR-21, miR-182, miR-183, and down-regulated miR-139, miR-144, miR-101-2, miR-451, miR-486 [Figure 3]. Interestingly, the expression of miR-142 and miR-455 were significantly down-regulated in HCC, but significantly upregulated in 5 other solid tumors. The down-regulated expression pattern of miR-142 and miR-455 were confirmed in the CUMC HCC validation set [Supplementary Table 5]. These results suggest that miR-142 and miR-455 may be potential HCC “tumor specific” markers, but they should be considered as “tumor common” markers for 5 other tumor types.
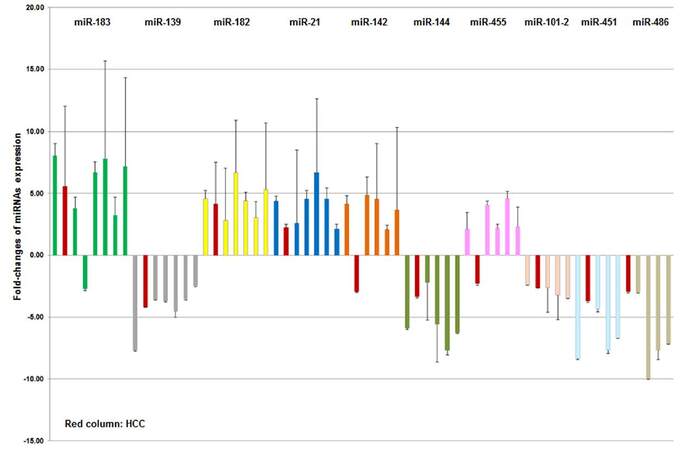
Figure 3. Expression patterns (fold-changes and standard errors) of 10 "tumor common" miRNAs in different types of solid tumors. MiR-182, miR-182, miR-21, miR-142 and miR-455 were significantly up-regulated, and miR-139, miR-144, miR-101-2, miR-451 and miR-486 were significantly down-regulated for most of tumor types. Interestingly, miR-142 and miR-455 were significantly down-regulated in HCC and is in the opposite direction from the five other solid tumors, suggesting their potential as "HCC tumor specific" markers. HCC: hepatocellular carcinoma
Searching for target genes and biologically enriched pathways
Mirsystem was used to search for target genes of the 5 HCC “tumor specific” miRNAs (miR-24-1, miR-130a, miR-505, miR-142 and miR-455). A total of 2,270 genes (1,937 unique genes) were obtained as the targets of at least one miRNA [Supplementary Table 6]. Among them, 619 genes were identified by 5 out of 7 predictive tools or are experimentally validated miRNA-targets including 577 unique genes because some might be targeted by 2 or 3 miRNAs [Supplementary Figure 10]. Among target genes, 130 genes have been associated with HCC in at least one previous report by comparing with Liverome database (http://liverome.kobic.re.kr/).[21] Upon further evaluation for enriched biological function, we identified several important biologic pathways, including transforming growth factor beta (TGFβ) receptor signaling pathway, endocytosis, signaling by epidermal growth factor receptor, signaling by nerve growth factor (NGF), NGF signaling via tropomyosin receptor kinase A from the plasma membrane, BMAL1: CLOCK/NPAS2 activates circadian expression and adherens junction [Supplementary Table 7], which confirmed the potential biological role of HCC specific miRNAs involved in tumorigenesis.
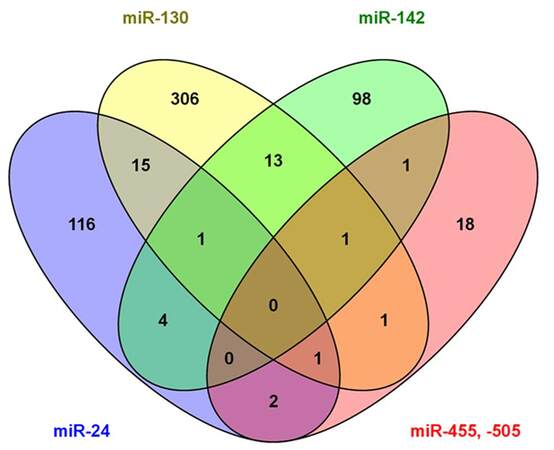
Supplementary Figure 10. Unique genes predicted by 5 out of 7 bioinformatics tools or functional experiments. A total of 577 unique genes were identified as the concordant targets in the current study
Discussion
The most interesting finding in the current study was significant down-regulation of miRNAs (miR-24-1, miR-130a, miR-505, miR-142 and miR-455) in HCC tumor tissue that showed “tumor type specificity” [Figure 2], [Supplementary Table 5]. Additionally, a panel of miRNAs (miR-21, miR-182, miR-183, miR-139, miR-144, miR-101-2, miR-451 and miR-486) was first identified as “tumor common” markers that are significantly altered in most solid tumors by comparing RNA-seq data from 9 different cancer types [Figure 3], [Supplementary Table 5]. A few miRNAs were also significantly dysregulated in etiology-specific (alcohol drinking, HBV- or HCV- infection) HCC [Table 3, Supplementary Figures 5 and 6], suggesting the potential impact of different etiologies in addition to tumorigenesis itself. However, most etiologic relevant miRNAs were also consistently observed in overall HCC, indicating similar fundamental mechanisms involved in hepatocarcinogenesis regardless of different etiologies. These candidate miRNAs may be applied to improve clinical early diagnosis of HCC and more precise prevention and therapy. A similar research strategy can be adopted to discover and verify other “tumor type specific” miRNAs and promote early detection and precise treatment of different types of cancer.
Accumulating evidence based on genome-wide and candidate miRNA approaches have uncovered miRNAs dysregulation in HCC acting as either oncogenes or tumor suppressors.[22,23] A few but not all studies of the 5 “HCC tumor specific” miRNAs are consistent. The expression of miR-24 was significantly reduced in HCC with cirrhosis compared to adjacent cirrhotic tissue, suggesting an influence on hepatocyte carcinogenic transformation of cirrhotic tissues.[24] Significant down-regulation of miR-130a was observed in over 75% (78/102) of HCC tumor tissues.[25] The same repression pattern of miR-130a was also found in HCV-infected human HCC cells[26] and rat liver tissue after treatment with AFB1, a strong hepatocarcinogen.[27] A recent study found that miR-142-3p and miR-142-5p were significantly downregulated in HCC.[28] The ectopic expression of miR-142 significantly reduced HCC cell migration and invasion, and overexpression both miR-142-3p and miR-142-5p synergistically inhibited HCC cell migration, indicating their cooperative regulatory role.[28] This result is supported by a mechanistic study demonstrating that miR-142-3p can directly repress the expression of RAC1 (Ras-Related C3 Botulinum Toxin Substrate 1), which regulates a diverse array of cellular events including increased colony formation, migration and invasion in HCC cell lines.[29] Only one study observed miR-455 significantly down-regulated in HCC tissue and serum in HCC related to type I glycogen storage disease.[30] The different up-regulation patterns of miR-130a and miR-505 in other solid tumors (bladder,[31] breast,[32] gastric,[33] ovarian,[34] colorectal[35] and non-small cell lung cancers[36]) provide further evidence for their potential as HCC specific biomarkers.
However, inconsistent results were also observed in previous studies that suggest significant up-regulation for some of “HCC specific” miRNAs. For example, miR-24 was found significantly up-regulated in HCC tumor tissue, cell lines,[37,38] and serum compared with healthy controls and/or chronic liver disease patients.[39,40] Serum miR-505 level was also increased in HCC cases compared to controls.[41] The expression of miR-130a was significantly higher in HCV-infected hepatocytes and liver biopsy specimens.[42] While in prostate cancer[43] and glioblastoma,[44] miR-130a was significantly down-regulated showing the same pattern as in HCC, indicating possible non-specificity as HCC biomarker. It is known that certain miRNAs may act as both tumor suppressor and oncogene in a cell/tissue specific manner or vary by etiology and cancer stage because they simultaneously regulate multiple target genes involved in different biological pathways. The function of miR-24 as a tumor suppressor can inhibit cell proliferation, migration and invasion by regulating cMyc and E2F2 in HCC-derived HepG2 cell line,[45] and Fascin homologue 1 (FSCN1) in nasopharyngeal carcinoma cell lines.[46] On the other hand, miR-24 acted as an oncogene directly repressing SOX7 (Sex Determining Region Y-Box 7), a putative tumor suppressor,[47] and overexpressed miR-24 led to inhibition of hepatocyte nuclear factor 4α and initiated hepatocellular transformation through an epigenetic positive feedback circuit in the absence of genetic alterations.[48] Tumor suppressor gene (p16)[49] and pro-apoptotic protein FAF1[50] can be negatively regulated by miR-24 in cervical carcinoma, prostate, gastric and HeLa cells. These data strongly suggested the complicated network of miRNA alterations in tumorigenesis that needs further clarification.
Several “tumor common” miRNAs have been extensively studied in HCC, as well as in various other cancers, but have not been recognized for their generalizability as “tumor type non-specific” biomarkers. The over-expression of miR-21 has been commonly observed in HCC tumor compared to adjacent non-tumor tissues, as well as in the circulation of patients with HCC. The overall pooled results from a diagnostic meta-analysis of miR-21 revealed a sensitivity of 74% and a specificity of 78%[51] for HCC classification that is far from ideal for clinical application. Meanwhile, miR-21 was also significantly up-regulated in other cancer types (breast, colorectal, esophageal, gastric, lung, pancreatic and prostate), and the overall predictive sensitivity and specificity were, respectively 76% and 79%,[52] which were similar to HCC. The cluster of miR-182/miR-96/miR-183 located within 2-4 kb at chromosome 7q32 functions as micro-oncogenes in carcinogenesis and the metastatic cascade. Two members (miR-182, miR-183) of this cluster showed frequent up-regulation in HCC.[53] The expression of miR-182 was also consistently increased in 14 other cancer types and miR-183 was up-regulated in 9 others.[53] Down-regulation of miR-139, miR-144, miR-101-2, miR-451 and miR-486 was also reported in various cancer types besides HCC. These observations are biologically plausible because the “tumor common” miRNAs and their target genes participate in general carcinogenic processes and tumorigenic pathways, such as p53, phosphatase and tensin homolog, fibroblast growth factor receptor 3, DNA damage/repair, apoptosis, angiogenesis, cell cycle, phosphoinositide 3-kinase, mitogen-activated protein kinase, TGFβ, NOTCH, and Wnt signaling pathways, etc.[54-56] Therefore, “tumor common” miRNAs aberrantly expressed in various tumors may provide clues to further investigate their common similar underlying mechanisms in tumorigenesis. If verified, the miRNA signatures may be promising targets for precision cancer prevention and therapy. However, these miRNAs may have limited power as diagnostic tools to detect specific cancer type because of their “non-specificity”.
The advantages of the current research include a two-phase study design using a discovery and independent validation sample sets, paired tumor/non-tumor tissues, and unpaired tissues to verify promising miRNAs; simultaneously analyzing miRNA sequencing data in multiple cancer types that allows us to identify “HCC specific” and “tumor common” miRNAs panels. We used the most stringent criteria to select miRNAs for the final data analyses, i.e., RPMM ≥ 10 in at least 90% of samples and P-value < 0.0001 as the significant level to adjust for multiple comparisons. Other studies, such as Wojcicka et al.[57] analyzed miRNAs (GSE63046) passing the criteria of RPMM ≥ 5 in samples with over 50% detectable rate; Zhang et al.[58] excluded miRNAs with missing data exceeding 10% of all subjects but without precluding unreliable sequencing reads less than 10, which may lead to biased results or identify miRNAs with too much missing data, and are unable to be applied in clinical samples.
In interpreting the results, some drawbacks need to be recognized. First, for some miRNAs, the results are not in agreement with previous studies. For example, miR-122 was identified as the most abundant miRNAs in liver tissue previously,[59,60] but is only the 7th in the TCGA data; miR-3591 has been reported as abundant in liver tissue,[60] but is not even detectable in TCGA data. So we may miss a few important candidates due to different detection techniques (RNA-seq, microarrays and RT-qPCR); different approaches and criteria for data processing and analysis, and the heterogeneity of tumor tissue itself. Second, 5 identified “HCC specific” miRNAs were all down-regulated in tumor tissue, which requires more sensitive methods of detection for future clinical application. It is known that RNA-seq has a better sensitivity than RT-qPCR,[61] but the latter is more accurate and usually used for the validation of candidate miRNAs.[62] We also observed that the changes of miRNAs in tumor tissue detected by RT-qPCR were minor compared to those by RNA-seq [Supplementary Table 3]. Even more challenging is to measure these miRNAs in circulation in pre-diagnostic samples, which strongly suggests a dire need for development of more sensitive PCR-based assays that can be used in large population studies.
In conclusion, our study identified 33 miRNAs significantly aberrantly expressed in HCC tumors with over 2-fold changes, and for the first time distinguished 5 of them as having “HCC tumor type specificity”, while another 8 are “tumor common” alterations. We also found several etiology-related miRNA panels, but most overlap with those observed in overall HCC. These findings have promising applications to better understand the common mechanisms underline tumorigenesis and improve precision prevention and therapy for specific cancers by targeting tumor specific miRNAs. Large retrospective and prospective studies to evaluate miRNA changes in circulation and trends during cancer development are warranted.
Acknowledgments
We used public available miRNAs and clinical data from TCGA in this manuscript. Information about TCGA and the investigators and institutions that constitute the TCGA research network can be found at http://cancergenome.nih.gov.
Financial support and sponsorship
This work is supported by NIH grants R01 ES005116, P30 ES009089, P30 CA013696, R03 CA156629. The Cancer Genome Atlas (TCGA) project is supported by the NCI and NHGRI.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
2. Lyra-Gonzalez I, Flores-Fong LE, Gonzalez-Garcia I, Medina-Preciado D, Armendariz-Borunda J. MicroRNAs dysregulation in hepatocellular carcinoma: insights in genomic medicine. World J Hepatol 2015;7:1530-40.
3. Altekruse SF, McGlynn KA, Reichman ME. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol 2009;27:1485-91.
5. El Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 2007;132:2557-76.
6. Cabibbo G, Maida M, Genco C, Antonucci M, Camma C. Causes of and prevention strategies for hepatocellular carcinoma. Semin Oncol 2012;39:374-83.
7. Wogan GN. Aflatoxins as risk factors for hepatocellular carcinoma in humans. Cancer Res 1992;52:s2114-8.
8. Li J, Shi W, Gao Y, Yang B, Jing X, Shan S, Wang Y, Du Z. Analysis of microRNA expression profiles in human hepatitis B virus-related hepatocellular carcinoma. Clin Lab 2013;59:1009-15.
9. Park KU, Seo YS, Lee YH, Park J, Hwang I, Kang KJ, Nam J, Kim SW, Kim JY. Altered microRNA expression profile in hepatitis B virus-related hepatocellular carcinoma. Gene 2015;573:278-84.
10. Diaz G, Melis M, Tice A, Kleiner DE, Mishra L, Zamboni F, Farci P. Identification of microRNAs specifically expressed in hepatitis C virus-associated hepatocellular carcinoma. Int J Cancer 2013;133:816-24.
11. Edge SB, Compton CC. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol 2010;17:1471-4.
12. Chu A, Robertson G, Brooks D, Mungall AJ, Birol I, Coope R, Ma Y, Jones S, Marra MA. Large-scale profiling of microRNAs for The Cancer Genome Atlas. Nucleic Acids Res 2016;44:e3.
13. Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Muertter RN, Muertter RN. NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res 2009;37:D885-90.
14. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 2008;3:1101-8.
15. Eminaga S, Christodoulou DC, Vigneault F, Church GM, Seidman JG. Quantification of microRNA expression with next-generation sequencing. Curr Protoc Mol Biol 2013;Chapter 4:Unit.
16. Rice J, Roberts H, Burton J, Pan J, States V, Rai SN, Galandiuk S. Assay reproducibility in clinical studies of plasma miRNA. PLoS One 2015;10:e0121948.
17. Tibshirani R, Hastie T, Narasimhan B, Chu G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc Natl Acad Sci U S A 2002;99:6567-72.
18. Simon R, Lam A, Li MC, Ngan M, Menenzes S, Zhao Y. Analysis of gene expression data using BRB-ArrayTools. Cancer Inform 2007;3:11-7.
19. Lu TP, Lee CY, Tsai MH, Chiu YC, Hsiao CK, Lai LC, Chuang EY. miRSystem: an integrated system for characterizing enriched functions and pathways of microRNA targets. PLoS One 2012;7:e42390.
20. Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 2009;37:W305-11.
21. Lee L, Wang K, Li G, Xie Z, Wang Y, Xu J, Sun S, Pocalyko D, Bhak J, Kim C, Lee KH, Jang YJ, Yeom YI, Yoo HS, Hwang S. Liverome: a curated database of liver cancer-related gene signatures with self-contained context information. BMC Genomics 2011;12 Suppl 3:S3.
22. Hou W, Bonkovsky HL. Non-coding RNAs in hepatitis C-induced hepatocellular carcinoma: dysregulation and implications for early detection, diagnosis and therapy. World J Gastroenterol 2013;19:7836-45.
23. Pogribny IP, Rusyn I. Role of epigenetic aberrations in the development and progression of human hepatocellular carcinoma. Cancer Lett 2014;342:223-30.
24. Salvi A, Abeni E, Portolani N, Barlati S, De Petro G. Human hepatocellular carcinoma cell-specific miRNAs reveal the differential expression of miR-24 and miR-27a in cirrhotic/non-cirrhotic HCC. Int J Oncol 2013;42:391-402.
25. Li B, Huang P, Qiu J, Liao Y, Hong J, Yuan Y. MicroRNA-130a is down-regulated in hepatocellular carcinoma and associates with poor prognosis. Med Oncol 2014;31:230.
26. Zhang X, Daucher M, Armistead D, Russell R, Kottilil S. MicroRNA expression profiling in HCV-infected human hepatoma cells identifies potential anti-viral targets induced by interferon-alpha. PLoS One 2013;8:e55733.
27. Liu C, Yu H, Zhang Y, Li D, Xing X, Chen L, Zeng X, Xu D, Fan Q, Xiao Y, Chen W, Wang Q. Upregulation of miR-34a-5p antagonizes AFB1-induced genotoxicity in F344 rat liver. Toxicon 2015;106:46-56.
28. Tsang FH, Au SL, Wei L, Fan DN, Lee JM, Wong CC, Ng IO, Wong CM. MicroRNA-142-3p and microRNA-142-5p are downregulated in hepatocellular carcinoma and exhibit synergistic effects on cell motility. Front Med 2015;9:331-43.
29. Wu L, Cai C, Wang X, Liu M, Li X, Tang H. MicroRNA-142-3p, a new regulator of RAC1, suppresses the migration and invasion of hepatocellular carcinoma cells. FEBS Lett 2011;585:1322-30.
30. Chiu LY, Kishnani PS, Chuang TP, Tang CY, Liu CY, Bali D, Koeberl D, Austin S, Boyette K, Weinstein DA, Murphy E, Yao A, Chen YT, Li LH. Identification of differentially expressed microRNAs in human hepatocellular adenoma associated with type I glycogen storage disease: a potential utility as biomarkers. J Gastroenterol 2014;49:1274-84.
31. Du M, Shi D, Yuan L, Li P, Chu H, Qin C, Yin C, Zhang Z, Wang M. Circulating miR-497 and miR-663b in plasma are potential novel biomarkers for bladder cancer. Sci Rep 2015;5:10437.
32. Matamala N, Vargas MT, Gonzalez-Campora R, Mi-ambres R, Arias JI, Menéndez P, Andrés-León E, Gómez-López G, Yanowsky K, Calvete-Candenas J, Inglada-Pérez L, Martínez-Delgado B, Benítez J. Tumor microRNA expression profiling identifies circulating microRNAs for early breast cancer detection. Clin Chem 2015;61:1098-106.
33. Jiang H, Yu WW, Wang LL, Peng Y. miR-130a acts as a potential diagnostic biomarker and promotes gastric cancer migration, invasion and proliferation by targeting RUNX3. Oncol Rep 2015;34:1153-61.
34. Li N, Yang L, Wang H, Yi T, Jia X, Chen C, Xu P. MiR-130a and MiR-374a function as novel regulators of cisplatin resistance in human ovarian cancer A2780 cells. PLoS One 2015;10:e0128886.
35. Liu L, Nie J, Chen L, Dong G, Du X, Wu X, Tang Y, Han W. The oncogenic role of microRNA-130a/301a/454 in human colorectal cancer via targeting Smad4 expression. PLoS One 2013;8:e55532.
36. Acunzo M, Visone R, Romano G, Veronese A, Lovat F, Palmieri D, Bottoni A, Garofalo M, Gasparini P, Condorelli G, Chiariello M, Croce CM. miR-130a targets MET and induces TRAIL-sensitivity in NSCLC by downregulating miR-221 and 222. Oncogene 2012;31:634-42.
37. El-Halawany MS, Ismail HM, Zeeneldin AA, Elfiky A, Tantawy M, Kobaisi MH, Hamed I, Abdel Wahab AH. Investigating the pretreatment miRNA expression patterns of advanced hepatocellular carcinoma patients in association with response to TACE treatment. Biomed Res Int 2015;2015:649750.
38. Liu YX, Long XD, Xi ZF, Ma Y, Huang XY, Yao JG, Wang C, Xing TY, Xia Q. MicroRNA-24 modulates aflatoxin B1-related hepatocellular carcinoma prognosis and tumorigenesis. Biomed Res Int 2014;2014:482926.
39. Meng FL, Wang W, Jia WD. Diagnostic and prognostic significance of serum miR-24-3p in HBV-related hepatocellular carcinoma. Med Oncol 2014;31:177.
40. Oksuz Z, Serin MS, Kaplan E, Dogen A, Tezcan S, Aslan G, Emekdas G, Sezgin O, Altintas E, Tiftik EN. Serum microRNAs; miR-30c-5p, miR-223-3p, miR-302c-3p and miR-17-5p could be used as novel non-invasive biomarkers for HCV-positive cirrhosis and hepatocellular carcinoma. Mol Biol Rep 2015;42:713-20.
41. Lin XJ, Chong Y, Guo ZW, Xie C, Yang XJ, Zhang Q, Li SP, Xiong Y, Yuan Y, Min J, Jia WH, Jie Y, Chen MS, Chen MX, Fang JH, Zeng C, Zhang Y, Guo RP, Wu Y, Lin G, Zheng L, Zhuang SM. A serum microRNA classifier for early detection of hepatocellular carcinoma: a multicentre, retrospective, longitudinal biomarker identification study with a nested case-control study. Lancet Oncol 2015;16:804-15.
42. Chowdhury JB, Shrivastava S, Steele R, Di Bisceglie AM, Ray R, Ray RB. Hepatitis C virus infection modulates expression of interferon stimulatory gene IFITM1 by upregulating miR-130A. J Virol 2012;86:10221-5.
43. Boll K, Reiche K, Kasack K, Mörbt N, Kretzschmar AK, Tomm JM, Verhaegh G, Schalken J, von Bergen M, Horn F, Hackermüller J. MiR-130a, miR-203 and miR-205 jointly repress key oncogenic pathways and are downregulated in prostate carcinoma. Oncogene 2013;32:277-85.
44. Qiu SW, Lin S, Hu D, Feng YM, Tan Y, Peng Y. Interactions of miR-323/miR-326/miR-329 and miR-130a/miR-155/miR-210 as prognostic indicators for clinical outcome of glioblastoma patients. J Transl Med 2013;11:10.
45. Lal A, Navarro F, Maher CA, Maliszewski LE, Yan N, O'Day E, Chowdhury D, Dykxhoorn DM, Tsai P, Hofmann O, Becker KG, Gorospe M, Hide W, Lieberman J. miR-24 inhibits cell proliferation by targeting E2F2, MYC, and other cell-cycle genes via binding to "seedless" 3'UTR microRNA recognition elements. Mol Cell 2009;35:610-25.
46. Li YQ, Lu JH, Bao XM, Wang XF, Wu JH, Hong WQ. MiR-24 functions as a tumor suppressor in nasopharyngeal carcinoma through targeting FSCN1. J Exp Clin Cancer Res 2015;34:130.
47. Ma Y, She XG, Ming YZ, Wan QQ. miR-24 promotes the proliferation and invasion of HCC cells by targeting SOX7. Tumour Biol 2014;35:10731-6.
48. Hatziapostolou M, Polytarchou C, Aggelidou E, Drakaki A, Poultsides GA, Jaeger SA, Ogata H, Karin M, Struhl K, Hadzopoulou-Cladaras M, Iliopoulos D. An HNF4alpha-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell 2011;147:1233-47.
49. Lal A, Kim HH, Abdelmohsen K, Kuwano Y, Pullmann R Jr, Srikantan S, Subrahmanyam R, Martindale JL, Yang X, Ahmed F, Navarro F, Dykxhoorn D, Lieberman J, Gorospe M. p16(INK4a) translation suppressed by miR-24. PLoS One 2008;3:e1864.
50. Qin W, Shi Y, Zhao B, Yao C, Jin L, Ma J, Jin Y. miR-24 regulates apoptosis by targeting the open reading frame (ORF) region of FAF1 in cancer cells. PLoS One 2010;5:e9429.
51. Yan SR, Liu ZJ, Yu S, Bao YX. Investigation of the value of miR-21 in the diagnosis of early stage HCC and its prognosis: a meta-analysis. Genet Mol Res 2015;14:11573-86.
52. Wang Y, Gao X, Wei F, Zhang X, Yu J, Zhao H, Sun Q, Yan F, Yan C, Li H, Ren X. Diagnostic and prognostic value of circulating miR-21 for cancer: a systematic review and meta-analysis. Gene 2014;533:389-97.
53. Zhang QH, Sun HM, Zheng RZ, Li YC, Zhang Q, Cheng P, Tang ZH, Huang F. Meta-analysis of microRNA-183 family expression in human cancer studies comparing cancer tissues with noncancerous tissues. Gene 2013;527:26-32.
54. Zhang HD, Jiang LH, Sun DW, Li J, Tang JH. MiR-139-5p: promising biomarker for cancer. Tumour Biol 2015;36:1355-65.
55. Mizuguchi Y, Takizawa T, Yoshida H, Uchida E. Dysregulated microRNAs in progression of hepatocellular carcinoma: a systematic review. Hepatol Res 2015; doi: 10.1111/hepr.12606.
56. He Y, Lin J, Kong D, Huang M, Xu C, Kim TK, Etheridge A, Luo Y, Ding Y, Wang K. Current state of circulating microRNAs as cancer biomarkers. Clin Chem 2015;61:1138-55.
57. Wojcicka A, Swierniak M, Kornasiewicz O, Gierlikowski W, Maciag M, Kolanowska M, Kotlarek M, Gornicka B, Koperski L, Niewinski G, Krawczyk M, Jazdzewski K. Next generation sequencing reveals microRNA isoforms in liver cirrhosis and hepatocellular carcinoma. Int J Biochem Cell Biol 2014;53:208-17.
58. Zhang J, Chong CC, Chen GG, Lai PB. A seven-microRNA expression signature predicts survival in hepatocellular carcinoma. PLoS One 2015;10:e0128628.
59. Hou J, Lin L, Zhou W, Wang Z, Ding G, Dong Q, Qin L, Wu X, Zheng Y, Yang Y, Tian W, Zhang Q, Wang C, Zhang Q, Zhuang SM, Zheng L, Liang A, Tao W, Cao X. Identification of miRNomes in human liver and hepatocellular carcinoma reveals miR-199a/b-3p as therapeutic target for hepatocellular carcinoma. Cancer Cell 2011;19:232-43.
60. Lopez JP, Diallo A, Cruceanu C, Fiori LM, Laboissiere S, Guillet I, Fontaine J, Ragoussis J, Benes V, Turecki G, Ernst C. Biomarker discovery: quantification of microRNAs and other small non-coding RNAs using next generation sequencing. BMC Med Genomics 2015;8:35.
61. Knutsen E, Fiskaa T, Ursvik A, Jørgensen TE, Perander M, Lund E, Seternes OM, Johansen SD, Andreassen M. Performance comparison of digital microRNA profiling technologies applied on human breast cancer cell lines. PLoS One 2013;8:e75813.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Shen J, Siegel AB, Remotti H, Wang Q, Santella RM. Identifying microRNA panels specifically associated with hepatocellular carcinoma and its different etiologies. Hepatoma Res 2016;2:151-62. http://dx.doi.org/10.20517/2394-5079.2015.66
AMA Style
Shen J, Siegel AB, Remotti H, Wang Q, Santella RM. Identifying microRNA panels specifically associated with hepatocellular carcinoma and its different etiologies. Hepatoma Research. 2016; 2: 151-62. http://dx.doi.org/10.20517/2394-5079.2015.66
Chicago/Turabian Style
Shen, Jing, Abby B. Siegel, Helen Remotti, Qiao Wang, Regina M. Santella. 2016. "Identifying microRNA panels specifically associated with hepatocellular carcinoma and its different etiologies" Hepatoma Research. 2: 151-62. http://dx.doi.org/10.20517/2394-5079.2015.66
ACS Style
Shen, J.; Siegel AB.; Remotti H.; Wang Q.; Santella RM. Identifying microRNA panels specifically associated with hepatocellular carcinoma and its different etiologies. Hepatoma. Res. 2016, 2, 151-62. http://dx.doi.org/10.20517/2394-5079.2015.66
About This Article
Copyright

Author Biographies

Data & Comments
Data


 Cite This Article 3 clicks
Cite This Article 3 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.