
Aggressive primary hepatic histiocytic sarcoma: case report and literature review

Abstract
Histiocytic sarcoma is an uncommon non-Langerhans histiocyte disorder of mature tissue histiocytes. The authors presented an example of this rare tumor in a 14-year-old girl who presented with left upper quadrant pain, loss of appetite, and weight loss. A large 18 cm × 10 cm heterogeneous solid and cystic enhancing mass was found in the left lobe of the liver. Based on the histomorphology and positivity for histiocyte-specific markers in a needle biopsy, a diagnosis of histiocytic sarcoma was made. Chemotherapy was initiated, but the tumor did not respond well, and she died about 7 weeks following initial diagnosis with multi-organ failure. At autopsy, the tumor showed extensive necrosis, with no evidence of metastatic spread. In conclusion, the diagnosis of histiocytic sarcoma is challenging, and requires a high index of suspicion, with an appropriate panel of confirmatory immunohistochemical stains. Recognition of this rare tumor is important because of its poor response to chemotherapy and high mortality.
Keywords
Introduction
Histiocytic sarcoma (HS) is an extremely uncommon neoplasm with morphologic and immunophenotypic characteristics of mature histiocytes. It is believed that HS originates from monocytes/macrophages, which are critical in the processing and presentation of antigens to T cells or B cells.[1]
Before the application of immunohistochemical techniques and the availability of molecular genetic tools, HS was occasionally diagnosed. It is now generally recognized however that the majority of the previously diagnosed HS cases are actually examples of non-Hodgkin lymphomas, most of which are diffuse large B-cell lymphoma and anaplastic large cell lymphoma.[2]
HS is an extremely aggressive neoplasm and responds poorly to regular therapy; most patients die of progressive disease, in part related to the high clinical stage (stage III/IV) at presentation in the majority of patients.[3] The most common affected site of HS is in lymph nodes, followed by different extranodal locations such as the gastrointestinal tract, spleen, soft tissue and skin.
Herein, we describe a child with an aggressive primary hepatic histiocytic sarcoma who responded poorly to multiple rounds of chemotherapy and passed away 7 weeks after initial diagnosis.
To the best of our knowledge, only three primary hepatic HS cases have been previously reported in the English literature.[4-6] Recognition of this type of rare tumor is important, due to its limited responsiveness to conventional chemotherapy and high mortality.
Case report
A 14-year-old previously healthy young girl presented with left upper quadrant pain, loss of appetite and weight loss for one month. An abdominal ultrasound showed a large heterogeneous solid and cystic appearing mass in the mid abdomen, measuring 18.3 cm × 10 cm, arising from the liver with associated mass effect and surrounding increased vascularity. Computed tomography (CT) scan of the abdomen and pelvis and magnetic resonance imaging revealed similar findings, with marked regional mass effect, including on the hepatic inferior vena cava and stomach, with extracapsular extension into the left upper quadrant [Figure 1].
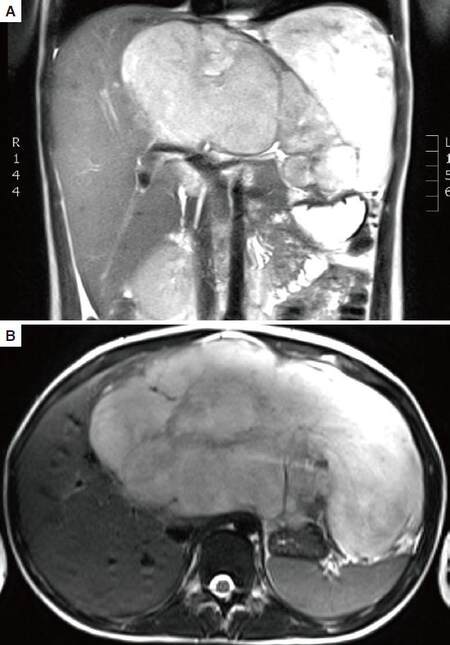
Figure 1. Abdominal magnetic resonance imaging (A: coronal; B: axial) revealed an 18.3 cm large heterogeneous enhancing mass with cystic components arising from the left hepatic lobe with associated mass effect
Laparoscopic liver needle biopsy was performed and showed a tumor of primarily epithelioid cells with areas of spindling also present. The lesional cells contained abundant eosinophilic cytoplasm and nuclei with open vesicular chromatin and prominent nucleoli. Prominent nuclear pleomorphism and elevated mitotic index were also identified [Figure 2]. A subsequent perihepatic fine needle aspiration biopsy and cell block of the tumor similarly showed a poorly differentiated neoplasm with epithelioid and spindle cell features in a background of fibrin and blood [Figure 3].
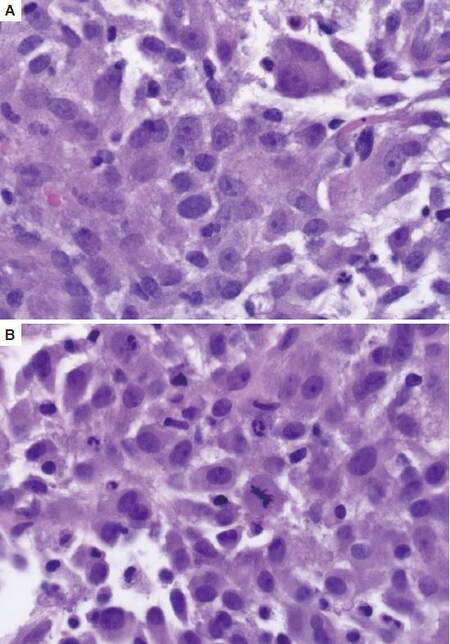
Figure 2. (A) Tumor cells comprising of primarily epithelioid cells with abundant eosinophilic cytoplasm, pleomorphic nuclei with open vesicular chromatin and prominent nucleoli; (B) Mitotic figures were easily found in the tumor. HE, ×40
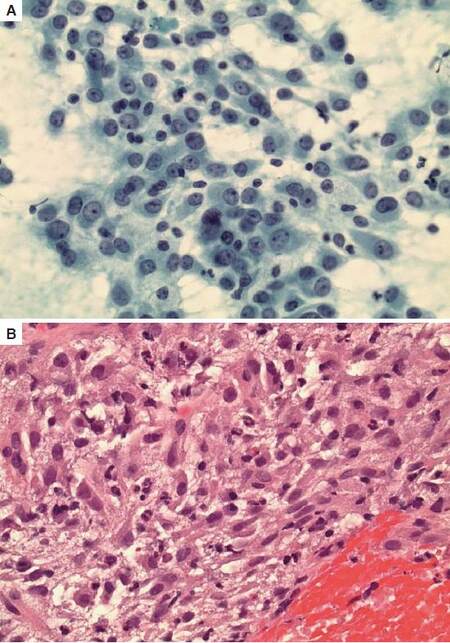
Figure 3. (A) Fine needle aspiration showed pleomorphic nuclei with prominent nucleoli (Papanicolaou, ×40); (B) Cell block of the tumor showed a poorly differentiated neoplasm with epithelioid and spindle cell features in a background of fibrin and blood (HE, ×40)
Immunohistochemical studies showed no staining with markers for epithelial, lymphomatous, germ cell, rhabdomyosarcoma, melanoma, gastrointestinal stromal or neuroendocrine tumors, or for Langerhans cell histiocytosis, including CK cocktail, CAM5.2, EMA, CD15, PAX-5, CD45, CD43, CD3, CD5, ALK1, Factor XIIIa, CD31, CD23, CD35, SALL-4, CD117, desmin, myogenin, SMA, melanoma cocktail, S-100, vimentin, CD34, chromogranin, synaptophysin, CD56, inhibin, and CD1a. Many lesional cells were strongly positive for CD163, with less distinct staining for CD68 [Figure 4]. Her bone marrow biopsy showed a mildly hypercellular marrow for age, with trilineage hematopoiesis, a left shift of the granulocytes, a moderate erythroid hyperplasia, and plasmacytosis. No morphologic or immunophenotypic evidence of malignancy was identified. A diagnosis of HS was made based on the combination of morphology and immunophenotype, and this diagnosis was supported by outside expert consultation.
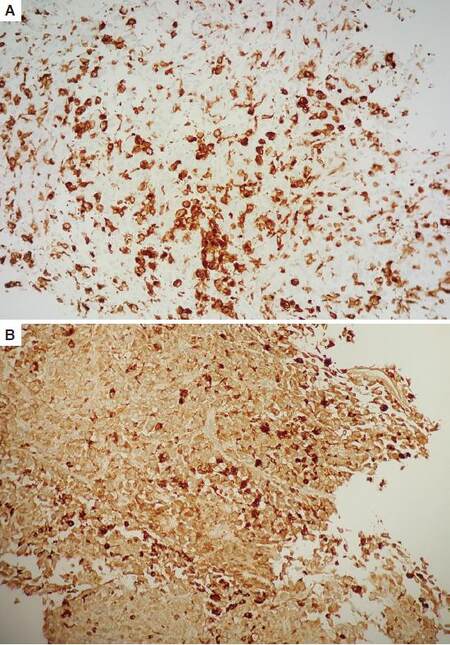
Figure 4. (A) CD163 immunostain; (B) CD68 immunostain. Tumor cells are strongly positive for CD163, with less distinct staining for CD68 (×10)
Clinical course
Because of the large size of this tumor and the non-availability of liver donor at that time, complete tumor resection and liver transplantation were not performed. The patient received two rounds of chemotherapy (1st round ifosfamide and doxorubicin, and 2nd round thalidomide) and intensive supportive measures. However, she gradually developed worsening multi-organ failure requiring mechanical ventilation and hemodialysis. Positron emission tomography CT scan suggested development of possible metastases at multiple sites. She developed abdominal distension and multi-organ failure, and passed away 7 weeks following initial diagnosis.
Autopsy findings
At autopsy, there was single large (20 cm × 17 cm × 9 cm) tumor arising from the left lobe of the liver, with extensive necrosis. No evidence of metastatic tumor was identified at any of the other suspected sites. Microscopic examination of the tumor was complicated by moderate autolysis with extensive necrosis, but otherwise showed similar morphology to the original biopsies.
Discussion
HS is a malignant proliferation of cells showing morphologic and immunophenotypic features of mature tissue histiocytes.[7] In 1970, "histiocytic sarcoma" was introduced for the first time to describe a collection of malignant tumor cells with histologic characteristics of large macrophages with abundant eosinophilic cytoplasm.[8] Now HS is defined as a malignant proliferation of cells showing morphologic and immunophenotypic characteristics of mature tissue histiocytes. Immunohistochemical studies are critical for the correct diagnosis of HS because it doesn’t have definitive morphologic features. As a matter of fact, many cases that were diagnosed as malignant histiocytosis and histiocytic medullary reticulosis in the past have been shown to be different subtypes of non-Hodgkin lymphoma.[9]
HS is extremely rare and accounts for less than 1% of all hematolymphoid neoplasms. It can occur over a wide range of ages (0.5-89 years, median age 46 years), showing bimodal age distribution with a small peak at 0-29 years and a larger peak at 50-69 years. HS is slightly more common in males than females.[1]
Although the etiology of HS remains unknown, some cases have occurred in patients with mediastinal germ cell tumor, raising the consideration that HS may arise from pluripotential germ cells. Associations between HS and follicular lymphoma, myelodysplastic syndrome, and acute lymphoblastic leukemia have also been made. Moreover, a study has reported trans-differentiation in patients with HS and follicular lymphoma and reported the presence of t(14;18) and immunoglobulin heavy chain (IGH) gene rearrangements in all of the patients, suggesting a common clonal origin of follicular lymphoma and HS. Another study reported that 2 patients with HS had a clonal immunoglobulin rearrangement, suggesting a clonal evolution of HS from chronic lymphocytic leukemia/small lymphocytic leukemia. Further research is needed to confirm these findings.[10]
Clinically, HS has been found to involve lymph nodes, skin, and at many extranodal locations, especially the gastrointestinal tract, often with the presentation of clinically advanced disease and aggressive clinical course. Cases arising primarily at extranodal sites often appear to go unsuspected and unrecognized.[2] Both localized and disseminated forms of HS exist. Systemic symptoms are relatively common and include fever, fatigue, night sweats, weight loss and weakness. Additionally, depending on the sites of involvement, HS can also present as skin rash, intestinal obstruction, hepatosplenomegaly, lytic bone lesions, and pancytopenia.[1] Although the liver is the most common site of murine HS,[11] human primary hepatic HS is very rare. To the best of our knowledge, the present case is the fourth HS primarily arising from the liver. The clinical features of these four reported cases are presented in Table 1.
Clinical features of primary hepatic histiocytic sarcoma cases
| Case | Age/gender | Initial presentation | Tumor location/size | Clinical course | Ref. |
|---|---|---|---|---|---|
| 1 | 55 years/male | Right shoulder and right upper quadrant pain for about 3 months | Right lobe 15 cm × 11 cm × 9 cm | Right hepatic lobectomy and partial diaphragmectomy | [9] |
| 2 | 68 years/male | Cutaneous langerhans cell histiocytosis and rosai-dorfman disease and splenic marginal zone lymphoma. Patient was stable for a few years; then suddenly developed remarkable enlargement of spleen and liver without lymphadenopathy or skin lesions | Remarkable enlargement of spleen (24 cm, compared to 14 cm from 2 years earlier) and liver (size not available) | Died 24 h after presentation with sudden hepatosplenomegaly, despite treatment with systemic chemotherapy combined with prednisone | [6] |
| 3 | Unknown | Unknown | Liver, not further specified | Unknown | [8] |
| 4 | 14 years/female | Left upper quadrant pain, anorexia and weight loss for 1 month | Left lobe 20 cm × 17 cm × 9 cm | Died 7 weeks following initial diagnosis despite aggressive systemic chemotherapy without tumor resection | Current study |
Morphologically, HS tumor consists of diffuse sheets of medium to large epithelioid cells with abundant, pale eosinophilic or foamy cytoplasm. The nuclei are generally irregular, vesicular with prominent nucleoli; binucleated or large multinucleated forms are commonly seen. Mitotic activity is usually high and cellular pleomorphism can occasionally be seen. Necrosis is common and an admixed inflammatory infiltrate of small lymphocytes and neutrophils may be seen. In some cases, focal areas of spindle cell morphology may be found.[12]
In terms of immunohistochemical studies, HS tumor cells are typically positive for one or more histiocytic markers, such as CD163, CD68 and lysozyme, with typical absence of markers for lymphocytes, Langerhans cells, follicular dendritic cells, epithelial cells, melanocytes and myeloid cells. The Ki-67 index is variable. S-100 and CD1a can occasionally be positive but usually only with weak and patchy staining. However, none of the antibodies are specific for histiocytic differentiation; therefore, it is important to evaluate with a panel of antibodies.[1] BRAF exon 15 mutational analysis shows that 62% of HS cases have BRAF V600E mutations. Clonal antigen receptor gene rearrangement for T-cell receptor gamma, T-cell receptor beta, and IGH genes may be seen and do not exclude the diagnosis of HS. However, when HS is associated with lymphoma, identical clonal gene rearrangements may be present.[13]
HS most commonly presents at an advanced clinical stage, with poor response to chemotherapy and a high mortality rate. Although some patients may respond to chemotherapy with or without radiotherapy, the majority of the patients die of progressive disease within two years. Important prognostic factors include stage at presentation and tumor size. There are no accepted staging or treatment guidelines due to the rarity of the disease.[1]
In conclusion, we present a rare case of primary hepatic HS. The diagnosis is challenging, and requires a high index of suspicion. The diagnosis should be based on a combination of compatible histomorphology and positivity for histiocyte-specific markers, and also requires exclusion of more common neoplasms by extensive immunophenotypic studies.
Acknowledgments
The authors thank Mary Dennis and Maria Rabina (MHS PA) for assistance in specimen grossing, and Michael Mitchell for assistance in autopsy.
Financial support and sponsorship
None.
Conflicts of interest
There are no conflicts of interest.
Patient consent
The patient's parent(s) signed consents and documents are available for review upon request.
Ethics approval
Case report is waived and does not require further approval by Institutional Review Board at Loma Linda University Medical Center.
REFERENCES
1. Takahashi E, Nakamura S. Histiocytic sarcoma: an updated literature review based on the 2008 WHO classification. J Clin Exp Hematop 2013;53:1-8.
2. Hornick JL, Jaffe ES, Fletcher CD. Extranodal histi ocytic sarcoma: clinicopathologic analysis of 14 cases of a rare epithelioid malignancy. Am J Surg Pathol 2004;28:1133-44.
3. Mounajjed T, Chandan VS, Torbenson MS, editors. Surgical Pathology of Liver Tumors. Springer; 2015. pp. 360-1.
4. Llamas-Velasco M, Cannata J, Dominguez I, García-Noblejas A, Aragües M, Fraga J, Arranz R. Coexistence of Langerhans cell histiocytosis, Rosai-Dorfman disease and splenic lymphoma with fatal outcome after rapid development of histiocytic sarcoma of the liver. J Cutan Pathol 2012;39:1125-30.
5. Rubtsova NA, Sidorov DV, Petrov LO, Zhukov NV, Vernyuk MA, Pavlova OA. Diagnosis and treatment of primary sarcoma of the liver (case review). Radiology 2014;1:43.
6. Chun HJ, Kee KH, Suh CH, Lim SC, Song HS. Primary malignant lymphoma of true histiocytic origin of the liver: histiocytic sarcoma, kupffer cell sarcoma: a case report with immunohistochemical and ultrastructural studies. Korean J Pathol 1989;23:165-80.
7. Swerdllow SH, Campo E, Harris NL. WHO classification of tumours of haematopoietic and lymphoid tissues. France: IARC Press; 2008. pp. 356-7.
8. Mathé G, Gerard-Marchant R, Texier JL, Schlumberger JR, Berumen L, Paintrand M. The two varieties of lymphoid tissue "reticulosarcomas", histiocytic and histioblastic types. Br J Cancer 1970;24:687-95.
9. Pakravan A, Bhatia R, Oshima K, Chen G, Fesler M, Prather C, Taylor JR. Histiocytic sarcoma: the first reported case of primary esophageal involvement. Am J Gastroenterol 2014;109:291-2.
10. Dalia S, Shao H, Sagatys E, Cualing H, Sokol L. Dendritic cell and histiocytic neoplasms: biology, diagnosis, and treatment. Cancer Control 2014;21:290-300.
11. Ohnishi K, Tanaka S, Oghiso Y, Takeya M. Immunohistochemical detection of possible cellular origin of hepatic histiocytic sarcoma in mice. J Clin Exp Hematop 2012;52:171-7.
12. Lindberg MR. Diagnostic Pathology: Soft Tissue Tumors. Elsevier Health Sciences; 2015. pp. 310-1.
13. Vasef MA, Auerbach A. Diagnostic Pathology: Molecular Oncology. Elsevier Health Sciences; 2016. pp. 2-4.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Yang G, Deisch J, Qin H, Zuppan C, Raza AS. Aggressive primary hepatic histiocytic sarcoma: case report and literature review. Hepatoma Res 2016;2:300-4. http://dx.doi.org/10.20517/2394-5079.2016.10
AMA Style
Yang G, Deisch J, Qin H, Zuppan C, Raza AS. Aggressive primary hepatic histiocytic sarcoma: case report and literature review. Hepatoma Research. 2016; 2: 300-4. http://dx.doi.org/10.20517/2394-5079.2016.10
Chicago/Turabian Style
Yang, Guang, Jeremy Deisch, Haixia Qin, Craig Zuppan, Anwar S. Raza. 2016. "Aggressive primary hepatic histiocytic sarcoma: case report and literature review" Hepatoma Research. 2: 300-4. http://dx.doi.org/10.20517/2394-5079.2016.10
ACS Style
Yang, G.; Deisch J.; Qin H.; Zuppan C.; Raza AS. Aggressive primary hepatic histiocytic sarcoma: case report and literature review. Hepatoma. Res. 2016, 2, 300-4. http://dx.doi.org/10.20517/2394-5079.2016.10
About This Article
Copyright

Data & Comments
Data


 Cite This Article 1 clicks
Cite This Article 1 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.