
Function of myeloid cell leukaemia-1 and its regulative relations with hepatocellular carcinoma


Abstract
Hepatocellular carcinoma (HCC) remains a challenging disease with a high recurrence rate after surgery and there is an imminent need to identify new treatments. Currently, adjuvant therapy like chemotherapeutics arises to counteract the malignant trait escaping from apoptosis of tumors induced by overexpressed anti-apoptotic factors in HCC. Myeloid cell leukaemia-1 (Mcl-1) as an anti-apoptotic member of Bcl-2 is highly expressed in diverse human cancers, which contributes to cancer cell survival and the resistance to diverse chemotherapeutic agents. It is confirmed that Mcl-1 protein expression is quite enhanced in human HCC tissue compared to adjacent non-tumor tissue. Correspondingly, forced Mcl-1 down-regulation leads to prominent apoptosis of HCC cells and a sensitization towards chemotherapeutic drug-induced apoptosis, which indicates Mcl-1 is indeed a crucial regulatory factor of HCC. Hence, this review highlights the function of Mcl-1 on HCC progression, how it is regulated in HCC and the recent anti-hepatoma drug research and development down-regulation of Mcl-1 or targeting on Mcl-1. Meanwhile, the authors discuss Mcl-1 as an essential regulatory factor in HCC can be designed as target for drugs to improve the survival of HCC patients.
Keywords
Introduction
Hepatocellular carcinoma (HCC), being the sixth most common cancer worldwide, represents the second most common cause of cancer-related mortality in the world[1] and the incidence and mortality rates continue to rise all over the world. The main reasons may be late diagnosis and poor treatment options. To date, surgical resection, local treatment and liver transplantation can only cure a small number of patients whose disease were at early stage, whereas the majority of patients with advanced disease undergone the torture of illness and were not suitable to receive surgery due to various reasons.[2,3] Although chemotherapy is known as a vital management for advanced HCC, inherent resistance to chemotherapeutics by HCC cells makes it hard to have a good effect on the disease. Therefore, identification of new drugs targeting different signaling pathways is urgently needed to improve the survival of HCC patients.[4] This review summarizes the current advances in the relationship between HCC treatment and anti-apoptotic molecule Mcl-1 and suggests that Mcl-1 is a potential target in abolishing the HCC cells’ malignant proliferation.
Bcl-2 is a well established family of proteins and has a significant impact on mitochondrial integrity by influencing the permeability of the mitochondrial membrane. Bcl-2 is localized to the outer membrane of mitochondria, where it plays a part by regulating the progression of apoptosis. According to the structures and the functional contribution, Bcl-2 family members can be divided into two subfamilies: pro-apoptotic members and anti-apoptotic members.[5] And it is the balance in activity between the two opposing groups which determines a cell’s progression towards apoptosis.
Mcl-1 as an antiapoptotic Bcl-2 family protein, is playing a pivotal role in the intrinsic apoptosis pathway and mitotic regulators.[6] As reported, Mcl-1 expresses extensively in the normal tissue of human and its overexpression is observed in many types of human tumors. In addition, Mcl-1 expression involves in disease grade and survival in human malignancies e.g. in patients with multiple myeloma or B-cell non-Hodgkin’s lymphoma.[7,8] It is also one of the pervasive recognized anti-apoptosis factor in HCC and mainly participate in maintenance of mitochondrial membrane stability and suppresses cytochrome c release from mitochondria to promote cell survival and inhibit cell apoptosis.[9] In addition, Mcl-1, serves as one of the important antiapoptotic factors in HCC, is involved in the development and progression of HCC. According to a research made by Sieghart et al.,[10] there were 51% liver tumor tissue appeared highly expression of Mcl-1 in 149 HCC patients, while the adjacent normal liver tissue presented a lower expression, which indicates the overexpression of Mcl-1 is one of the characteristics of specific changes of tumor. Additionally, silencing Mcl-1 gene gives rise to apoptosis of tumor cells with no effect to biological character in normal hepatocytes.[11] Hence, it is indeed escapable and essential to discuss the relationship between Mcl-1 and HCC progression.
Structural and functional properties of Mcl-1
Structure
Mcl-1, one of the antiapoptotic members of the Bcl-2 family protein, was first identified by Kozopas et al.[12] from a human myeloid leukemia cell line in 1993. The human locus of Mcl-1 gene is on chromosome 1q21. With 6502 bp full-length gene, Mcl-1 coding region comprises 3 exons and 2 introns. Bae et al.[13] verified that alternative splicing occurred in the transcription of Mcl-1 and eventually generated 2 different transcript variants. The one including 3 exons encodes Mcl-1L isoform while the other lack of exon 2 encodes Mcl-1S isoform. Sequence analysis revealed that Mcl-1L contains 350 residues which is larger than Bcl-2 (237 residues) and Bcl like protein X (Bcl-xl) (233 residues) and has 3 homo domains BH1, BH2, BH3 and C-terminal transmembrane (TM) domains but lack the N-terminal BH4 domain compared to Bcl-2 and Bcl-xl. The TM domain could anchor Mcl-1L to the outer mitochondrial membrane (OMM).[14] By contrast, Mcl-1S comprises 271 residues and retains only BH3 domains just like other BH3-only members of Bcl-2 family and is primarily localized to the cytosol. Surprisingly, Mcl-1L inhibits apoptosis while Mcl-1S exhibits an opposite role and promotes apoptosis.[13,15] Different from other proteins of Bcl-2 family, the N-terminal region of Mcl-1 (Mcl-1L will be simply called as Mcl-1 hereafter), affecting Mcl-1’s function and localization, is larger than that of other Bcl-2 family members which contains PEST sequences rich in proline (P), gluatamic acid (E), serine (S), and threonine (T). As characteristic sequences of Mcl-1, the PEST regions are rich in putative regulatory motifs that have been shown to target proteins for degradation, which are thought to be as the main reasons of the short half-life of Mcl-1 protein.[14,16] There are also multiple phosphorylation sites in Mcl-1 PEST region, and it is likely that multiple proteins resulting in different fates of Mcl-1 mediate the phosphorylation of these sites. Moreover, with a surface-exposed hydrophobic groove formed by BH1, BH2, and BH3, Mcl-1 can integrate with other pro-apoptotic protein containing BH3-domain to impede apoptosis [Figure 1].[17]

Figure 1. The structure of myeloid cell leukaemia-1 (Mcl-1) protein sequence. A schematic of the wild-type Mcl-1 protein, highlighting the relative location of functional domains of Mcl-1. PEST: proline, glutamicacid, serine, and threonine domain; BH1, 3, 2: Bcl-2 homology domains 1, 3, 2; TM: transmembrane domain
Function
Mainly, Mcl-1 protein is located in OMM, which enables Mcl-1 to interact with other proteins to play a part in anti-apoptosis. Immunoblot analysis revealed that both Mcl-1’s C-terminal and N-terminal domains are necessary for its mitochondrial localization. There is a mitochondrial targeting sequence at Mcl-1’s amino-terminus which anchors Mcl-1 at outer membrane or matrix. Particularly, the anti-apoptotic activities of Mcl-1 require outer membrane-localized Mcl-1.[18] Besides, the first 79 amino acids of Mcl-1 regulate its subcellular localization and overexpression of the N terminus of Mcl-1 recruit more Mcl-1 at mitochondria and as a result, promote cell survival.[19] Furthermore, Mcl-1 with an internal domain containing an EELD motif (at residue 124-127) interacts with the mitochondrial import receptor Tom70, which facilitates Mcl-1’s import onto mitochondrial membrane.[20] Moreover, the C-terminal transmembrane domain of approximately 20 amino acids is required for targeting Mcl-1 to mitochondria.[21]
Normally, in order to maintain the inhibition of the pro-apoptotic proteins Bcl-2 homologous antagonist killer (Bak), Mcl-1 often prevents Bak from forming dimer with Bcl-2-associated protein X (Bax) via combination with it. At the same time, Mcl-1 binds to Bax to make sure sequestering Bak and Bax, and then blocks forming pores in the mitochondrial membrane caused by conformational change and homologous oligomerization, and eventually stops the release of cytochrome c into the cytoplasm, which means blocking the subsequent caspase cascade reaction of apoptosis. In addition, Mcl-1 also binds and sequesters BH3 only proteins which act to induce the polymerisation of Bak and Bax to play its antiapoptosis role effectively [Figure 2].[22] As for the function of Mcl-1’s different isoforms induced by alternative splicing, Mcl-1 and Mcl-1S are capable of forming heterodimers and thus neutralize either the pro-apoptotic function of Mcl-1S or the anti-apoptotic function of Mcl-1. Owing to alternative splicing mechanisms and interactions of the resulting Mcl-1 and Mcl-1S proteins, the fate of cells expressing the Mcl-1 gene may be closely related to the ratio of Mcl-1/Mcl-1S.[23] It is noteworthy that Mcl-1 plays the leading role in the regulation of apoptosis induced by Mcl-1/Mcl-1S and is expressed at higher levels than Mcl-1S.[13,24,25] In cancer, Mcl-1S is expressed at much lower levels than Mcl-1 that it was even hardly undetectable.[26] Some cancer cells such as human lung cancer cell lines A549, Chinese hamster ovary cells and multiple myeloma MOLP-8 cells show high level of Mcl-1S.[13,15,27] Hence, this review mainly discusses Mcl-1.
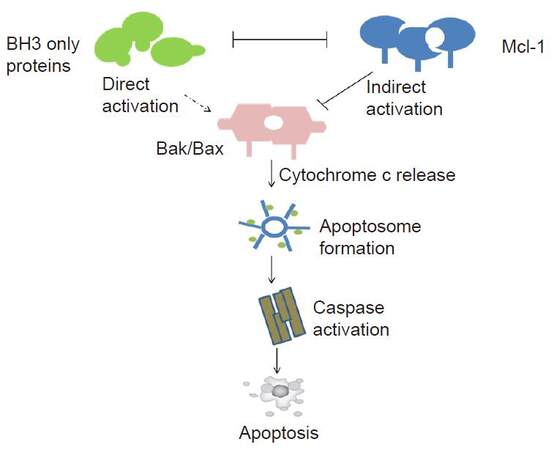
Figure 2. Myeloid cell leukaemia-1 (Mcl-1) regulates initiation of apoptosis through interaction with other Bcl-1 family members. Under normal circumstances, Mcl-1 prevents the activation of Bax and Bak to protect mitochondrial outer membrane integrity and cell survival. Under conditions of stress, the BH3 only proteins are activated and induce apoptosis either by releasing Bak/Bax from Mcl-1 or by BH3 only proteins binding to Mcl-1 directly. Bak/Bax form pores on mitochondrial outer membrane and cytochrome C is relieved into the cytoplasm. In the cytoplasm, cytochrome C activates a family of cysteine proteases named caspases which subsequently induce cell apoptosis
Paradoxically, it is possible that Mcl-1 also plays an important role in delaying cell cycle progression for the existence of Mcl-1 in nucleus have been reported as well.[28] The first 79 amino acids of Mcl-1 promotes its association with mitochondria, the N terminus of Mcl-1 also plays a regulatory role in regulating nuclear (anti-proliferative) functions of Mcl-1 and has an antagonistic effect on proliferation. There seems to be a balance between anti-apoptotic and anti-proliferative functions of Mcl-1 regulated by the N terminus of Mcl-1.[19] In addition to antiapoptosis, Mcl-1 is capable of interacting with proliferating-cell nuclear antigen (PCNA)[29] and cyclin depndent kinase 1 (CDK1),[4] which may inhibit cell cycle progression.[29,30] On the basis of co-immunoprecipitation experiments, Jamil et al.[30] showed that endogenous Mcl-1 interacted with CDK1. The interaction involved a truncated form of Mcl-1, which was termed snMcl-1 as a result of proteolysis at the C-terminus that regulated cell-cycle progression by an inhibitory effect on CDK1 activity. The snMcl-1 was presented during S and G2 phases. The authors proposed that the Mcl-1-CDK1 interaction associated with a protein containing a nuclear localization signal that mediated rapid translocation to the nucleus.[30] Mcl-1 can also regulate the S-phase of the cell cycle through interaction with PCNA, and such interaction may be through Mcl-1’s binding to PCNA.[29] Nonetheless, the binding between PCNA and the Mcl-1 can not be detected in solution studied by NMR, which suggests that the interaction occurs very weakly, or with other unidentified factors in cells.[31] Of note, the interaction with PCNA represses cell cycle progression, but it is not related to Mcl-1’s anti-apoptotic activity.[29] And it’s not clear that whether such two kinds of interaction are mechanistically linked.
Mcl-1 is highly expressed in a variety of human hematopoietic, lymphoid cancers and solid tumors including leukemia,[32,33] lymphoma,[8] cervical carcinoma,[34] HCC,[10,35] breast carcinoma,[36] lung cancer[37] and multiple myeloma.[7,38,39] In addition, its expression is often implicated in the chemotherapeutic resistance and relapse of certain malignancies. For instance, it is crucial for Mcl-1 to survive human myeloma cells in vitro and it has been showed that Mcl-1 is overexpressed in vivo in multiple myeloma, which seems to be related to relapse and shorter survival.[7] Expression of Mcl-1 was also bound up with high tumor grade and reduced survival of patient in human breast cancer samples.[40] Immunohistochemistry and western blotting analysis showed that Mcl-1 was overexpressed in cervical cancer tissue in comparison with normal tissue and the author confirmed Mcl-1 expression was positively correlated with poor prognosis.[34] As for acute myeloid leukemia, Mcl-1 served as a critical molecule to develop and maintain malignant tumor.[41,42] Moreover, Campbell et al.[43] reported that elevated Mcl-1 promotes Myc-induced lymphomagenesis and enhances drug resistance. And also, in human HCC, it has been concluded that Mcl-1 expression was prominently enhanced in diseased tissue as well as in various HCC cell lines.[10,35] On the contrary, in mice lacking the anti-apoptotic protein Mcl-1 specifically in hepatocytes not only increased hepatocyte apoptosis, but also resulted in hepatocarcinogenesis, which is related to compensatory hyper-proliferation induced by Mcl-1 deficiency.[44] Besides, another mouse model indicates that Mcl-1 is stabilized by interleukin (IL)-6 and obesity and thus apoptosis of damaged hepatocytes was inhibited, which eventually promoted HCC progression.[45]
Regulative relations with HCC
Combining unrestrained cell proliferation and damaged apoptosis was found as a main feature of tumor. And as mentioned before, the anti-apoptotic member Mcl-1 was overexpressed in HCC endowing tumor cells with ability to escape from programmed cell death. Consequently, it is of great necessary to make clear the regulation and execution of apoptosis in HCC so that people can find a new way to confront malignant tumor. The regulative relation between Mcl-1 and HCC is listed in Table 1.
Overview of regulation of Mcl-1 in HCC
| Transcriptional regulation | Translational regulation | Post-translational regulation | Interacting proteins | ||
|---|---|---|---|---|---|
| Phosphorylation sites | Kinases | ||||
| Cytokines | STATs, IL-3, HIF-1 | mir-29b | Thr92 | ERK(-) | Mule |
| Thr163 | ERK(-), JNK(+) | CDK | |||
| Signaling ways | PI3K/AKT, P38/MAPK, | mTORC1 | Ser121, Thr163 | JNK(-) | PCNA |
| P53, Wnt/β-actein, Notch | Ser155, Ser159 | GSK3(+) | TCTP | ||
Transcriptional regulation
Mcl-1 can be regulated at transcriptional level by a variety of cytokines including signal transducers and activators of transcription (STAT), cAMP-response element binding protein (CREB), purine-rich nucleic acid binding protein 1 (PU.1), and hypoxia-inducible factor-1 (HIF-1), etc. The STATs, a family of transcription factors, has been shown to bind to Mcl-1 promoter. Al Zaid Siddiquee and Turkson[46] reported that constitutively activated STAT3 participate in oncogenesis of the liver through up-regulating STAT3-targeted genes encoding apoptosis inhibitors including Mcl-1 and subsequently inhibiting pro-apoptotic molecules such as Bax, Bad, and Bid. Additionally, sorafenib was affirmed for its efficacy against Janus Kinase (JAK)-STAT signaling in HCC cells and downregulation of pSTAT3 and its target genes including Mcl-1 by immunblotting.[47] Irophic factor IL-3 also involves in transcriptional upregulation of Mcl-1. Through activation of the PU.1 transcription factor, IL-3 activates Mcl-1 transcription by the P38 mitogen-activated protein kinase (MAPK)-dependent pathway.[48] On the other hand, Mcl-1 transcription can also be activated by IL-3 increasing of the DNA binding activity of the CRE-2 binding complex through phosphatidylinositol-3-kinase (PI3K)/Akt signaling pathway.[49] HIF-1 is a putative key transcription factor which can regulate cells under hypoxia undergoing different transcriptional adaptations.[50] Through analysis of the Mcl-1 promoter sequence in hepatoma HepG2 cells incubated under hypoxia, Jean-Pascal Piret et al.[51] demonstrated that there was a hypoxia-responsive element in Mcl-1 promoter fragment that was able to bind HIF-1 in vitro. Detailed results revealed that HIF-1 showed a potential anti-apoptotic role and could protect cells against apoptosis as a result of hypoxia by up-regulation of the Mcl-1 protein.[51] Luciferase reporter assay revealed that overexpression of periostin enhanced HIF-1α–dependent transcriptional activity and induced multiple HIF-1α target genes including Mcl-1, and Bcl-xL in HCC cells.[52] Moreover, the ternary complex factor-serum response factor complex are also involved in regulating Mcl-1 expression and protecting cells from apoptotic cell death.[53] After activating cells with a variety of cytokines, Mcl-1 expression can be regulated transcriptionally in several signaling pathways. A recent report describes that after treatment of HCC SK-Hep-1 cells with exposure to ursolic acid (UA), western blot results showed decreased expression of the Mcl-1 and that treatment with UA induces apoptosis by inhibition of PI3K/Akt and P38/MAPK signaling pathway.[54] Yu et al.[55] found that Mcl-1 protein expression was downregulated via inhibition PI3K by LY294002 in HepG2 cells, which indicates that PI3K/Akt signaling pathway regulates Mcl-1 expression. Data from two human HCC cell lines, SMMC7721 and HepG2, indicated that exogenous rhHPPCn (Hepatopoietin Cn) suppressed trichostatin A-induced apoptosis of HCC cells and up-regulated Mcl-1 expression in HCC-derived cells via the MAPK or sphingosine kinase-1.[56] Real-time polymerase chain reaction analysis and western blot results demonstrated that aspirin induced Mcl-1 expression at mRNA level as well as protein level through Akt/extracellular regulated kinase (ERK) 1/2 and stimulates AMPK-Akt/ERK1/2-Mcl-1 axis in HepG2 cells.[57] P53 as a tumor suppressor protein also involves the regulation of Mcl-1. It has been reported that mutation in the P53 frequently occurred in HCC and contributed to hepatocarcinogenesis as well as apoptosis resistance.[58] Additionally, Leu et al.[59] demonstrated that P53 antagonized the interactions between Mcl-1 and Bak. Once mutation happens in HCC, Mcl-1 couldn’t be dissociated from Bak and the final result is apoptosis resistance of hepatoma cells. Data from human samples showed that P53 protein was also overexpressed in HCC tissues and its expression was significantly correlated with Mcl-1 expression. Further research indicated that silencing Mcl-1 sensitizes hepatoma cells towards chemotherapy may be attributed to the dysfunction of P53 through Mcl-1/P53 interaction.[55] According to combination of ICG-001, a small molecule which blocks the interaction of β-catenin with its transcriptional coactivator CREB-binding protein, and sorafenib to treat several HCC cell lines, the effect was a significant downregulation of Mcl-1 which was the most consistent change across tested HCC cell lines. The author concluded that the sorafenib-sensitizing effect of Wnt/β-catenin pathway inhibition was closely associated with Mcl-1 downregulation in HCC cells.[60] In addition, recent reports described that Wnt/β-catenin signaling could regulate Mcl-1 expression indirectly, involving genes regulated by Wnt/β-catenin pathway or other transcriptional factors.[61,62] Moreover, a panel of HCC cell lines has been on treatment with Xanthohumol (XN), a prenylated chalcone having anti-proliferative effects in various cancers types in vitro, and growth suppression due to apoptosis was evidenced by reduced expression of anti-apoptotic proteins including Mcl-1. Importantly, XN treatment decreased the expression of Notch1 and hairy and enhancer of split-1 proteins while ectopic expression of Notch1 in HCC cells abolished the anti-proliferative effect of XN. In brief, XN mediated growth suppression of HCC through inhibition of the Notch signaling pathway.[63]
Translational regulation
The same as Mcl-1 protein, Mcl-1 mRNA have very short half-lives. Translationally, mir-29b binding to the 3’-untranslated region of Mcl-1 mRNA inhibits expression of Mcl-1.[64] Northern blot and real-time quantitative reverse transcription polymerase chain reaction showed that downregulation of mir-29 was a frequent event in HCC tissues. Further study implicated that mir-29 may promote apoptosis of HCC cells through directly targeting Bcl-2 and Mcl-1. Besides, the ability of HCC cells to form tumor in nude mice was dramatically repressed by induction of mir-29. These results indicated that Bcl-2 and Mcl-1 were predominant mediators of mir-29 promoted apoptosis in HCC cells.[65] The mammalian target of rapamycin complex 1 (mTORC1) is a protein complex whose role is to activate translation of proteins just like a nutrient sensor controlling protein synthesis and a downstream target of PI3K/Akt.[66] There was a report that described that activation of mTORC1 was of vital importance to be a potent antiapoptotic signal through Mcl-1 which is a translationally regulated genetic determinant of mTORC1-dependent survival.[67] And a recent study demonstrated that metformin-induced apoptosis in HCC was mediated by the downstream mTORC1 effectors eukaryotic initiation factor 4E (eIF4E) and eIF4E-binding proteins who were required to induce apoptosis by metformin in HCC and to repress Mcl-1 expression.[68]
Post-translational regulation
There is variety of modes regulating Mcl-1 at post-translational level. In the preceding part of this review, we have mentioned that the PEST region of Mcl-1 was rich in putative phosphorylation sites which made Mcl-1 different from other Bcl-2 family members. Here, we detail those phosphoresidues of Mcl-1 and the influence of phosphorylation in HCC. Both of the phosphoresidues Threonine 92 and Threonine 163 of Mcl-1 were identified by Ding et al.[69] using ERK-1 kinase assay. ERK-1 phosphorylation of Thr 92 and Thr 163 stabilizes Mcl-1 and then promotes Mcl-1’s anti-apoptosis. It has been demonstrated that heat shock protein 90 inhibitor 17-allylaminogeldanamycin (17-AAG) partially inversed (-)-gossypol-induced Mcl-1 accumulation by inhibiting ERK phosphorylation in HCC cells.[70] Of note, Inoshita et al.[71] concluded that phosphorylation of Thr 163 by JNK destabilized Mcl-1, whereas the results showed by Kodama et al.[72] suggested that C-Jun N-terminal kinase (JNK) as the kinase contributed to phosphorylation of Thr 163 and Serine 121 of Mcl-1, prolonged the half-life of the Mcl-1 protein and protected hepatocytes against apoptosis induced by tumor necrosis factor alpha (TNFα). And data from Wang et al.[73] demonstrated that the Bcl-2/xL inhibitor ABT-263 increased Mcl-1 stability in HCC cells while activation of ERK and JNK involved in ABT-263-mediated Mcl-1 protein stabilization through phosphorylation of Mcl-1 Thr163. And also it has been reported that the new tubulin inhibitor MT189 (2-(6-fluoro-3-((4-methoxybenzyl)amino) imidazo [1,2- α] pyridin-2-yl) phenol)-mediated JNK activation caused degradation of Mcl-1 protein via facilitating its phosphorylation in the SMMC-7721 cells.[74]
Glycogen synthase kinase-3 (GSK-3) inactivated by Akt plays a crucial role in the regulation of apoptosis. It has been demonstrated that the control of Mcl-1 stability by GSK-3 is an important mechanism for the regulation of apoptosis by growth factors, PI3K, and Akt.[75] Deeper research indicated that GSK-3 was conducive to degradation of Mcl-1 by means of phosphorylation of its Serine 155 and Serine 159 and the latter inhibited the interaction of Mcl-1 with the pro-apoptotic protein, Bim, thus impairing its anti-apoptotic function.[75,76] What’s more, Wang et al.[73] indicated that Akt-mediated GSK-3β inactivation also implicated in ABT-263-induced Mcl-1 stabilization, possibly through regulating the phosphorylation of Mcl-1 Ser159 in HCC cells.
Mcl-1 interacting proteins
The majority of proteins interacting with Mcl-1 belong to the Bcl-2 protein family including multidomain pro-apoptotic members and the BH3-only proteins.[77-79] In this review, we just discuss other proteins interacting with Mcl-1. The Mcl-1 protein level can be downregulated by adenovirus infection through proteasome-mediated turnover of Mcl-1.[80] Mcl-1 ubiquitin ligase E3 (Mule) contains a region similar to BH3 domain that enables Mule to interact with Mcl-1. It has been demonstrated that Mule was required for the polyubiquitination of Mcl-1 in the ubiquitin dependent proteasome degradation pathway.[81] According to a research treatment of HepG2 cells with glycochenodeoxycholate (GCDA), one of the major human bile salts, the author reported that GCDA facilitated Mcl-1 dissociation from E3 ligase Mule and increased the half-life of Mcl-1.[82]
Cyclin-dependent kinase (CDK) could combine and activate cyclin, and thus lead to phosphorylation of target protein. The phosphoresidue serine 64 of Mcl-1 was identified by Kobayashi et al.[83] through MS analysis of a threonine 163 to alanine mutant of Mcl-1 and then CDK1 and CDK2, proteins related to cell cycle, and JNK were affirmed to phosphorylate this residue, which plays a negatively role on the progression of apoptosis. Moreover, it was reported that both protein and mRNA levels of Mcl-1 were down-regulated by a novel synthetic CDK inhibitor ibulocydine in HCC cells.[84]
Another protein impacting Mcl-1’s roles in cell cycle is PCNA. It has been mentioned before that Mcl-1 can bind to PCNA and CDK1 in the nucleus, which participate in repression of cell cycle progression. When transfection of Huh7 and HepG2 cells with glypican 3-specific siRNA, cell proliferation detected by PCNA immunohistochemistry was inhibited, cell cycle was arrested at the G1 phase and anti-apoptotic proteins (Bcl-2, Bcl-xL, and Mcl-1) were down-regulated.[85] Besides, different from other reports about interaction between PCNA and the Mcl-1 or the CDK2 protein in biochemical assays, De Biasio et al.[31] detected no binding between them and suggested that the interaction, if any, occurs with very low affinity or is mediated by other factors. Lately, a report described that following the inhibition of RNA polymerase II phosphorylation, ibulocydine caused down-regulation of Mcl-1, survivin, and X-linked inhibitor of apoptosis protein (XIAP), thus inducing apoptosis in HCC cells.[84]
Owing to the extremely labile nature of Mcl-1, it is as important as those that regulate Mcl-1 synthesis for cellular processes to regulate Mcl-1 stability. Recently, a Mcl-1 interacting protein the translationally controlled tumor protein (TCTP) was identified to upregulate the expression levels of Mcl-1 through modulating Mcl-1 stability and eventually modulate Mcl-1’s antiapoptotic activity by the ubiquitin-dependent proteasome degradation pathway. Detailed analysis revealed that TCTP overexpression inhibited apoptosis by binding to Mcl-1 and antagonizing Bax.[86] It has been well documented that TCTP was implicated in many cellular functions including human allergic response,[87] apoptosis[88] and cell growth.[89] Chen et al.[90] described that Sann-Joong-Kuey-Jian-Tang (SJKJT), a traditional medicinal prescription, could downregulate the protein expression level of Mcl-1 and TCTP in Hep-G2 cells, thus they considered that decreasing TCTP and Mcl-1 expression may be one of the molecular mechanisms by which SJKJT inhibits Hep-G2 cells. It has also been reported that curcumin inhibited the proliferation of human HCC J5 cells and induced mitochondrial dysfunction by decreasing the expressions of TCTP, Mcl-1 and Bcl-2.[91] Similarly, after treatment of HCC SK-Hep-1 cells with ursolic acid, the western blot results were associated with decreased expression of Mcl-1, TCTP and Bcl-2.[54]
Drug R&D for regulating Mcl-1 of HCC
Drugs that down-regulate Mcl-1
Cyclin-dependent kinase inhibitors
Flavopiridol is a semisynthetic compound that functions as a CDK inhibitor though inhibiting CDKs and thus inducing cell cycle arrest at the G1 or the G2/M transition point.[92] In a recent study, flavopiridol augmented TNF-related apoptosis-inducing ligand (TRAIL) sensitivity of human HCC cells by up-regulation of TRAIL receptors and down-regulation of survivin, FLICE-inhibitory protein and Bcl-xL.[93] Flavopiridol has also been shown to induce apoptosis in a P53-independent manner and to down-regulate XIAP, Mcl-1, Bcl-2, survivin in kinds of cancer cells.[94,95]
Another CDK inhibitor, as mentioned above, is ibulocydine - a novel isobutyrate prodrug inhibitor of CDK7/9. In comparison, ibulocydine inhibited the growth of HCC cells more effectively than other CDK inhibitors via prolonged inhibition of CDK7/9 leading to induction of apoptosis by down-regulating the cellular levels of anti-apoptotic proteins such as Mcl-1 and XIAP. Besides, data from human HCC xenografts indicated that ibulocydine selectively induced apoptosis but has no cytotoxic effects on normal tissues. Consequently, ibulocydine is a strong candidate for the treatment of HCC.[84]
Deubiquitinase inhibitors
Mcl-1 is degraded rapidly in the cell via a proteasome-dependent pathway, whereas deubiquitinases (DUBs) are capable of removing ubiquitin from ubiquitinated Mcl-1 to rescue Mcl-1 from degradation. WP1130, a small molecule that was initially identified as JAK and STAT inhibitors, can also inhibit activity of DUBs. And it has been demonstrated that DUBs ubiquitin-specific protease 9X (DUB USP9X) was one of the proteins to co-immunoprecipitates with Mcl-1.[96] In a recent study, it has been found that combined treatment with WP1130 sensitized HCC cells to doxorubicin via USP9X-depedent P53 degradation.[97]
STAT protein inhibitors
As mentioned previously, STAT could regulate Mcl-1 at transcriptional level, thus attenuating the activity of STAT protein by agents is a fine choice to down-regulate the expression of Mcl-1 proteins. Surprisingly, ethanol extracts from Sedum sarmentosum have been reported to inhibit STAT-3 signaling, down-regulate Mcl-1 and Bcl-2 expressions, and finally inhibit proliferation of HepG2 cells, and induce HepG2 cells apoptosis.[98]
PI3K/Akt signaling inhibitors
LY294002 functions as a PI3K/Akt signaling inhibitor which is capable of repressing the activation of AKT-1 to down-regulate the expression of Mcl-1 protein and to induce the apoptosis of macrophage. In the research of HCC therapies, Yang et al.[99] found that a disintegrin and metalloproteinase 10 (ADAM10) overexpression conferred resistance to doxorubicin-induced apoptosis in HCC, whereas the pretreatment with the PI3K inhibitor LY294002 significantly enhanced doxorubicin-induced apoptosis and diminished the Mcl-1 expression in ADAM10-overexpressing Huh7 cells. And also LY294002 could down-regulate the expression of Mcl-1 rapidly in HCC cells and increase the sensitivity of HCC cells to chemotherapeutics.[35]
MEK/ERK signaling inhibitors
Sorafenib actually inhibits multiple other kinases. It is the first and only orally administered drug to treat advanced HCC. One of the molecular mechanisms of sorafenib in HCC cells is that sorafenib induces apoptosis by reducing elF4E phosphorylation and blocking the initiation of Mcl-1 translation.[100] Chen et al.[47] demonstrated that sorafenib downregulated phospho-STAT3 and subsequently reduced the expression levels of STAT3-related proteins including Mcl-1 in a dose- and time-dependent manner in TRAIL-treated HCC cells.
Antisense oligonucleotide treatment
Antisense oligonucleotide (ASO)[10] is a kind of synthetic oligonucleotides fragment expressed by antisense expression plasmid. Recently, it has been found that Mcl-1 ASO could downregulate Mcl-1 efficiently in various tumor cells and animal models. According to ASO treatment as monotherapy in the HCC cell lines HepG2 and Snu398, the result showed that ASO targeting Mcl-1 specifically downregulated Mcl-1 protein expression and led to significant dose and time dependent single agent activity in HCC cells characterized by increased apoptosis and decreased cell viability. And Upon combination with cisplatin, Mcl-1 ASO revealed a significant chemosensitizing effect.[10] However, there is no report about Mcl-1 ASO treatment used in clinic.
BH3 mimetics
BH3 mimetics mainly play a part through the interaction of proteins to inhibit Mcl-1’s function. As discussed, there was a surface-exposed hydrophobic groove contributing to the anti-apoptosis function of Mcl-1 [Figure 2].[17] Consequently, BH3 mimetics were designed to fit into the hydrophobic groove and block Mcl-1’s ability to bind pro-apoptotic proteins, inhibiting the anti-apoptosis function of Mcl-1. ABT-737, a small-molecule cell-permeable Bcl-2/Bcl-XL antagonist, is a novel cancer therapeutic agent because it potently induces apoptosis in certain cancer cells. Nevertheless, owing to low affinity with Mcl-1, ABT-737-mediated apoptosis signaling was inhibited in HCC cells due to the elevated expression of Mcl-1 which may contribute to HCC resistance to ABT737.[101,102] Consequently, it is unlikely to be effective as a single agent in solid tumors and thereby a great many research about combining ABT-737 with other agents to abolish the resistance has emerged recently such as norcantharidin, celastrol, etc.[103,104]
Polyphenols derivatives
The mother nucleus structure of polyphenols derivatives has polyhydroxyphenol. Gossypol as a typical BH3 mimetic that inhibits the Bcl-2 anti-apoptotic proteins Bcl-2, Bcl-XL, and Mcl-1 by binding to them.[105] Because of the ability to target Mcl-1, gossypol shows toxicity against variety of cancer types in comparison to ABT-737. Since it was shown that HCC cells were relatively resistant to Bcl-2 inhibitors, then co-treatment of Bcl-2 inhibitor (-)-gossypol and Hsp90 inhibitor 17-AAG attenuated (-)-gossypol-induced protective autophagy by inhibiting ERK-mediated Bcl-2 phosphorylation and downregulated (-)-gossypol-triggered Mcl-1 accumulation by suppressing Mcl-1 Thr163 phosphorylation.[70] Apogossypolone (ApoG2) was the first derivative of gossypol for the potential non-specific reactivity related to the 2 aldehyde groups in gossypol.[106] In order to investigate the in vitro and in vivo activities and related mechanism of ApoG2 against HCC, Mi et al.[107] found that the ApoG2 induced apoptosis in SMMC-7721 cells by downregulation of anti-apoptotic proteins Bcl-2, Mcl-1, and Bcl-XL and up-regulation of pro-apoptotic protein Noxa, which indicated ApoG2 was a potential pan Bcl-2 family protein inhibitor, targeting Bcl-2, Mcl-1, and Bcl-XL, and inducing apoptosis in HCC. Moreover, several gossypol analogues arose to inactivate Mcl-1 such as TM-1206,[108] BI-33[109] and TM-179.[110]
Indole dipyrrole derivatives
Obatoclax is a synthetic indole dipyrrole derivative derived from prodigiosin and acts as a BH3 mimetic which binds the anti-apoptotic Bcl-2 proteins, releases proapoptotic proteins and thus triggers caspase activation. And SC-2001 was originally derived from the Mcl-1 inhibitor obatoclax, that was suggested better antitumor effects than obatoclax in HCC cell lines, including HepG2, PLC5 and Huh-7.[111]
Acenaphthene heterocyclic derivatives
S-1 (one mixed formulation containing 5-FU prodrug and dihydropyrimidine dehydrogenase inhibitor) inhibits both Bcl-2 and Mcl-1 and is capable of disturbing interaction between Mcl-1 and Bak, resulting in apoptosis.[112] Furuse et al.[113] reported that S-1 was effective and had an acceptable toxicity profile in patients with advanced HCC, which indicates S-1 is a potential candidate for antitumor agent.
Conclusion
With highly expressed Mcl-1, HCC cells tend to escape from apoptosis and thus proliferate at an increasingly high speed. Mcl-1 is a critical survival factor for malignant tissues of HCC and its expression is regulated via multiple mechanisms. Hence, it is a promising target for HCC treatment. Over the past several decades, there has been significant progress towards relevant molecular interacting with Mcl-1. On the one hand, Mcl-1’s expression in HCC is regulated at transcriptional by a variety of cytokines and signaling pathways, including the P38/MAPK, PI3K/AKT, STAT, P53, ERK, JNK, Wnt/β-catenin, Notch signaling ways. On the other hand, the role of microRNAs in Mcl-1 regulation has been highlighted at the translational level and multiple phosphorylation sites in Mcl-1’s PEST region regulate Mcl-1 expression at post-translational level. Other Mcl-1 interacting proteins such as Mule, CDK1, CDK2, PCNA, TCTP, etc. also involve in Mcl-1 regulation through interaction with it. According to these molecular mechanisms, numerous of chemotherapeutic agents have been reported to decrease the level of Mcl-1 towards HCC treatment including agents not specifically targeting Mcl-1 but involving downregulation of Mcl-1 and those drugs targeting Mcl-1 directly. Thereinto, BH3 mimetics are the most studied among all the chemotherapies. Of note, HCC with high levels of Mcl-1 are resistant to apoptosis induction by some compounds, posing a major problem for its potential utility. Thus, combination of multiple targets agents for HCC chemotherapy, production of good drug delivery system, and designing novel interventions specifically targeting Mcl-1 will be a major tendency in the future.
Authors’ contributions
Design and performing the research, manuscript review: Y.M. Zhang
Manuscript drafting: M. Zhu
Financial support and sponsorship
This work was supported by the National Natural Science Foundation of China (Grant no. 81370088), the Fundamental Research Funds for the Central Universities of Zhuizong.
Conflicts of interest
There are no conflicts of interest.
Patient consent
Not applicable.
Ethics approval
Not applicable.
REFERENCES
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011;61:69-90.
2. Burroughs A, Hochhauser D, Meyer T. Systemic treatment and liver transplantation for hepatocellular carcinoma: two ends of the therapeutic spectrum. Lancet Oncol 2004;5:409-18.
3. Marrero JA, Kudo M, Bronowicki JP. The challenge of prognosis and staging for hepatocellular carcinoma. Oncologist 2010;15 Suppl 4:23-33.
4. Zheng Y, Gery S, Sun H, Shacham S, Kauffman M, Koeffler HP. KPT-330 inhibitor of XPO1-mediated nuclear export has anti-proliferative activity in hepatocellular carcinoma. Cancer Chemother Pharmacol 2014;74:487-95.
5. Kang MH, Reynolds CP. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res 2009;15:1126-32.
6. Hiraki M, Suzuki Y, Alam M, Hinohara K, Hasegawa M, Jin C, Kharbanda S, Kufe D. MUC1-C stabilizes MCL-1 in the oxidative stress response of triple-negative breast cancer cells to BCL-2 inhibitors. Sci Rep 2016;6:26643.
7. Wuilleme-Toumi S, Robillard N, Gomez P, Moreau P, Le Gouill S, Avet-Loiseau H, Harousseau JL, Amiot M, Bataille R. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia 2005;19:1248-52.
8. Cho-Vega JH, Rassidakis GZ, Admirand JH, Oyarzo M, Ramalingam P, Paraguya A, McDonnell TJ, Amin HM, Medeiros LJ. MCL-1 expression in B-cell non-Hodgkin's lymphomas. Hum Pathol 2004;35:1095-100.
9. Shimazu T, Degenhardt K, Nur EKA, Zhang J, Yoshida T, Zhang Y, Mathew R, White E, Inouye M. NBK/BIK antagonizes MCL-1 and BCL-XL and activates BAK-mediated apoptosis in response to protein synthesis inhibition. Genes Dev 2007;21:929-41.
10. Sieghart W, Losert D, Strommer S, Cejka D, Schmid K, Rasoul-Rockenschaub S, Bodingbauer M, Crevenna R, Monia BP, Peck-Radosavljevic M, Wacheck V. Mcl-1 overexpression in hepatocellular carcinoma: a potential target for antisense therapy. J Hepatol 2006;44:151-7.
11. Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno S, Akashi K, Korsmeyer SJ. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science 2005;307:1101-4.
12. Kozopas KM, Yang T, Buchan HL, Zhou P, Craig RW. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc Natl Acad Sci U S A 1993;90:3516-20.
13. Bae J, Leo CP, Hsu SY, Hsueh AJ. MCL-1S, a splicing variant of the antiapoptotic BCL-2 family member MCL-1, encodes a proapoptotic protein possessing only the BH3 domain. J Biol Chem 2000;275:25255-61.
14. Yang T, Kozopas KM, Craig RW. The intracellular distribution and pattern of expression of Mcl-1 overlap with, but are not identical to, those of Bcl-2. J Cell Biol 1995;128:1173-84.
15. Bingle CD, Craig RW, Swales BM, Singleton V, Zhou P, Whyte MK. Exon skipping in Mcl-1 results in a bcl-2 homology domain 3 only gene product that promotes cell death. J Biol Chem 2000;275:22136-46.
16. Rechsteiner M, Rogers SW. PEST sequences and regulation by proteolysis. Trends Biochem Sci 1996;21:267-71.
17. Day CL, Chen L, Richardson SJ, Harrison PJ, Huang DC, Hinds MG. Solution structure of prosurvival Mcl-1 and characterization of its binding by proapoptotic BH3-only ligands. J Biol Chem 2005;280:4738-44.
18. Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M, Temirov J, Cleland MM, Pelletier S, Schuetz JD, Youle RJ, Green DR, Opferman JT. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol 2012;14:575-83.
19. Germain M, Duronio V. The N terminus of the anti-apoptotic BCL-2 homologue MCL-1 regulates its localization and function. J Biol Chem 2007;282:32233-42.
20. Chou CH, Lee RS, Yang-Yen HF. An internal EELD domain facilitates mitochondrial targeting of Mcl-1 via a Tom70-dependent pathway. Mol Biol Cell 2006;17:3952-63.
21. Akgul C, Moulding DA, White MR, Edwards SW. In vivo localisation and stability of human Mcl-1 using green fluorescent protein (GFP) fusion proteins. FEBS Lett 2000;478:72-6.
22. Han J, Goldstein LA, Hou W, Rabinowich H. Functional linkage between NOXA and Bim in mitochondrial apoptotic events. J Biol Chem 2007;282:16223-31.
23. Shieh JJ, Liu KT, Huang SW, Chen YJ, Hsieh TY. Modification of alternative splicing of Mcl-1 pre-mRNA using antisense morpholino oligonucleotides induces apoptosis in basal cell carcinoma cells. J Invest Dermatol 2009;129:2497-506.
24. Kim JH, Bae J. MCL-1ES induces MCL-1L-dependent BAX- and BAK-independent mitochondrial apoptosis. PLoS One 2013;8:e79626.
25. Li P, Guo W, Du L, Zhao J, Wang Y, Liu L, Hu Y, Hou Y. microRNA-29b contributes to pre-eclampsia through its effects on apoptosis, invasion and angiogenesis of trophoblast cells. Clin Sci (Lond) 2013;124:27-40.
26. Palve V, Mallick S, Ghaisas G, Kannan S, Teni T. Overexpression of Mcl-1L splice variant is associated with poor prognosis and chemoresistance in oral cancers. PLoS One 2014;9:e111927.
27. Legartova S, Krejci J, Harnicarova A, Hajek R, Kozubek S, Bartova E. Nuclear topography of the 1q21 genomic region and Mcl-1 protein levels associated with pathophysiology of multiple myeloma. Neoplasma 2009;56:404-13.
28. Leuenroth SJ, Grutkoski PS, Ayala A, Simms HH. The loss of Mcl-1 expression in human polymorphonuclear leukocytes promotes apoptosis. J Leukoc Biol 2000;68:158-66.
29. Fujise K, Zhang D, Liu J, Yeh ET. Regulation of apoptosis and cell cycle progression by MCL1. Differential role of proliferating cell nuclear antigen. J Biol Chem 2000;275:39458-65.
30. Jamil S, Sobouti R, Hojabrpour P, Raj M, Kast J, Duronio V. A proteolytic fragment of Mcl-1 exhibits nuclear localization and regulates cell growth by interaction with Cdk1. Biochem J 2005;387:659-67.
31. De Biasio A, Campos-Olivas R, Sanchez R, Lopez-Alonso JP, Pantoja-Uceda D, Merino N, Villate M, Martin-Garcia JM, Castillo F, Luque I, Blanco FJ. Proliferating cell nuclear antigen (PCNA) interactions in solution studied by NMR. PLoS One 2012;7:e48390.
32. Aichberger KJ, Mayerhofer M, Krauth MT, Skvara H, Florian S, Sonneck K, Akgul C, Derdak S, Pickl WF, Wacheck V, Selzer E, Monia BP, Moriggl R, Valent P, Sillaber C. Identification of mcl-1 as a BCR/ABL-dependent target in chronic myeloid leukemia (CML): evidence for cooperative antileukemic effects of imatinib and mcl-1 antisense oligonucleotides. Blood 2005;105:3303-11.
33. Breitenbuecher F, Markova B, Kasper S, Carius B, Stauder T, Bohmer FD, Masson K, Ronnstrand L, Huber C, Kindler T, Fischer T. A novel molecular mechanism of primary resistance to FLT3-kinase inhibitors in AML. Blood 2009;113:4063-73.
34. Zhang T, Zhao C, Luo L, Zhao H, Cheng J, Xu F. The expression of Mcl-1 in human cervical cancer and its clinical significance. Med Oncol 2012;29:1985-91.
35. Fleischer B, Schulze-Bergkamen H, Schuchmann M, Weber A, Biesterfeld S, Muller M, Krammer PH, Galle PR. Mcl-1 is an anti-apoptotic factor for human hepatocellular carcinoma. Int J Oncol 2006;28:25-32.
36. Henson ES, Hu X, Gibson SB. Herceptin sensitizes ErbB2-overexpressing cells to apoptosis by reducing antiapoptotic Mcl-1 expression. Clin Cancer Res 2006;12:845-53.
37. Song L, Coppola D, Livingston S, Cress D, Haura EB. Mcl-1 regulates survival and sensitivity to diverse apoptotic stimuli in human non-small cell lung cancer cells. Cancer Biol Ther 2005;4:267-76.
38. Derenne S, Monia B, Dean NM, Taylor JK, Rapp MJ, Harousseau JL, Bataille R, Amiot M. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L) is an essential survival protein of human myeloma cells. Blood 2002;100:194-9.
39. Le Gouill S, Podar K, Harousseau JL, Anderson KC. Mcl-1 regulation and its role in multiple myeloma. Cell Cycle 2004;3:1259-62.
40. Ding Q, He X, Xia W, Hsu JM, Chen CT, Li LY, Lee DF, Yang JY, Xie X, Liu JC, Hung MC. Myeloid cell leukemia-1 inversely correlates with glycogen synthase kinase-3beta activity and associates with poor prognosis in human breast cancer. Cancer Res 2007;67:4564-71.
41. Glaser SP, Lee EF, Trounson E, Bouillet P, Wei A, Fairlie WD, Izon DJ, Zuber J, Rappaport AR, Herold MJ, Alexander WS, Lowe SW, Robb L, Strasser A. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev 2012;26:120-5.
42. Xiang Z, Luo H, Payton JE, Cain J, Ley TJ, Opferman JT, Tomasson MH. Mcl1 haploinsufficiency protects mice from Myc-induced acute myeloid leukemia. J Clin Invest 2010;120:2109-18.
43. Campbell KJ, Bath ML, Turner ML, Vandenberg CJ, Bouillet P, Metcalf D, Scott CL, Cory S. Elevated Mcl-1 perturbs lymphopoiesis, promotes transformation of hematopoietic stem/progenitor cells, and enhances drug resistance. Blood 2010;116:3197-207.
44. Weber A, Boger R, Vick B, Urbanik T, Haybaeck J, Zoller S, Teufel A, Krammer PH, Opferman JT, Galle PR, Schuchmann M, Heikenwalder M, Schulze-Bergkamen H. Hepatocyte-specific deletion of the antiapoptotic protein myeloid cell leukemia-1 triggers proliferation and hepatocarcinogenesis in mice. Hepatology 2010;51:1226-36.
45. Gruber S, Straub BK, Ackermann PJ, Wunderlich CM, Mauer J, Seeger JM, Buning H, Heukamp L, Kashkar H, Schirmacher P, Bruning JC, Wunderlich FT. Obesity promotes liver carcinogenesis via Mcl-1 stabilization independent of IL-6Ralpha signaling. Cell Rep 2013;4:669-80.
46. Al Zaid Siddiquee K, Turkson J. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res 2008;18:254-67.
47. Chen KF, Tai WT, Liu TH, Huang HP, Lin YC, Shiau CW, Li PK, Chen PJ, Cheng AL. Sorafenib overcomes TRAIL resistance of hepatocellular carcinoma cells through the inhibition of STAT3. Clin Cancer Res 2010;16:5189-99.
48. Wang JM, Lai MZ, Yang-Yen HF. Interleukin-3 stimulation of mcl-1 gene transcription involves activation of the PU.1 transcription factor through a p38 mitogen-activated protein kinase-dependent pathway. Mol Cell Biol 2003;23:1896-909.
49. Wang JM, Chao JR, Chen W, Kuo ML, Yen JJ, Yang-Yen HF. The antiapoptotic gene mcl-1 is up-regulated by the phosphatidylinositol 3-kinase/Akt signaling pathway through a transcription factor complex containing CREB. Mol Cell Biol 1999;19:6195-206.
51. Piret JP, Minet E, Cosse JP, Ninane N, Debacq C, Raes M, Michiels C. Hypoxia-inducible factor-1-dependent overexpression of myeloid cell factor-1 protects hypoxic cells against tert-butyl hydroperoxide-induced apoptosis. J Biol Chem 2005;280:9336-44.
52. Liu Y, Gao F, Song W. Periostin contributes to arsenic trioxide resistance in hepatocellular carcinoma cells under hypoxia. Biomed Pharmacother 2017;88:342-8.
53. Vickers ER, Kasza A, Kurnaz IA, Seifert A, Zeef LA, O'Donnell A, Hayes A, Sharrocks AD. Ternary complex factor-serum response factor complex-regulated gene activity is required for cellular proliferation and inhibition of apoptotic cell death. Mol Cell Biol 2004;24:10340-51.
54. Chuang WL, Lin PY, Lin HC, Chen YL. The apoptotic effect of ursolic acid on SK-Hep-1 cells is regulated by the PI3K/Akt, p38 and JNK MAPK signaling pathways. Molecules 2016;21:460.
55. Yu Q, Liu ZY, Chen Q, Lin JS. Mcl-1 as a potential therapeutic target for human hepatocelluar carcinoma. J Huazhong Univ Sci Technolog Med Sci 2016;36:494-500.
56. Chang J, Liu Y, Zhang DD, Zhang DJ, Wu CT, Wang LS, Cui CP. Hepatopoietin Cn suppresses apoptosis of human hepatocellular carcinoma cells by up-regulating myeloid cell leukemia-1. World J Gastroenterol 2010;16:193-200.
57. Gao M, Kong Q, Hua H, Yin Y, Wang J, Luo T, Jiang Y. AMPK-mediated up-regulation of mTORC2 and MCL-1 compromises the anti-cancer effects of aspirin. Oncotarget 2016;7:16349-61.
58. Zhang CZ, Chen GG, Merchant JL, Lai PB. Interaction between ZBP-89 and p53 mutants and its contribution to effects of HDACi on hepatocellular carcinoma. Cell Cycle 2012;11:322-34.
59. Leu JI, Dumont P, Hafey M, Murphy ME, George DL. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol 2004;6:443-50.
60. Lin HH, Feng WC, Lu LC, Shao YY, Hsu CH, Cheng AL. Inhibition of the Wnt/beta-catenin signaling pathway improves the anti-tumor effects of sorafenib against hepatocellular carcinoma. Cancer Lett 2016;381:58-66.
61. Yang L, Perez AA, Fujie S, Warden C, Li J, Wang Y, Yung B, Chen YR, Liu X, Zhang H, Zheng S, Liu Z, Ann D, Yen Y. Wnt modulates MCL1 to control cell survival in triple negative breast cancer. BMC Cancer 2014;14:124.
62. Iqbal S, Zhang S, Driss A, Liu ZR, Kim HR, Wang Y, Ritenour C, Zhau HE, Kucuk O, Chung LW, Wu D. PDGF upregulates Mcl-1 through activation of beta-catenin and HIF-1alpha-dependent signaling in human prostate cancer cells. PLoS One 2012;7:e30764.
63. Kunnimalaiyaan S, Sokolowski KM, Balamurugan M, Gamblin TC, Kunnimalaiyaan M. Xanthohumol inhibits Notch signaling and induces apoptosis in hepatocellular carcinoma. PLoS One 2015;10:e0127464.
64. Mott JL, Kobayashi S, Bronk SF, Gores GJ. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene 2007;26:6133-40.
65. Xiong Y, Fang JH, Yun JP, Yang J, Zhang Y, Jia WH, Zhuang SM. Effects of microRNA-29 on apoptosis, tumorigenicity, and prognosis of hepatocellular carcinoma. Hepatology 2010;51:836-845.
66. Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002;110:163-75.
67. Mills JR, Hippo Y, Robert F, Chen SM, Malina A, Lin CJ, Trojahn U, Wendel HG, Charest A, Bronson RT, Kogan SC, Nadon R, Housman DE, Lowe SW, Pelletier J. mTORC1 promotes survival through translational control of Mcl-1. Proc Natl Acad Sci U S A 2008;105:10853-8.
68. Bhat M, Yanagiya A, Graber T, Razumilava N, Bronk S, Zammit D, Zhao Y, Zakaria C, Metrakos P, Pollak M, Sonenberg N, Gores G, Jaramillo M, Morita M, Alain T. Metformin requires 4E-BPs to induce apoptosis and repress translation of Mcl-1 in hepatocellular carcinoma cells. Oncotarget 2016; doi: 10.18632/oncotarget.10671.
69. Ding Q, Huo L, Yang JY, Xia W, Wei Y, Liao Y, Chang CJ, Yang Y, Lai CC, Lee DF, Yen CJ, Chen YJ, Hsu JM, Kuo HP, Lin CY, Tsai FJ, Li LY, Tsai CH, Hung MC. Down-regulation of myeloid cell leukemia-1 through inhibiting Erk/Pin 1 pathway by sorafenib facilitates chemosensitization in breast cancer. Cancer Res 2008;68:6109-17.
70. Wang B, Chen L, Ni Z, Dai X, Qin L, Wu Y, Li X, Xu L, Lian J, He F. Hsp90 inhibitor 17-AAG sensitizes Bcl-2 inhibitor (-)-gossypol by suppressing ERK-mediated protective autophagy and Mcl-1 accumulation in hepatocellular carcinoma cells. Exp Cell Res 2014;328:379-87.
71. Inoshita S, Takeda K, Hatai T, Terada Y, Sano M, Hata J, Umezawa A, Ichijo H. Phosphorylation and inactivation of myeloid cell leukemia 1 by JNK in response to oxidative stress. J Biol Chem 2002;277:43730-4.
72. Kodama Y, Taura K, Miura K, Schnabl B, Osawa Y, Brenner DA. Antiapoptotic effect of c-Jun N-terminal Kinase-1 through Mcl-1 stabilization in TNF-induced hepatocyte apoptosis. Gastroenterology 2009;136:1423-34.
73. Wang B, Ni Z, Dai X, Qin L, Li X, Xu L, Lian J, He F. The Bcl-2/xL inhibitor ABT-263 increases the stability of Mcl-1 mRNA and protein in hepatocellular carcinoma cells. Mol Cancer 2014;13:98.
74. Wang W, Wang YQ, Meng T, Yi JM, Huan XJ, Ma LP, Tong LJ, Chen Y, Ding J, Shen JK, Miao ZH. MCL-1 degradation mediated by JNK activation via MEKK1/TAK1-MKK4 contributes to anticancer activity of new tubulin inhibitor MT189. Mol Cancer Ther 2014;13:1480-91.
75. Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell 2006;21:749-60.
76. Ding Q, He X, Hsu JM, Xia W, Chen CT, Li LY, Lee DF, Liu JC, Zhong Q, Wang X, Hung MC. Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol Cell Biol 2007;27:4006-17.
77. Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature 2003;426:671-6.
78. Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DC. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 2005;17:393-403.
79. Wang K, Gross A, Waksman G, Korsmeyer SJ. Mutagenesis of the BH3 domain of BAX identifies residues critical for dimerization and killing. Mol Cell Biol 1998;18:6083-9.
80. Cuconati A, Mukherjee C, Perez D, White E. DNA damage response and MCL-1 destruction initiate apoptosis in adenovirus-infected cells. Genes Dev 2003;17:2922-32.
81. Zhong Q, Gao W, Du F, Wang X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 2005;121:1085-95.
82. Liao M, Zhao J, Wang T, Duan J, Zhang Y, Deng X. Role of bile salt in regulating Mcl-1 phosphorylation and chemoresistance in hepatocellular carcinoma cells. Mol Cancer 2011;10:44.
83. Kobayashi S, Lee SH, Meng XW, Mott JL, Bronk SF, Werneburg NW, Craig RW, Kaufmann SH, Gores GJ. Serine 64 phosphorylation enhances the antiapoptotic function of Mcl-1. J Biol Chem 2007;282:18407-17.
84. Cho SJ, Kim YJ, Surh YJ, Kim BM, Lee SK. Ibulocydine is a novel prodrug Cdk inhibitor that effectively induces apoptosis in hepatocellular carcinoma cells. J Biol Chem 2011;286:19662-71.
85. Sun CK, Chua MS, He J, So SK. Suppression of glypican 3 inhibits growth of hepatocellular carcinoma cells through up-regulation of TGF-beta2. Neoplasia 2011;13:735-47.
86. Liu H, Peng HW, Cheng YS, Yuan HS, Yang-Yen HF. Stabilization and enhancement of the antiapoptotic activity of mcl-1 by TCTP. Mol Cell Biol 2005;25:3117-26.
87. MacDonald SM, Rafnar T, Langdon J, Lichtenstein LM. Molecular identification of an IgE-dependent histamine-releasing factor. Science 1995;269:688-90.
88. Li F, Zhang D, Fujise K. Characterization of fortilin, a novel antiapoptotic protein. J Biol Chem 2001;276:47542-9.
89. Gachet Y, Tournier S, Lee M, Lazaris-Karatzas A, Poulton T, Bommer UA. The growth-related, translationally controlled protein P23 has properties of a tubulin binding protein and associates transiently with microtubules during the cell cycle. J Cell Sci 1999;112:1257-71.
90. Chen YL, Yan MY, Chien SY, Kuo SJ, Chen DR, Cheng CY, Su CC. Sann-Joong-Kuey-Jian-Tang inhibits hepatocellular carcinoma Hep-G2 cell proliferation by increasing TNF-alpha, Caspase-8, Caspase- 3 and Bax but by decreasing TCTP and Mcl-1 expression in vitro. Mol Med Rep 2013;7:1487-93.
91. Cheng CY, Lin YH, Su CC. Curcumin inhibits the proliferation of human hepatocellular carcinoma J5 cells by inducing endoplasmic reticulum stress and mitochondrial dysfunction. Int J Mol Med 2010;26:673-8.
92. Shapiro GI. Preclinical and clinical development of the cyclin-dependent kinase inhibitor flavopiridol. Clin Cancer Res 2004;10:s4270-5.
93. Miyashita K, Shiraki K, Fuke H, Inoue T, Yamanaka Y, Yamaguchi Y, Yamamoto N, Ito K, Sugimoto K, Nakano T. The cyclin-dependent kinase inhibitor flavopiridol sensitizes human hepatocellular carcinoma cells to TRAIL-induced apoptosis. Int J Mol Med 2006;18:249-56.
94. Wittmann S, Bali P, Donapaty S, Nimmanapalli R, Guo F, Yamaguchi H, Huang M, Jove R, Wang HG, Bhalla K. Flavopiridol down-regulates antiapoptotic proteins and sensitizes human breast cancer cells to epothilone B-induced apoptosis. Cancer Res 2003;63:93-9.
95. Jung C, Motwani M, Kortmansky J, Sirotnak FM, She Y, Gonen M, Haimovitz-Friedman A, Schwartz GK. The cyclin-dependent kinase inhibitor flavopiridol potentiates gamma-irradiation-induced apoptosis in colon and gastric cancer cells. Clin Cancer Res 2003;9:6052-61.
96. Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, Maecker H, O'Rourke K, Bazan F, Eastham-Anderson J, Yue P, Dornan D, Huang DC, Dixit VM. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 2010;463:103-7.
97. Liu H, Chen W, Liang C, Chen BW, Zhi X, Zhang S, Zheng X, Bai X, Liang T. WP1130 increases doxorubicin sensitivity in hepatocellular carcinoma cells through usp9x-dependent p53 degradation. Cancer Lett 2015;361:218-25.
98. Zeng JY, Li SH, Wu XJ, Liu D, Wan X. Study on impact of ethanol extracts from Sedum sarmentosum in inhibiting STAT-3 signaling and inducing apoptosis of human hepatocellular carcinoma cell line HepG2. Zhongguo Zhong Yao Za Zhi 2014;39:3349-52. (in Chinese).
99. Yang CL, Jiang FQ, Xu F, Jiang GX. ADAM10 overexpression confers resistance to doxorubicin-induced apoptosis in hepatocellular carcinoma. Tumour Biol 2012;33:1535-41.
100. Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, Wilhelm S, Lynch M, Carter C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res 2006;66:11851-8.
101. Cragg MS, Harris C, Strasser A, Scott CL. Unleashing the power of inhibitors of oncogenic kinases through BH3 mimetics. Nat Rev Cancer 2009;9:321-6.
102. Khaw SL, Huang DC, Roberts AW. Overcoming blocks in apoptosis with BH3-mimetic therapy in haematological malignancies. Pathology 2011;43:525-35.
103. Zhang S, Li G, Ma X, Wang Y, Liu G, Feng L, Zhao Y, Zhang G, Wu Y, Ye X, Qin B, Lu J. Norcantharidin enhances ABT-737-induced apoptosis in hepatocellular carcinoma cells by transcriptional repression of Mcl-1. Cell Signal 2012;24:1803-9.
104. Zhu H, Yang W, He LJ, Ding WJ, Zheng L, Liao SD, Huang P, Lu W, He QJ, Yang B. Upregulating Noxa by ER stress, celastrol exerts synergistic anti-cancer activity in combination with ABT-737 in human hepatocellular carcinoma cells. PLoS One 2012;7:e52333.
105. Wei J, Kitada S, Rega MF, Emdadi A, Yuan H, Cellitti J, Stebbins JL, Zhai D, Sun J, Yang L, Dahl R, Zhang Z, Wu B, Wang S, Reed TA, Wang HG, Lawrence N, Sebti S, Reed JC, Pellecchia M. Apogossypol derivatives as antagonists of antiapoptotic Bcl-2 family proteins. Mol Cancer Ther 2009;8:904-13.
106. Stein RC, Joseph AE, Matlin SA, Cunningham DC, Ford HT, Coombes RC. A preliminary clinical study of gossypol in advanced human cancer. Cancer Chemother Pharmacol 1992;30:480-2.
107. Mi JX, Wang GF, Wang HB, Sun XQ, Ni XY, Zhang XW, Tang JM, Yang DJ. Synergistic antitumoral activity and induction of apoptosis by novel pan Bcl-2 proteins inhibitor apogossypolone with adriamycin in human hepatocellular carcinoma. Acta Pharmacol Sin 2008;29:1467-77.
108. Tang G, Yang CY, Nikolovska-Coleska Z, Guo J, Qiu S, Wang R, Gao W, Wang G, Stuckey J, Krajewski K, Jiang S, Roller PP, Wang S. Pyrogallol-based molecules as potent inhibitors of the antiapoptotic Bcl-2 proteins. J Med Chem 2007;50:1723-6.
109. Tang G, Ding K, Nikolovska-Coleska Z, Yang CY, Qiu S, Shangary S, Wang R, Guo J, Gao W, Meagher J, Stuckey J, Krajewski K, Jiang S, Roller PP, Wang S. Structure-based design of flavonoid compounds as a new class of small-molecule inhibitors of the anti-apoptotic Bcl-2 proteins. J Med Chem 2007;50:3163-6.
110. Tang G, Nikolovska-Coleska Z, Qiu S, Yang CY, Guo J, Wang S. Acylpyrogallols as inhibitors of antiapoptotic Bcl-2 proteins. J Med Chem 2008;51:717-20.
111. Chen KF, Su JC, Liu CY, Huang JW, Chen KC, Chen WL, Tai WT, Shiau CW. A novel obatoclax derivative, SC-2001, induces apoptosis in hepatocellular carcinoma cells through SHP-1-dependent STAT3 inactivation. Cancer Lett 2012;321:27-35.
112. Zhang Z, Song T, Zhang T, Gao J, Wu G, An L, Du G. A novel BH3 mimetic S1 potently induces Bax/Bak-dependent apoptosis by targeting both Bcl-2 and Mcl-1. Int J Cancer 2011;128:1724-35.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Zhu M, Zhang YM. Function of myeloid cell leukaemia-1 and its regulative relations with hepatocellular carcinoma. Hepatoma Res 2017;3:129-40. http://dx.doi.org/10.20517/2394-5079.2017.14
AMA Style
Zhu M, Zhang YM. Function of myeloid cell leukaemia-1 and its regulative relations with hepatocellular carcinoma. Hepatoma Research. 2017; 3: 129-40. http://dx.doi.org/10.20517/2394-5079.2017.14
Chicago/Turabian Style
Zhu, Man, Yan-Min Zhang. 2017. "Function of myeloid cell leukaemia-1 and its regulative relations with hepatocellular carcinoma" Hepatoma Research. 3: 129-40. http://dx.doi.org/10.20517/2394-5079.2017.14
ACS Style
Zhu, M.; Zhang Y.M. Function of myeloid cell leukaemia-1 and its regulative relations with hepatocellular carcinoma. Hepatoma. Res. 2017, 3, 129-40. http://dx.doi.org/10.20517/2394-5079.2017.14
About This Article
Copyright

Data & Comments
Data


 Cite This Article 8 clicks
Cite This Article 8 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.