
Hepatoma Research









Lewis R. Roberts
Special Interview with Prof. Lewis R. Roberts from Mayo Clinic
NaN
Giuseppe Guido Maria Scarlata , ... Ludovico Abenavoli
, ... Ludovico Abenavoli
, ... Ludovico Abenavoli
Views: Downloads:
Masayuki Ohtsuka, ... Shinichiro Nakada
, ... Shinichiro Nakada
Views: Downloads:
Carmen Colaci, ... Ludovico Abenavoli
Views: Downloads:
Views: Downloads:
Views: Downloads:
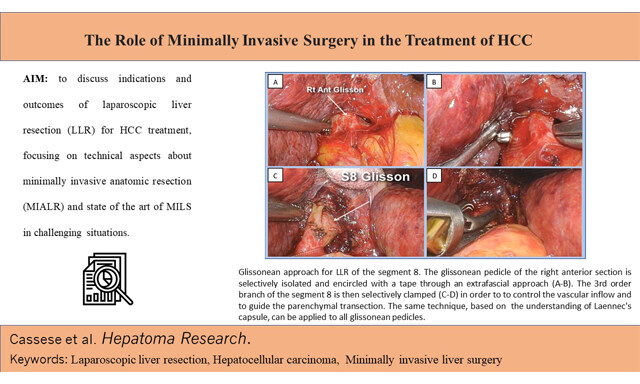
Gianluca Cassese, ... Roberto Ivan Troisi
, ... Roberto Ivan Troisi
Views: Downloads:
Views: Downloads:







Data
2368
Authors
997
Reviewers
2015
Published Since
2,481,110
Article Views
482,079
Article Downloads
For Reviewers
For Readers
Add your e-mail address to receive forthcoming Issues of this journal:
Themed Collections
Hepatocellular carcinoma
HCC
Liver Cancer
Liver tumors
Hepatoblastoma
Cholangiocarcinoma
Nonalcoholic fatty liver disease
Nonalcoholic steatohepatitis
Hepatitis B; Hepatitis C
Immunotherapy
Systemic treatment
Liver transplantation
Liver resection
Surgical
Management
Surveillance
Epidemiology
Molecular mechanisms
Tumor microenvironment
Biomarker
Stem cell

Related Journals


Lewis R. Roberts
Special Interview with Prof. Lewis R. Roberts from Mayo Clinic
NaN
Related Journals
Data
2368
Authors
997
Reviewers
2015
Published Since
2,481,110
Article Views
482,079
Article Downloads


