
Cancer stem cell-mediated therapeutic resistance in hepatocellular carcinoma


Abstract
Hepatocellular carcinoma (HCC) is a highly heterogeneous malignancy. In the clinic, therapeutic resistance is largely attributed to tumor heterogeneity. Growing evidence indicates that cancer stem cells (CSCs) are the major source of tumor heterogeneity. Hence, uncovering the resistance mechanisms associated with CSC properties is essential for developing effective therapeutics. CSCs resemble embryonic stem cells. Embryonic development-related genes and signaling pathways are usually abnormally active and function as oncofetal drivers in HCC. Multiple strategies have been applied to identify oncofetal drivers. The mechanisms of CSC resistance could also provide reliable biomarkers to predict treatment failure. Precisely targeting these specific CSC properties may be effective in preventing or annihilating therapy resistance. This review provides an overview of drug resistance mechanisms associated with CSC traits and summarize therapeutic strategies against drug resistance.
Keywords
INTRODUCTION
Globally, hepatocellular carcinoma (HCC) is one of the most common and deadliest cancer types. Liver resection, ablation, and transplantation are potentially curative procedures but require an early diagnosis. Despite diligent surveillance, a large percentage of HCC patients present with intermediate or advanced stages of the disease, and treatments are often ineffective. To date, various molecular targeted drugs for advanced HCC have been developed. Both sorafenib and lenvatinib are inhibitors of multityrosine kinases that have been approved as a first-line treatment for advanced HCC management. Regorafenib has a molecular target spectrum similar to that of sorafenib and is applied as an alternative for sorafenib-resistant HCC patients. Cabozantinib and ramucirumab are novel tyrosine kinases used as second-line HCC treatments. Additionally, immune checkpoint inhibitors (ICIs) are also being considered as systemic HCC strategies. Some of the ICIs approved or in clinical research include agents (nivolumab, pembrolizumab, tislelizumab) against programmed cell death protein 1 (PD1), agents (atezolizumab, durvalumab, sintilimab) against PD-1 ligand (PDL1), agents (ipilimumab, tremelimumab) against cytotoxic T-lymphocyte associated protein 4 (CTLA4) and other agents [cobolimab against T-cell immunoglobulin and mucin domain-containing protein 3 (TIM3), and relatlimab against lymphocyte activation
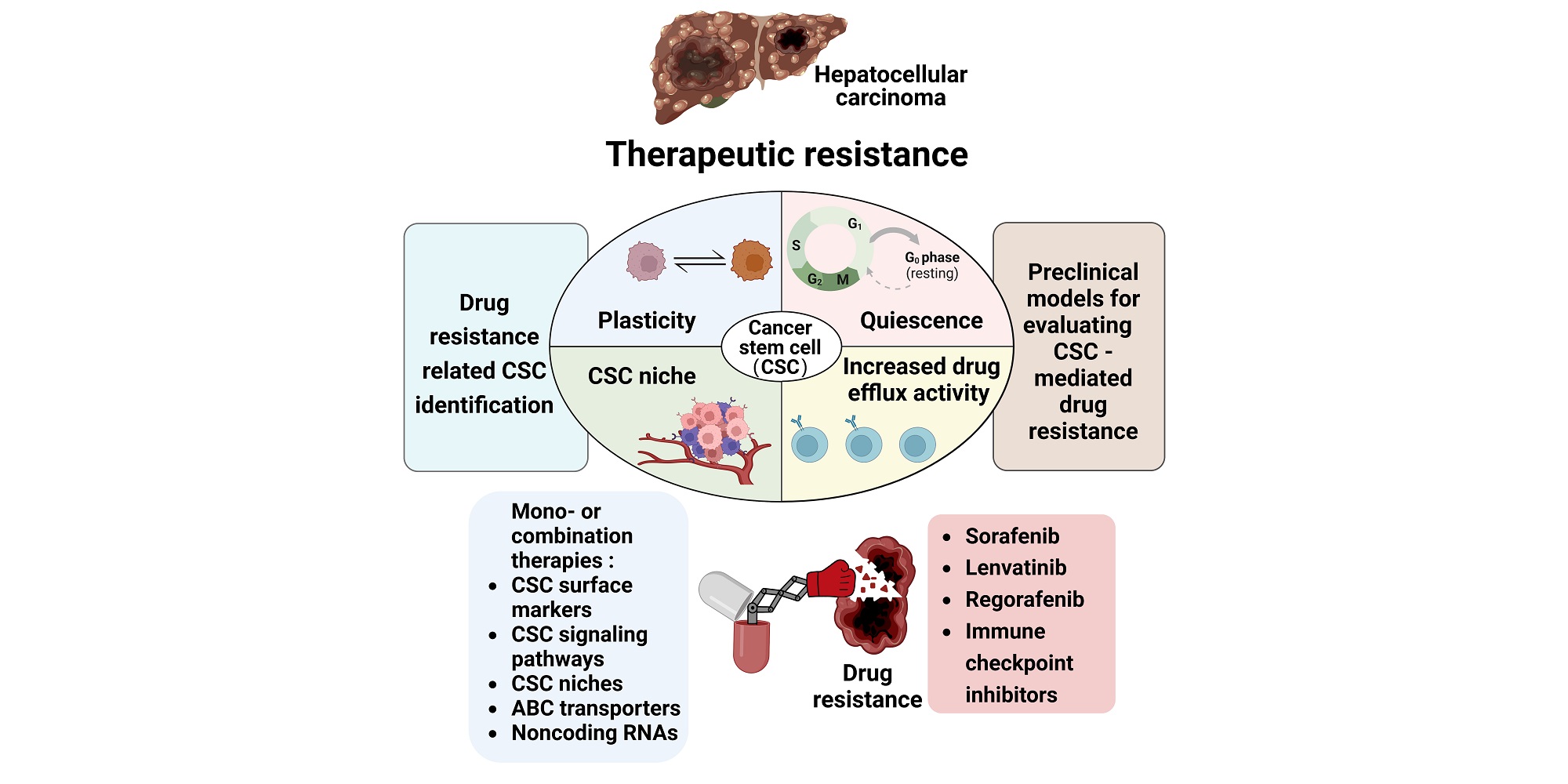
CSCs are a profoundly heterogeneous subpopulation of “stem-like" cancer cells that behave like "tumor-initiating cells" or "sphere-forming cells". Unlimited self-renewal, multilineage differentiation, and maintenance of pluripotency are key characteristics of CSCs, which are similar to those of embryonic stem cells (ESCs)[5]. Accumulating evidence suggests that drug resistance is largely attributed to CSCs, which account for much of the intratumor heterogeneity within each tumor population[6-9]. Integrating CSC properties into our understanding of drug resistance is crucial and may not only allow a better understanding of the mechanisms of chemoresistance but also facilitate the identification of potential biomarkers to predict treatment failure, as well as druggable targets for developing novel pharmacological strategies for improving the sensitivity of HCC to anticancer drugs. The present review summarizes recent studies on drug resistance mechanisms from the perspective of CSCs [Figure 1], provides an overview of therapeutic strategies for CSC-mediated drug resistance, and finally focuses on mono- or combination therapies against drug resistance [Table 1].
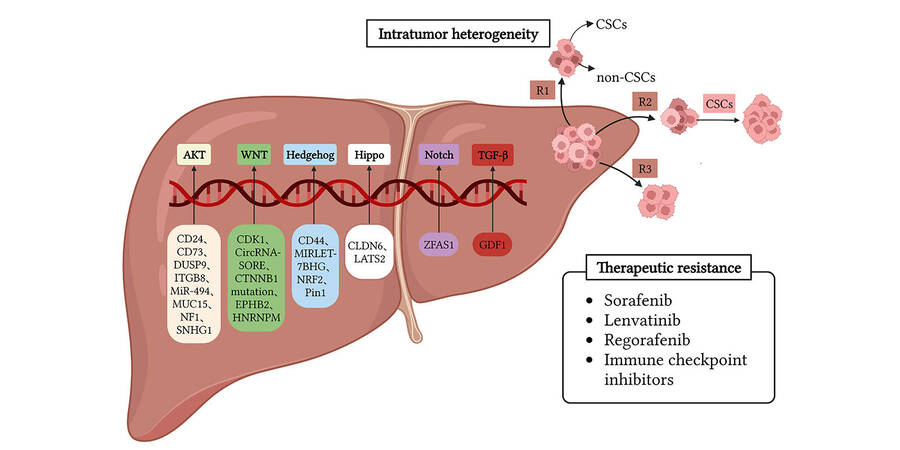
Figure 1. The mechanisms of heterogeneous CSCs that are responsible for the acquisition of therapeutic resistance. Selected mechanisms include CSC-associated potential biomarkers and developmental pathways.
Targeted therapies for resistant HCC
| Clinical drug | Resistance mechanisms | Candidate biomarker | Therapeutic strategies | Reference |
| Sorafenib | Suppress the DNA damage repair signaling through CHD1L | PARP1 | Olaparib | [22] |
| CD13 activates HDAC5/LSD1/NF-κB signaling | CD13 | CD13 shRNA, ubenimex | [36] | |
| CD24 increases PP2A protein production and induces the deactivation of the mTOR/AKT pathway | CD24 | CD24 shRNA | [37] | |
| Activation of JAK/STAT pathway in the SP/CD44+ fraction of the Akt/β-catenin tumorspheres | AKT/β-catenin | TG101209, AZ960 | [38] | |
| LGR5+ compartment | LGR5 | Diphtheria toxin+5-FU | [39] | |
| Increasing IL-10 and IL-35 expression and suppressing CD8+ T cells | CCR4+ Tregs | N-CCR4-Fc | [40] | |
| CLDN6/TJP2/YAP1 signaling regulatory axis | CLDN6 | CLDN6-DM1 | [21] | |
| CDK1/PDK1/β-Catenin signaling regulatory axis | CDK1 | RO3306 | [41] | |
| Activation of Hedgehog signaling pathway | CD44 | GANT61 | [42] | |
| NRF2/SHH/GLI signaling regulatory axis | NRF2 | NRF shRNA | [43] | |
| EPHB2 drives SRC/AKT/GSK3β/β-catenin signaling cascade, EPHB2 activates TCF1 to form a positive Wnt/β-catenin feedback loop | EPHB2 | rAAV8-shEPHB2 | [44] | |
| MiR-494 activates AKT/mTOR pathway | miR-494 | Anti-miR-494 | [45] | |
| circRNA-SORE sequesters miR-103a-2-5p and miR-660-3p and competitively activates the Wnt/β-catenin pathway | CircRNA-SORE | circRNA-SORE shRNA | [46] | |
| miR-21 promotes SNHG1, resulting in upregulation of SLC3A2, leading to the activation of AKT pathway | SNHG1 | SNHG1-Smart Silencer | [47] | |
| TRERNA1 activates the NRAS/Raf/MEK/ERK pathway by modulating miR-22-3p | TRERNA1 | TRERNA1 shRNA | [48] | |
| ZFAS1 activates stemness genes (eg., EpCAM, CD24, CD90, CD133,DLK1, Krt18/19), EMT markers (eg., S100A4/A6, Twist, Vimentin) and notch signaling pathway related genes (eg., Notch1-3, Delta1-4, Jagged2, Hes1/Hey1, DLL1, DLL4) | ZFAS1 | ZFAS1 siRNA | [49] | |
| Lenvatinib | CD73 upregulates SOX9 by AKT signaling, CD73 activates SOX9 transcription through c-Myc, CD73 prevents SOX9 ubiquitination andproteasome degradation by inhibiting GSK3β, GSK3β/SOX9/AKT signaling | CD73 | CD73 shRNA | [51] |
| CD73 activates AKT signaling via the Rap1/P110β cascade | CD73 | APCP | [52] | |
| EGFR/PAK2/ERK5 signaling regulatory axis | EGFR | Geftinib | [54] | |
| Activation of EGFR and IGF1R/INSR | EGFR | Erlotinib | [55] | |
| NF1 and DUSP9 loss activate PI3K/AKT and MAPK/ERK signaling pathways | NF1, DUSP9 | Trametinib | [56] | |
| ITGB8/HSP90/AKT signaling regulatory axis | ITGB8 | 17‐AAG, MK-2206 | [57] | |
| FGF19 enhances ST6GAL1 via STAT3 phosphorylation | ST6GAL1 | ST6GAL1 siRNA | [59] | |
| ADAMTSL5 upregulates RTKs, such as MET, EGFR, GAB1, PDGFRβ, IGF1Rβ, and FGFR4 | ADAMTSL5 | ADAMTSL5 shRNA | [60] | |
| miR-183-5p.1/MUC15/c-MET/PI3K/AKT/SOX2 regulatory circuit | MUC15 | ShMUC15 | [61] | |
| circMED27/miR-665-3p/USP28 signaling regulatory axis | circMED27 | circMED27 shRNA | [62] | |
| Regorafenib | Gankyrin activates β-catenin/c-Myc signaling | Gankyrin | 10058-F4 | [64] |
| Pin1 regulates the EMT via the Gli1/Snail/E-cadherin pathway | Pin1 | Pin1 shRNA | [65] | |
| LAST2 activates Hippo signaling pathway | LATS2 | Verteporfin | [66] | |
| RAB27A-dependent exosome secretion induces Nanog expression | RAB27A | CRISPR-Cas9-sgRAB27A | [67] | |
| Activation of TGF-β and Wnt/β-catenin signaling pathways | TGF-β signaling, Wnt/β-catenin signaling | TGFβ-R1 inhibitor | [68] | |
| ATF3 binds to the IL-6Rα promoter and induces IL-6Rα expression | IL-6Rα | Sarilumab | [69] | |
| Immune checkpoint inhibitors | Wnt/CTNNB1 mutation alters Wnt-β-catenin pathway | Wnt/CTNNB1 mutation | Not Applicable | [71] |
| CTNNB1 mutation alters Wnt-β-catenin pathway | CTNNB1 mutation | Ccl5 | [72] | |
| GDF1 induces ALK7-SMAD2/3 signalling cascade and suppresses LSD1 to boost CTA expression | GDF1 | HY-100546A | [23] | |
| SOX2, OCT4, HNRNPM, MBD2a, and FZD3 comprise a positive feedback loop | HNRNPM | HNRNPM-specific LNA modified ASOs | [73] | |
| PKCα phosphorylates ZFP64 at S226 and induces its nuclear translocation, leading to transcriptional activation of macrophage CSF1 | PKCα | Gö6976 | [74] | |
| circMET activates miR-30-5p/Snail/DPP4/CXCL10 axis | circMET | Sitagliptin | [75] |
CSC AND HCC HETEROGENEITY
HCC is a highly heterogeneous disease with unique biological features and molecular backgrounds that may respond differently to different treatments. A well-documented study links heterogeneity to clinical outcomes. HCC heterogeneity consists of intertumor (within the tumor cell population) and intratumor (tumors from the same patient) heterogeneity. Convincing evidence shows that CSC heterogeneity is the major source of intratumor heterogeneity that induces chemoresistance and subsequent tumor relapse. Different CSC subpopulations function differently, exhibit different developmental states or express different gene expression profiles[10,11]. Currently, studies of liver CSCs have mainly focused on identifying liver CSC surface markers via cell-sorting and xenotransplantation analyses in immunodeficient mice. A number of the liver CSC markers in HCC appear to be oncofetal markers, as the development of HCC is similar to that of the development of fetal, normal, and regenerating livers. Classical stemness markers, including Nanog, SRY-box transcription factor 2 (SOX2), and Oct4, and liver CSC markers, including CD13, CD24, CD44, CD47, CD90, CD133, intercellular adhesion molecule 1 (ICAM1), epithelial cell adhesion molecule (EPCAM), Leucine-rich repeat-containing G protein-coupled receptor 5 (LGR5), OV6, and Cytokeratin 19 (CK19), have been extensively applied[9].
CSCs exhibit features similar to those of ESCs due to their ability to retain the activity of vital and highly conserved developmental signaling pathways involved in managing the features of embryonic cells, normal organogenesis, and cell lineage differentiation, which may lead to the initiation or progression of poorly differentiated HCC[5]. Several important and well-characterized signaling pathways, including the protein kinase B (AKT), Hedgehog, Hippo, nuclear factor kappa-B (NF-κB), Notch, TGF-β and Wnt/β-catenin pathways, are involved in acquiring and maintaining stem-like traits, such as self-renewal, plasticity, and quiescence[8]. Interestingly, tumor cells expressing distinct CSC markers often exhibit activation of CSC pathways. For example, Notch and Jagged are highly expressed in CD133+ HCC CSCs[12]. Specifically, the molecular subtypes are usually indicative of tumor heterogeneity. The expression levels as well as the activation degree of CSC markers and signaling pathways vary among subtypes, which is a major challenge in preventing or abolishing CSC resistance[13].
MAJOR HINDRANCE OF RESISTANCE
Growing evidence shows that tumor cells harboring stem cell-like characteristics are more resistant to conventional therapeutic approaches than nonstem-like populations. The mechanisms of resistance occurrence and the acquisition of stem-like cell traits are tightly linked to the properties of CSCs, including plasticity, quiescence, CSC niches and the increased drug efflux activity of CSCs.
Plasticity
Triggered by microenvironmental cues (e.g., oncogenic stress, inflammation, and senescence), cancer cells in a nontransformed differentiated state can shift to a tumorigenically transformed undifferentiated or CSC state; this process is referred to as “plasticity”[14]. Epithelial-to-mesenchymal transition (EMT) is the most typical example of plasticity. During tumor initiation, tumor cells with a differentiated epithelial phenotype lose apicobasal polarity and acquire a stem-like gene expression program and self-renewal capacity. In turn, the elevation of EMT not only enforces the tumor-initiating capacity but also exacerbates metastatic and therapeutic resistance potential[15,16].
CSC plasticity is usually a result of multilineage interconversion, transdifferentiation and dedifferentiation[17]. Aberrantly activated plasticity enables tumors to adapt to growth constraints and resist therapy[18]. In our recently published studies, we developed a hepatocyte differentiation model that mimics liver development by inducing human ESCs to differentiate into adult hepatocytes along hepatic lineages[19]. Inspired by this model, we found that chromodomain-helicase-DNA-binding protein 1-like gene (CHD1L)[20], Claudin6 (CLDN6)[21], poly (ADP-ribose) polymerase 1 (PARP1)[22] and growth differentiation factor 1 (GDF1)[23] are potential therapeutic targets linked to HCC lineage plasticity. These four oncofetal drivers are expressed in the embryonic stage, diminish during terminal differentiation, and reamplify abnormally in HCC. This dynamic change is accompanied by lineage plasticity as a shift from a hepatic lineage to a biliary or progenitor lineage. Notably, the phenotypic shift caused by a high abundance of CLDN6 and PARP1 confers sorafenib resistance. Further suppression of our candidate targets may adversely affect poorly differentiated HCC and sensitize patients to chemotherapeutic agents.
Quiescence
Quiescence or dormancy is another characteristic of CSCs that hinders standard therapy. Upon microenvironmental alterations, such as oxidative stress, hypoxia, nutrient deprivation or chemotherapeutic pressure, CSCs can temporarily transition into the Go phase of the cell cycle and remain dormant. Most standard cancer treatments are directed against proliferating cells, and quiescent or dormant CSCs can evade therapies and revert to the proliferative state when favorable conditions appear[24]. Quiescence may be triggered by altered CSC signaling pathways in response to unfavorable microenvironmental stimuli. For example, in HCC, the Smad-independent TGF-β pathway can activate the dormancy program. Restoration of miR122 expression can direct HCC stem-like cells toward cell differentiation and tumor dormancy[25]. TGF-β signaling participates in switching quiescent hepatic stellate cells (HSCs) to a myofibroblast (MFB) phenotype through the process of activation to transdifferentiation[26]. In addition, MIRLET7BHG induces the activation of quiescent HSCs by provoking the SMO-involved Hedgehog pathway[27].
CSC niche
The tumor microenvironment (TME) consists of multiple types of benign cells, extracellular matrix, and signaling molecules. By inducing inflammation, angiogenesis, hypoxia, and fibrosis, particularly in chronically damaged liver tissue, the TME contributes to tumor progression and response to therapy. HCC harbors CSCs in dedicated niches. The extracellular matrix (ECM) component residing in the TME is made up of glycosaminoglycans, fibronectins, collagens, proteoglycans, elastins, and other glycoproteins, which are required for the development of stemness-related CSC phenotypes. An ECM niche is a complex network of macromolecules produced by cancer-associated fibroblasts (CAFs), macrophages, and endothelial cells (ECs) that regulates tissue architecture and intracellular signaling. The remodeling of the ECM in CSC niches allows CSCs and the ECM to interact abnormally, which is crucial for CSC senescence, quiescence, and phenotypic plasticity. CAFs display enhanced remodeling of the ECM and alter extracellular signaling, producing cytokines and growth factors that fuel cancer cell self-renewal. Activation of stem cell signals has been linked to CAF-mediated therapy resistance in some cases. By allowing tumor cells to disseminate, a soft matrix stiffness promotes tumor outgrowth. As HCC progresses, higher ECM stiffness favors proliferation and self-renewal of CSCs, whereas soft ECM may facilitate metastasis of CSCs[7,9].
Hypoxia is critical for the formation and maintenance of CSCs. In the hypoxic TME, HCC cells remain undifferentiated, clone faster, and express specific biomarkers for CSCs. Hypoxia can also enhance HCC stemness via hypoxia-inducible factor (HIF)-dependent mechanisms. Although hypoxic conditions enrich CSC subsets in HCC, biologically distinct liver CSC subpopulations with differing single-cell surface markers and hypoxia-responsive traits have been identified[1].
An increasing number of studies have shown a strong interplay between CSCs and angiogenesis. Angiogenesis nurtures CSCs by secreting a variety of instructive angiocrine factors to establish a vascular niche and promote the CSC phenotype and regulate CSC stemness. In turn, CSCs preferentially secrete exosomes or angiogenic factors to activate ECs. As CSCs differentiate, they become cancer vascular stem cells/progenitor cells and mimic ECs by forming blood vessels. This phenomenon is termed vasculogenic mimicry (VM), which directly affects tumor microcirculation. In addition, as a result of the vascular microenvironment, stem cells preserve undifferentiated dormancy and protect themselves from injury[7,24].
It is becoming increasingly apparent that CSCs and immune cells that infiltrate tumors communicate reciprocally, resulting in immune evasion and tumor progression, as well as having therapeutic implications. The polarization and activation of cells, including tumor-associated macrophages (TAMs), neutrophils (TANs), myeloid-derived suppressor cells (MDSCs), B cells, and subsets of T lymphocytes, suppress adaptive immunity and signaling directly to cancer cells. By activating downstream stem cell pathways, growth factors and cytokines released from immune cells can boost self-renewal and increase treatment resistance. Clinical sensitivity to checkpoint blockade is variable, and many patients exhibit acquired or primary resistance. A patient's response to immunotherapy depends on the expression of cell surface molecules, antigenicity, and T-cell infiltration and function. CSCs can modulate these factors to determine a patient's sensitivity. As CSCs have been found to preferentially activate immune suppressive signaling, disrupting immune suppressive signaling could block self-renewal and improve therapeutic sensitivity[9,28].
Increased drug efflux activity of CSCs
ATP-binding cassette (ABC) transporters are capable of exporting a wide range of toxin-producing substances from cells and thus directly contribute to the acquisition of resistance. CSCs overexpress ABC transporters and dysregulate signaling pathway networks, leading to the acquisition of multidrug resistance and maintenance of self-renewal properties, respectively. Side population (SP) cells can be identified as CSCs. ABCG2 was the first ABC transporter to be identified as having phenotypic significance for SP cells. SP cells sorted from HCCLM3, MHCC97-H, MHCC97-L and Hep3B cells display CSC characteristics, remarkable levels of chemoresistance and ABCG2 expression[29,30]. TGF-β signaling contributes to drug resistance in liver cancer cells by inducing the expression of the xenobiotic nuclear receptor PXR[31]. The ATP-binding cassette transporter ABCF1 functions as a hepatic oncofetal protein and modulates chemoresponse, migration, EMT and cancer stemness properties[32]. SOX9 enhances sorafenib resistance by modulating ABCG2 expression. Thus, ABC transporter blockade may attenuate CSC-mediated chemoresistance[33].
THERAPEUTIC RESISTANCE IN HCC: FROM THE PERSPECTIVE OF CSC TRAITS
Currently, molecular targeted therapies and immunotherapy are commonly used to treat HCC. Molecular targeted drugs consist of first-line sorafenib and lenvatinib and second-line regorafenib, cabozantinib and ramucirumab. Immunotherapy is a novel management option for HCC and principally includes ICIs against PD-1, PD-L1 and CTLA-4, such as nivolumab, pembrolizumab, MED14736, MPDL3280a, ipilimumab and tremelimumab[34]. The indications, contraindications and interactions for the first- and second-line drugs cited in the review are listed in Table 2. Although these strategies have an increased survival benefit for advanced and metastatic HCC patients, anticancer efficacy remains unsatisfactory, as most patients will eventually acquire drug resistance due to the existence of CSCs; therefore, elucidation of the mechanisms of drug resistance from the CSC perspective is essential for developing effective therapies. Here, the review focuses on related articles published in the past five years concerning CSC-mediated therapeutic resistance in HCC.
Indications, contraindications and interactions for the first- and second-line drugs cited in the review
| First/second-line treatment | Drugs | Indications | Contraindications | Interactions | References |
| First-line | Sorafenib | • Advanced renal cell carcinoma (RCC) • Unresectable HCC • Locally recurrent or metastatic, progressive, differentiated thyroid carcinoma refractory to radioactive iodine treatment | • Patients who are hypersensitive to sorafenib or any other component of this drug • Patients have squamous cell lung cancer and receive carboplatin and paclitaxel | • Decrease efficacy: strong CYP3A4 inducers (e.g. carbamazepine, dexamethasone, phenobarbital, phenytoin, rifampin, rifabutin, St. John’s wort); Neomycin | [95,96] |
| Lenvatinib | • Radioactive iodine-refractory differentiated thyroid cancer (DTC) • Unresectable or advanced HCC • Advanced RCC • Endometrial carcinoma | None | • Increase toxicity: CYP3A, P-gp, and BCRP inhibitors (e.g. Ketoconazole); P-gp inhibitors: [e.g. Rifampicin (600 mg as a single dose)] • Decrease efficacy: CYP3A and P-gp inducers [e.g. Rifampicin (600 mg daily for 21 days)] • Prolong the QT/QTc interval: Drugs known to prolong QT/QTc intervals (e.g. Everolimus) | [97,98] | |
| Atezolizumab and bevacizumab combination | • Treatment of advanced or unresectable HCC not amenable to curative or locoregional therapies, who have not received prior systemic therapy • Barcelona clinic liver cancer (BCLC) stage B or C • Child-Pugh A classification of liver function • Oesophagogastroduodenoscopy for varices within the last 6 months • ECOG performance status 0 or 1. | None | • There are no known drug interactions with atezolizumab bevacizumab | [99,100] | |
| Second-line | Regorafenib | • Colorectal cancer • Gastrointestinal stromal tumors • HCC | None | • Decrease efficacy: strong CYP3A4 inducers (e.g. rifampin, phenytoin, carbamazepine, phenobarbital, and St. John’s Wort) • Increase toxicity: strong CYP3A4 inhibitors (e.g. clarithromycin, grapefruit juice, itraconazole, ketoconazole, nefazodone, posaconazole, telithromycin, and voriconazole) • Increase breast cancer resistance protein (BCRP) substrates toxicity: BCRP substrates | [101] |
| Cabozantinib | • Advanced RCC • HCC | None | • Increase the risk of exposure-related adverse reactions: strong CYP3A4 inhibitors • Reduce efficacy: strong CYP3A inducers | [102] | |
| Ramucirumab | • Gastric cancer • Non-small cell lung cancer (NSCLC) • Colorectal cancer • HCC | None | No information provided | [103] | |
| Pembrolizumab | • Unresectable or metastatic melanoma • NSCLC • Head and neck squamous cell cancer • Relapsed or refractory hodgkin lymphoma • Primary mediastinal large B-cell lymphoma • Urothelial carcinoma • Microsatellite instability-high or mismatch repair deficient cancer • Gastric cancer • Esophageal cancer • Recurrent or metastatic cervical cancer • HCC • RCC • Merkel cell carcinoma • Endometrial carcinoma • Tumor mutational burden-high cancer • Cutaneous squamous cell carcinoma • Triple-negative breast cancer | Hypersensitivity to pembrolizumab or any component of the formulation. | Thalidomide analogues: Pembrolizumab may enhance the adverse/toxic effect of thalidomide analogues. | [104,105] | |
| Nivolumab | • Hodgkin lymphoma, classical • HCC • Colorectal cancer, metastatic (microsatellite instability-high or mismatch repair deficient cancer) • Head and neck squamous cell cancer (recurrent or metastatic) • Melanoma • NSCLC, metastatic, progressive • Small cell lung cancer, metastatic • Advanced RCC | Hypersensitivity to nivolumab or any component of the formulation | • Belimumab: Monoclonal antibodies may enhance the adverse/toxic effect of Belimumab • Immunosuppressants: May diminish the therapeutic effect of nivolumab | [106] | |
| Ipilimumab | • Metastatic colorectal cancer • Unresectable or metastatic melanoma • Advanced RCC | Hypersensitivity to ipilimumab or any component of the formulation; active life-threatening autoimmune disease, or with organ transplantation graft where further immune activation is potentially imminently life-threatening | • Vemurafenib: Ipilimumab may enhance the hepatotoxic effect of vemurafenib • Disconinue breastfeeding during treatment and for 3 months following the final dose | [107] |
Sorafenib
Sorafenib is a multikinase inhibitor that targets the tyrosine kinases vascular endothelial growth factor receptor (VEGFR)/platelet-derived growth factor receptor (PDGFR) and RAF serine/threonine kinases in the RAS/RAF/MEK/extracellular signal-regulated kinase (ERK) pathway[35]. Recent studies have revealed that enriched CSCs contribute to sorafenib resistance. CD13 not only provokes HCC carcinogenesis but also induces sorafenib resistance by activating histone deacetylase 5 (HDAC5), lysine-specific demethylase 1 (LSD1)-NF-κB oncogenic signaling[36]. CD24 regulates resistance to sorafenib by deactivating the mammalian target of rapamycin (mTOR)/AKT pathway[37]. AKT/β-catenin HCC tumors harbor a subpopulation of cells that overexpress the cancer stem cell-like marker CD44, which may contribute to tumor sustenance and therapy resistance[38]. The LGR5+ cell population, which harbors features of tumor-initiating cells (TICs), is resistant to sorafenib treatment[39]. Tumor-infiltrating CCR4+ regulatory T cells (Tregs) display PD-1+TCF1+ stem-like properties and enhance immunosuppressive resources in the TME. CCR4+ Tregs promote sorafenib resistance by increasing IL-10 and IL-35 expression and suppressing CD8+ T cells[40].
In addition to liver CSC markers, CSC signaling pathways are another key factor involved in sorafenib-mediated resistance. Cyclin-dependent kinase 1 (CDK1), which is frequently overexpressed in HCC, reduces sorafenib efficacy via CDK1/PDK1/β-catenin signaling[41]. The CD44-positive HCC subpopulation exhibits sorafenib resistance; however, Hedgehog signaling inhibition can sensitize this subpopulation to the drug[42]. Nuclear factor (erythroid-derived 2)-like 2 (NRF2) is a transcription factor that is highly enriched in liver TICs and regulates HCC TIC properties and sorafenib resistance through regulation of the SHH (sonic hedgehog)/GLI (glioma-associated oncogene homolog) signaling cascade[43]. Our published oncofetal driver CLDN6 is more refractory to sorafenib treatment because it enhances tumor lineage plasticity via the CLDN6/TJP2 (tight junction protein 2)/YAP1 (yeast aspartyl protease) interaction axis and further activates the Hippo signaling pathway[21]. The Wnt target gene EPHB2 (EPH receptor B2) enhances CSC properties and drives sorafenib resistance via the TCF1/EPHB2/β-catenin positive feedback loop[44].
Increasing evidence suggests that noncoding RNAs (ncRNAs), mainly miRNAs and lncRNAs, are key factors in the development of drug resistance in HCC. miR-494 is associated with stem cell-like characteristics and decreases the sorafenib response by regulating the AKT/mTOR pathway and decreasing PARP levels[45]. N6-methyladenosine-modified circRNA-SORE functions as a miRNA sponge to sequester miR-103a-2-5p and miR-660-3p and maintains sorafenib resistance by competitively activating the Wnt/β-catenin pathway[46]. Overexpression of lncRNA SNHG1 (small nucleolar RNA host gene 1) supports sorafenib resistance by activating the AKT pathway via upregulation of SLC3A2 (solute carrier family 3 member 2), and its increased nuclear expression is promoted by miR-21 in HCC cells[47]. Translation regulatory lncRNA 1 (TRERNA1) acts as a miR-22-3p sponge to positively regulate NRAS expression at the posttranscriptional level. TRERNA1 enhances HCC sorafenib resistance by activating the RAS/Raf/MEK/ERK signaling pathway[48]. Under long-term sorafenib exposure, HCC cells enrich a fraction of quiescent stem-like cells. Single-cell RNA analysis has demonstrated the vital role of stemness and EMT in causing sorafenib resistance. ZNFX1 antisense RNA 1 (ZFAS1) is a novel regulator lncRNA found to be overexpressed in higher stages (III/IV) of HCC and in poorly (G3)/undifferentiated HCC (G4) compared with levels at early stages (I/II) and in highly (G1)/moderately (G2) differentiated HCC. Overexpression of ZFAS1 increases the expression of stemness genes (e.g., EpCAM, CD24, CD90, CD133, DLK1, and CK18/19) and EMT markers (e.g., S100A4/A6, Twist, and Vimentin). Thus, ZFAS1is regarded as a critical mediator of sorafenib resistance via the induction of stemness and EMT phenotypes[49].
Lenvatinib
Lenvatinib is another oral multikinase inhibitor that selectively inhibits the tyrosine kinases VEGFR1-3, fibroblast growth factor receptor (FGFR)-1-4, PDGFR-α, c-Kit and RET and is recommended as a first-line systematic therapy in patients with advanced HCC[50]. CD73 is a novel biomarker and poor prognosis indicator of HCC, and overexpression of CD73 is essential for the sustainment of CSC features, thereby promoting HCC progression and metastasis. CD73 boosts lenvatinib resistance by upregulating SOX9 (sex determining region Y-box 9), and the increase in SOX9 expression induced by CD73 is achieved through reactivation of AKT signaling, followed by enhancement of SOX9 transcription via the downstream AKT signaling target c-Myc. Moreover, CD73 hinders SOX9 ubiquitination and proteasome degradation by abolishing GSK3β[51,52]. By applying a kinome-centered clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated system (CRISPR-Cas9) genetic screen, EGFR was found to be negatively correlated with HCC sensitivity to lenvatinib. Notably, EGFR is an essential receptor tyrosine kinase regulator of CSCs[53]. In terms of mechanism, lenvatinib inhibits FGFR and then triggers EGFR-PAK2-ERK5 signaling activation[54]. In addition, IGF1R is responsible for the acquisition of further lenvatinib resistance in HCC cells[55]. Another genome-scale CRISPR-Cas9 knockout screening identified two important resistance genes, neurofibromin 1 (NF1) and dual specificity phosphatase 9 (DUSP9). Removal of NF1 and DUSP9 limits the response of HCC to lenvatinib. The absence of NF1 activates both the PI3K/AKT and mitogen-activated protein kinase (MAPK)/ERK signaling pathways, while DUSP9 loss activates the MAPK/ERK but not the PI3K/AKT pathway, thereby downregulating FOXO3 activity, followed by FOXO3 degradation[56]. Integrin subunit beta 8 (ITGB8) is a novel contributor to lenvatinib resistance. Elevated expression of ITGB8 may be regulated via the transcription factor NF-κB. ITGB8-mediated resistance to lenvatinib occurs through an induced increase in the expression of HSP90, which inhibits AKT ubiquitination and promotes AKT stabilization, thereby activating the AKT signaling pathway[57]. ST6 beta-galactoside alpha-2,6-sialyltransferase 1 (ST6GAL1) is a tumor-derived secreted protein that is positively regulated upstream of fibroblast growth factor (FGF)-19 in HCC cells. FGF19 is a critical oncogenic driver gene that facilitates CSC-like properties in liver CSCs[58]. Intriguingly, low levels of FGF19 eliminated lenvatinib susceptibility. However, FGF19 was re-expressed in lenvatinib-resistant HCC cells; therefore, FGF19 is a potential biomarker of lenvatinib-susceptible HCC. Proteome and secretome analyses revealed that FGF19 transcriptionally activates ST6GAL1 via STAT3 phosphorylation, and serum ST6GAL may be a useful biomarker for the identification of lenvatinib-susceptible FGF19-driven HCC[59]. The HCC-secreted glycoprotein, a disintegrin and metalloprotease domain containing thrombospondin type 1 motif-like 5 (ADAMTSL5), is another master regulator of tumorigenicity. ADAMTSL5 depletion gives rise to loss of the expression of the HCC marker AFP and the CSC markers CD133, EPCAM and CDH1 and interferes with the self-renewal ability of HCC cells. Strikingly, ADAMTSL5 downregulation influences HCC cell sensitivity to clinically relevant drugs, such as sorafenib, crizotinib, lenvatinib, and regorafenib[60]. Mucin 15 (MUC15) is downregulated in CD24+ or EpCAM+ HCC cells as well as in liver tumor-initiating cells (TICs) and is essential for HCC cell self-renewal, tumorigenicity, and lenvatinib resistance. Mechanistically, MUC15 interacts with c-MET and then inhibits PI3K/AKT/SOX2 signaling. Additionally, miR-183-5p.1 targets the 3’-UTR of MUC15 directly. In turn, SOX2 transactivates miR-183-5p.1 to inhibit MUC15 expression in liver TICs. Importantly, the miR-183-5p.1/MUC15/c-MET/PI3K/AKT/SOX2 regulatory circuit triggers lenvatinib resistance[61].
The expression of the ncRNA circMED27 is elevated in HCC serum and is positively associated with a poor prognosis for HCC patients. circMED27 induces insensitivity to lenvatinib by sponging miR-655-3p, which acts as a tumor suppressor by inhibiting the β-catenin pathway[62].
Regorafenib
Regorafenib is an oral multikinase inhibitor approved as a second-line treatment for unresectable HCC. Regorafenib targets a broad range of relevant protein kinases, including VEGFR1-3, TIE2, FGFR1-2, PDGFR, KIT, RAF, and RET. Although regorafenib brings clinical benefits, drug resistance is inevitable[63]. Gankyrin augments β-catenin mRNA levels by increasing the expression of the RNA-binding protein HuR. The upregulation of β-catenin promotes the expression of c-Myc, which contributes to the tolerance to sorafenib and regorafenib mediated by Gankyrin in HCC[64]. Pin1 is a high-abundance gene in regorafenib-resistant HCC cells. Pin1 contributes to regorafenib resistance by affecting EMT traits. Inhibition of Pin1 suppresses EMT and metastasis by reducing the levels of the EMT regulators E-cadherin and Snail; additionally, Pin1 interacts with another EMT regulator, Gli1[65]. Through CRISPR/Cas9 screening, the Hippo signaling pathway, which is responsible for regorafenib resistance, was identified. Inactivation of large-tumor suppressor 2 (LATS2) leads to YAP dephosphorylation, and YAP inhibition resensitizes regorafenib-insensitive HCC cells to regorafenib[66]. RAB27A is a Rab GTPase that controls the release of exosomes. Exosomes secreted by liver CSCs transfer regorafenib resistance to differentiated HCC cells when RAB27A is activated. Moreover, RAB27A-derived exosomes promote Nanog expression to maintain CSC self-renewal ability. Downregulating RAB27A sensitizes HCC cells to regorafenib[67]. Interestingly, acute regorafenib administration enhances Wnt/β-catenin signaling in hepatoblast-like HCC cells, along with the activation of hepatic stem/progenitor markers. In turn, the activation of Wnt/β-catenin signaling caused by Wnt3a/R-Spo1 intervention avoids apoptosis from regorafenib stimuli. In addition, long-term regorafenib tolerance leads to enhanced TGF-β signaling activity and increased expression of the CSC markers CD24 and CD133[68]. Interleukin-6 receptor alpha (IL-6Rα) is induced in response to sorafenib and is essential for IL-6-mediated sorafenib resistance in HCC. Transcription factor 3 (ATF3) binds to the IL-6Rα promoter and induces IL-6Rα expression. The ATF3-IL-6Rα cascade is also activated in regorafenib-resistant HCC cells. Blockade of IL-6Rα sensitizes HCC to sorafenib and regorafenib both in vitro and in vivo[69].
Immune checkpoint inhibitors
Despite the promising outcomes of ICIs for HCC treatment, not all patients are sensitive to ICIs and inevitably acquire resistance to ICI therapy. A large number of studies have shown that CSCs are necessary for immunosuppressive microenvironment formation, resulting in immune evasion. The molecular mechanisms mainly include elevated expression of immunosuppressive factors and activation of CSC signaling pathways correlated with ICI resistance. The Wnt/β-catenin signaling pathway and TGF-β signaling pathway are closely connected with immune evasion, exclusion and resistance[70].
An HCC immune-excluded class (cold tumors), defined by Wnt/CTNNB1 mutations, is refractory to ICIs in patients treated with anti-PD-1/PD-L1 monoclonal antibodies (81%), anti-CTLA-4 monoclonal antibodies or combinations of anti-PD-1/PD-L1 antibodies with anti-CTLA-4, anti-KIR or anti-LAG3 antibodies (19%)[71]. MYC; p53-/- HCC tumors show highly active β-catenin signaling, which directly promotes immune escape. In addition, the β-catenin-driven MYC-lucOS;CTNNB1 or MYC-luc;CTNNB1 models are insensitive to nivolumab and pembrolizumab. Of particular interest, CTNNB1 mutation may be used as a biomarker for HCC patient exclusion[72]. In our recent studies, the secreted protein GDF1, which belongs to the TGF-β superfamily, was found to be highly expressed in embryonic stem cells and is gradually downregulated in the endoderm, liver progenitor cells, premature hepatocytes, and hepatocytes but reactivated in HCC. Overexpression of GDF1 leads to tumor dissemination and metastasis. Ectopic expression of GDF1 can induce tumor-lineage plasticity through the activin receptor-like kinase 7 (ALK7)-mothers against decapentaplegic homolog 2/3 (SMAD2/3) signaling cascade. Intriguingly, GDF1-mediated lineage plasticity might be an Achilles heel for HCC immunotherapy. A wide panel of cancer-testis antigens (CTAs) are activated in HCC by GDF1 via suppression of the epigenetic regulator lysine-specific demethylase 1 (LSD1). The inhibition of LSD1 is mediated by SMAD2/3 binding to its promoter region in a GDF1-dependent manner[23]. HNRNPM (heterogeneous nuclear ribonucleoprotein M) is an elevated oncofetal splicing factor that has the same expression pattern as GDF1 in HCC. HNRNPM is essential for maintaining the stem cell-like properties and tumorigenesis of HCC cells. Moreover, HNRNPM mediates the immunosuppressive tumor environment in HCC. Mechanistically, HNRNPM binds to the flanking introns of MBD2 to promote its alternative splicing. MBD2a, one of the isoforms of MBD2, enhances FZD3 expression and activates the Wnt/β-catenin signaling pathway. FZD3 and β-catenin further promote the expression of SOX2 and OCT4. Interestingly, ectopic expression of SOX2 and OCT4, in turn, upregulates HNRNPM expression via direct promoter binding[73]. Patients with anti-PD1-resistant HCC are frequently positive for zinc finger protein 64 (ZFP64). ZFP64 induces anti-PD1 resistance by shifting macrophage polarization to an abnormal activation phenotype (M2) and inhibiting the TME. Mechanistically, protein kinase C alpha (PKCα) phosphorylates ZFP64 at S226 and induces its nuclear translocation, leading to transcriptional activation of macrophage colony-stimulating factor (CSF1). This results in a shift of macrophages to the M2 phenotype. Clinically, anti-PD1 resistance is frequently observed in individuals with an active PKC/ZFP64/CSF1 axis[74].
circMET (hsa_circ_0082002) is an onco-circRNA that is upregulated in HCC, and overexpression of circMET promotes EMT and the formation of an immunosuppressive tumor microenvironment. Generally, the effect of immune tolerance triggered by circMET occurs through the Snail/dipeptidyl peptidase 4 (DPP4)/CXCL10 axis. In detail, circMET works as a sponge for miR-30-5p; Snail is a novel identified target of miR-30-5p, and the upregulation of Snail significantly promotes DPP4 expression. The miR-30-5p/Snail/DPP4 axis further degrades CXCL10 to trigger immunosuppression[75].
THERAPEUTIC STRATEGIES FOR CSC-MEDIATED DRUG RESISTANCE
Drug resistance-related CSC identification strategies
The identification of drug resistance-related CSCs can be achieved by multi-omics (e.g., transcriptomics, proteomics, metabolomics, lipidomics, glycomics) in publicly accessible repositories (e.g., gene expression omnibus (GEO), CancerDR, canSAR, Genomics of Drug Sensitivity in Cancer data, and Platinum) or institutional sources. However, due to intratumor heterogeneity, tissue biopsy sampling errors may occur. Strikingly, liquid biopsy has recently gained popularity as a noninvasive and repeatable means of detecting drug resistance indicators in body fluids. Liquid biopsy can alleviate the difficulties in collecting tissue biopsies, provide insight into spatial and/or temporal heterogeneity, and facilitate dynamic therapeutic response monitoring (detection of resistance mechanisms)[76,77].
Multiple techniques have been applied for analysis and post-analysis after sample collection and processing. To date, drug-resistant drivers have been measured via real-time quantitative polymerase chain reaction (qPCR), immunoblotting, immunohistochemistry, immunofluorescence, flow cytometry, mass spectrometry and next-generation sequencing (NGS). Recent achievements in NGS, such as bulk-cell RNA sequencing, genome-wide CRISPR sequencing, long-read RNA sequencing, circular RNA sequencing and single-cell RNA sequencing (scRNA-seq), have provided extraordinary insights into tumor heterogeneity, treatment resistance, tumor relapse, and metastasis. Ho et al. adopted single-cell genomics to unveil the landscape of intratumoral heterogeneity and identify a rare cell subpopulation of CD24+/CD44+ cells within the EPCAM+ population in HCC[78]. Zhou et al. applied lineage tracing and scRNA-seq to reveal the increasing tumorigenicity and the ability to form cancers of differential lineages of Prom1+ HCC cells, highlighting the heterogeneity and dynamics of Prom1+ HCC cells, which confer a dedifferentiated status and stem cell traits[79]. CD24, CD44, and PROM1 have already been verified to be responsible for drug resistance in HCC[37,42,80].
Preclinical models for evaluating CSC-mediated drug resistance
Tumor-derived cell lines and patient-derived xenografts (PDXs) are the two most common models used for drug resistance studies because they are able to retain the majority of the molecular characteristics of primary tumors. A large variety of tumor-derived cell lines are acceptable for studies on the molecular prediction of drug response and biomarker discovery. Caruso et al. performed whole-exome RNA and microRNA sequencing to screen the efficacy of 31 anticancer agents in 34 liver cancer cell lines and distinguished genetic alterations and gene expression patterns correlated with drug response[81]. Large-scale functional screening using RNAi or CRISPR/Cas9 is another promising technique for studying CSC-mediated drug resistance. For instance, Wei et al. utilized CRISPR/Cas9 library screening and identified phosphoglycerate dehydrogenase (PHGDH) as an essential gene for sorafenib resistance[82]. Overexpression of PHGDH leads to regorafenib and lenvatinib resistance in HCC. In particular, PHGDH is required for cancer stem cell maintenance[83]. Despite the ease of manipulation and suitability for high-throughput screening of tumor-derived cell lines, they are unable to fully reflect the native 3D environment of tumor cells. Instead, PDXs more closely mimic parental tumors. He et al. developed the first public liver cancer PDX model database, which contains 116 PDTXs generated from HCC[84]. The molecular and drug response data, as well as the clinical annotation, are available from: http://www.picb.ac.cn/PDXliver/. Despite this, long engraftment periods and low engraftment efficiency are still the major drawbacks hindering the application of the PDX model in large-scale drug response testing. To overcome the shortcomings of 2D culture and PDX, a 3D preclinical organoid model was developed. Coexisting CSC subclones display different sensitivities to drug therapies, but HCC organoids retain the histological architecture, genetic background and heterogeneity of the parent tumor; therefore, organoid models allow the personalized management of targeted therapies. Broutier et al. first successfully developed human primary liver cancer organoid lines to identify patient-specific drug sensitivities[85]. Nuciforo et al. generated long-term organoid cultures to test sorafenib sensitivity[86]. Wang et al. established patient-derived organoid models to examine how CD44 and Hedgehog signaling contribute to sorafenib tolerance and determine whether combination therapy with sorafenib and a compound targeting Hedgehog signaling is effective in overcoming resistance[42]. In addition to the above models, immunocompetent mice, including spontaneous, chemically induced and genetically engineered models, are also widely used in the study of drug resistance[87]. Overall, the development of novel preclinical models and the combined application of multiple preclinical models will undoubtedly improve the accuracy of drug sensitivity and/or resistance predictions in a patient-specific manner [Figure 2].
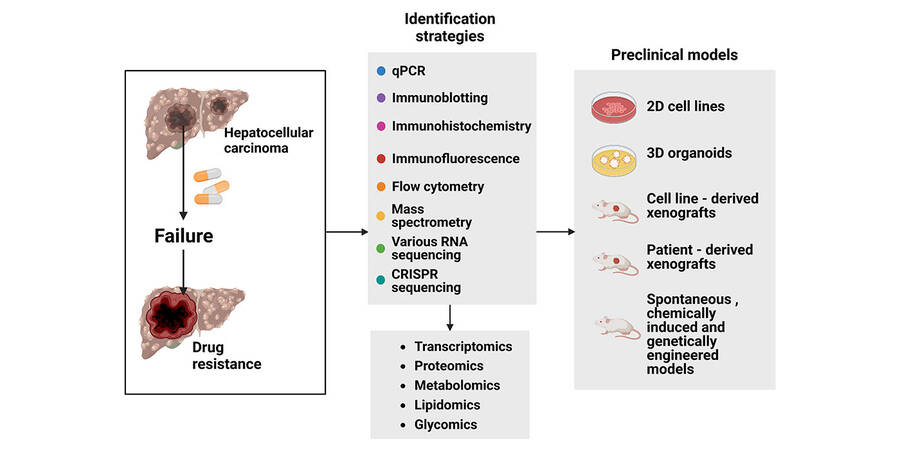
Figure 2. Scheme of therapeutic strategies for CSC-mediated drug resistance.
MONO- OR COMBINATION THERAPIES TO AGAINST DRUG RESISTANCE
Therapeutic strategies targeting CSCs in HCC
Recent advances in CSC biology have accelerated research on CSC eradication. Multiple therapeutic strategies are being developed to target CSCs. CSCs can be effectively targeted by targeting core signaling cascades. AKT, Hedgehog, Notch, TGF-β, and Wnt pathway inhibitors suppress CSC characteristics in HCC. Considering the crosstalk between CSCs and HCC cells, intricate CSC niches contribute to both tumor growth and resistance to therapy. Single or combined treatments that target the CSC niches are effective at treating CSCs. Targeting CSC surface markers provides a straightforward approach for addressing CSCs. It has been suggested that some ABC transporters are abundantly expressed in SP cells and positively associated with the acquisition of drug resistance; thus, targeting critical molecules in SP cells may be an effective strategy to eradicate CSCs. Epigenetic modifications, including chromatin remodeling, DNA methylation and histone modification, are involved in the regulation of CSC properties. However, epigenetic modulation is rarely targeted to specific genes, resulting in an alteration of gene function that is not specific. In light of the roles of miRNAs and lncRNAs in regulating the CSC phenotype and HCC progression, miRNA/lncRNA-targeted therapies are expected to have great therapeutic potential[88,89].
Sorafenib
Our published oncofetal driver CLDN6 strikingly enhances tumor lineage plasticity, resulting in activation of the Hippo signaling pathway via the CLDN6/TJP2/YAP1 axis. A de novo anti-CLDN6 monoclonal antibody conjugated with a cytotoxic agent (mertansine, DM1) (CLDN6-DM1) was established. Cotreatment with sorafenib and CLDN6-DM1 exhibited an even more dramatic synergistic effect in inhibiting tumor formation than treatment with sorafenib alone[21]. Another oncofetal driver, PARP1, is prone to induce stem cell pluripotency and sorafenib resistance in HCC. Sorafenib treatment stimulates DNA damage repair. The PARP inhibitor olaparib profoundly inhibits key pluripotent transcription factors and DNA damage repair signaling, potentially through CHD1L-mediated condensation of the chromatin structure at promotor regions. A synergistic combination of olaparib and sorafenib is more effective than sorafenib alone in eliminating HCC residual tumors[22]. CD13 is necessary for sorafenib-induced resistance, and the blockade of CD13 with its inhibitor ubenimex can suppress tumor growth and restore sorafenib sensitivity in HCC[36]. Multiple lines of evidence suggest that Jak/Stat signaling is activated in HCC CSCs. Jak/Stat inhibitors (AZ960 and TG101209) remarkably suppress CSC properties and delay the formation of AKT/β-catenin-driven HCC tumors, which may be an ideal method to overcome drug resistance[38]. LGR5 is a well-characterized CSC marker in HCC. The LGR5+ cell compartment can be ablated through diphtheria toxin (DT). Simultaneous administration of 5-FU and DT obviously improves the efficacy of 5-FU in combatting HCC, but this is not the case with the combination of sorafenib and DT[39]. Studies have shown that the CDK1/PDK1/β-catenin signaling pathway is activated in HCC. Abrogation of CDK1 with its inhibitor RO3306 confers sensitivity to sorafenib in HCC cells; a 50% tumor incidence was reported in the sorafenib-treated group, while only 17% incidence was observed in the combinatorial treatment group[41]. CD44+ populations in HCC are more refractory to sorafenib. For CD44-positive HCC patient-derived organoids, restricting Hedgehog signaling improves sensitivity to sorafenib. Cotreatment with sorafenib and GANT61 (a Hedgehog signaling inhibitor) has a highly synergistic effect on inhibiting malignant properties in CD44-positive HCC, both in vitro and in vivo[43]. The Wnt target gene EPHB2 has been identified to form an EPHB2/β-catenin/TCF1 positive feedback loop that elevates HCC stemness and sorafenib resistance. Currently, there are no available EPHB2 kinase inhibitors. Intravenous injection of rAAV-8-EPHB2 in an immunocompetent mouse model enhanced the efficacy of sorafenib treatment and further blocked tumorigenicity[44]. The lncRNA SNHG1 participates in sorafenib resistance by activating the AKT pathway via regulation of SL3A2. SNHG1 depletion boosts sorafenib antitumor responses. Compared with control tumors, anti-SNHG1 treatment reduced tumor size by 54.5%, while combinational therapy reduced tumor size by 76.4%[47]. ZFAS1 is a novel regulatory IncRNA that mediates sorafenib resistance via the induction of stemness and EMT phenotypes, and silencing ZFAS1 was found to kill quiescent HCC stem-like cells (EpCAM+CD133+ cells) to overcome sorafenib resistance[50]. CCR4+ TIL-Tregs display enhanced immunosuppressive PD1+TCF1+ stem-like specificity; thus, targeting CCR4+ regulatory T cells using N-CCR4-Fc, a neutralizing pseudoreceptor, can eliminate sorafenib resistance and increase the tumor response to immune checkpoint blockade, which has been shown to suppress tumor growth, reduce tumor weight and prolong the survival of mice with liver cancer[40].
Lenvatinib
Treatment of 12 patients with EGFRhigh HCC who were unresponsive to lenvatinib with the widely used EGFR inhibitor erlotinib plus lenvatinib resulted in beneficial clinical outcomes (trial identifier NCT04642547). Lenvatinib plus gefitinib reduced the total tumor burden by 76.5% after four weeks. Erlotinib effectively reverses lenvatinib resistance[54,55]. Loss of NF1 and DUSP9 activates the PI3K/AKT and MAPK/ERK signaling pathways that confer lenvatinib resistance. In NF1 single-guide RNA (sgRNA) cells, lenvatinib displayed significantly weaker inhibition of tumorigenesis, indicating resistance. In addition, despite NF1 knockout, trametinib (a MEK inhibitor) is still capable of halting HCC growth, which provides evidence that trametinib may mediate the susceptibility of HCC cells to lenvatinib[56]. The ITGB8/HSP90/AKT axis offers an attractive strategy for treating lenvatinib-unresponsive HCC, and the AKT inhibitor MK-2206 or the HSP90 inhibitor 17-AAG could elevate the efficacy of lenvatinib treatment. Lenvatinib treatment completely retards tumor growth in mice raised from ITGB8 knockdown cells, while control small hairpin RNA (shRNA) cells grew steadily[57].
Regorafenib
Gankyrin has been reported to promote HCC tumorigenesis, metastasis and resistance to sorafenib or regorafenib through β-catenin/c-Myc signaling. c-Myc pathway inhibition by shRNA or 10058-F4 largely abolishes c-Myc-dependent metabolic reprogramming, and 10058-F4 in combination with sorafenib or regorafenib has more significant effects on the inhibition of tumor cell growth[64]. Pin1 is insensitive to regorafenib treatment and EMT properties via the Gli1/Snail/E-cadherin pathway in HCC. All-trans retinoic acid (ATRA) has recently been identified as a Pin1 inhibitor. ATRA reduces the number of metastatic nodules in lung tissue by downregulating Pinl and Snail and upregulating E-cadherin, which indicates that ATRA alleviates regorafenib resistance-induced metastasis by reversing EMT[65]. The Hippo signaling pathway could act as a mediator for the efficacy of regorafenib in HCC. The YAP inhibitor verteporfin significantly augments the cytotoxicity of regorafenib in regorafenib-tolerant HCC cells, which offers an attractive strategy for treating regorafenib-insensitive HCC[66]. RAB27A-dependent exosome secretion from CSCs supports regorafenib resistance by inducing Nanog expression. RAB27A depletion via CRISPR/Cas9 alleviates Nanog expression and strongly increases the efficacy of regorafenib, significantly restraining the growth of xenografts[67]. Long-term regorafenib tolerance contributes to enhanced Transforming growth factor (TGF-β) signaling activity, and TGF-β type 1 receptor (TGFβ-R1) inhibition leads to a decrease in colony formation and an increase in cell death in resistant spheroids. In addition, in the zebrafish model, HuH7 cells migrated in 21.6% of zebrafish, regorafenib-resistant cells migrated in 43% of zebrafish, and TGFβ-R1 inhibitor-treated regorafenib-resistant cells only migrated in 12.9% of zebrafish. The combination of a TGFβ-R1 inhibitor and regorafenib diminishes pSTAT3, pSMAD2 and pERK (44/42) expression and increases regorafenib sensitization[68]. The ATF4-ATF3-IL-6Rα cascade is responsible for sorafenib- and regorafenib-induced resistance. IL-6Rα can be blocked with sarilumab, an FDA-approved antibody drug, and the combination of sorafenib or regorafenib and sarilumab is even more effective, with a significant and pronounced reduction in tumor growth and tumor weight in HCC PDX models compared with sorafenib or regorafenib alone. Blockade of IL-6Rα might be a novel therapeutic approach to improve the efficacy of sorafenib/regorafenib in advanced HCC patients[69].
Immune checkpoint inhibitors
Our recent study showed that GDF1 suppresses LSD1 to enhance CTA expression. The combination of an LSD1 inhibitor and an anti-PD1 antibody is more effective than a single treatment in restraining tumor growth and metastasis and prolonging overall survival. Blockade of LSD1 may boost the immune system and broaden the therapeutic window for ICIs in HCC patients[23]. Studies have established links between HNRNPM and immunotherapeutic resistance. HNRNPM-specific antisense oligonucleotides (ASOs) can inhibit cancer stemness, recruit more CD8+ T cells and suppress the WNT/β-catenin signaling pathway; hence, HNRNPM inhibition can block immune evasion and elevate the efficacy of ICI therapy[73]. A recent study suggested that the PKCα/ZFP64/CSF1 axis is critical for triggering immune evasion and anti-PD1 resistance in HCC patients. Gö6976, an inhibitor of PKCα, inhibits activation of the PKCα/ZFP64/CSF1 axis in a dose- and time-dependent manner. The Gö6976-treated group had significantly lower tumor weights than the vehicle group. Gö6976 combined with anti-PD1 therapy significantly inhibits tumor growth and evidently improves the tumor immune microenvironment, thus overcoming anti-PD1 resistance[74]. The circMET/miR-30-5p/Snail/DPP4 axis has been reported to be involved in the immune tolerance of HCC. Sitagliptin is a selective DPP4 inhibitor that has been approved for diabetes treatment, and sitagliptin plus anti-PD1 therapy resulted in 100% tumor regression. The combination therapy with sitagliptin and anti-PD1 antibody increased CD8+ T lymphocyte trafficking and sensitized tumors to PD1 blockade in the HCC clinical setting[75].
For unbiased and easily accessed information on the management of liver injury attributable to prescription and nonprescription medications, the potential hepatotoxicity of indicated drugs for HCC treatment is shown in Table 3. The RUCAM (Roussel Uclaf Causality Assessment Method) causality assessment score yields total scores that can range from -9 to 14; drug causality can be “highly probable” (score > 8), “probable” (score 6-8), “possible” (score 3-5), “unlikely” (score 1-2) or “excluded” (score ≤ 0). For“highly probable”, RUCAM sometimes uses other terms, including “highly likely” and “definite” [90].
Hepatotoxicity of indicated drugs in case reports
| Drugs | RUCAM score | Causality assessment | References |
| Sorafenib | 7 | Probable | [108] |
| 9 | Highly probable | [109] | |
| NA | Probable | [110] | |
| 5-FU | 6 | Probable | [111] |
| Crizotinib | 8 | Probable | [112] |
| 9 | Highly probable | [113] | |
| Atezolizumab | 7 | Probable | [114] |
| Ipilimumab | 9 | Highly probable | [115] |
| Pembrolizumab | 9 | Highly probable | [116] |
| 8 | Probable | [117] | |
| Erlotinib | NA | Probable | [110] |
| Paclitaxel+Seggio+Sintilimab | 8 | Highly likely | [118] |
CONCLUSIONS
For many years, pharmaceutical therapies have largely relied on sorafenib, which has enabled some improvements in survival but not in the quality of life. Recently, a phase 3 randomized, open-label study, the IMbrave150 study (NCT03434379), showed that atezolizumab (anti-PD-L1 antibody) plus bevacizumab (anti-VEGF antibody) significantly improved overall survival (OS) and progression-free survival (PFS) versus sorafenib and had an acceptable safety profile in patients with unresectable HCC after longer follow-up, which confirmed atezolizumab plus bevacizumab as the first-line standard of care for advanced HCC[91,92]. Another phase III randomized trial, the HIALAYA trial (NCT03298451), compared a single priming dose of tremelimumab (anti-CTLA-4 antibody) with once-monthly durvalumab (anti-PDL1 antibody) to sorafenib. Tremelimumab plus durvalumab led to better OS than sorafenib as a first-line treatment for patients with unresectable HCC[93]. Other clinical trials are underway to improve the efficacy of combination therapy in advanced HCC. For example, a phase III randomized trial is examining apatinib plus camrelizumab (anti-PD1 antibody) versus sorafenib (NCT03764293), a phase II study is exploring nivolumab and regorafenib (NCT04310709), and a phase II study is investigating regorafenib plus tislelizumab (anti-PD1 antibody) (NCT04183088)[94].
There has been a dramatic improvement in our understanding of HCC biology. Biological features and clinical properties differ among HCC patients, which is likely due to heterogeneity. Clinically, heterogeneity is largely responsible for tumor progression, metastasis, relapse, and resistance to treatment. Because CSCs are the major source of tumor heterogeneity, CSC characteristics are undoubtedly tightly associated with drug resistance. Intriguingly, CSCs have features similar to those of ESCs, which suggests the importance of developmental signals in resistance to therapeutics. Hence, deep exploration of stemness properties will largely facilitate further insights into treatment resistance.
Although studies on CSCs are broadening our understanding, the implementation of effective precision medicine remains a challenge. Reliable biomarkers to predict treatment failure are still lacking. Precise targeting of specific CSC properties might serve as a novel strategy to eliminate resistant CSC subpopulations. In addition, combined application of multiple techniques and models or development of new effective approaches will allow detailed characterization of heterogeneity and elucidation of the mechanisms underlying therapeutic resistance and thus result in more favorable outcomes.
DECLARATIONS
AcknowledgmentsThe figures are created with BioRender.com and quoted with permission from BioRender.
Authors’ contributionsInitiated the study and fnalized the manuscript: Liu M
Reviewed the literatures and wrote the manuscript: Li MM
Reviewed the literatures and drew the pictures: He YT, Liang JK
Review and revised the manuscript: Guan XY, Ma NF
Read and approved the final manuscript: All authors
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported by the National Natural Science Foundation of China (No. 82003773).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Chen S, Cao Q, Wen W, Wang H. Targeted therapy for hepatocellular carcinoma: challenges and opportunities. Cancer Lett 2019;460:1-9.
2. Greten TF, Lai CW, Li G, Staveley-O'Carroll KF. Targeted and immune-based therapies for hepatocellular carcinoma. Gastroenterology 2019;156:510-24.
3. Huang A, Yang XR, Chung WY, Dennison AR, Zhou J. Targeted therapy for hepatocellular carcinoma. Signal Transduct Target Ther 2020;5:146.
4. Sangro B, Sarobe P, Hervás-Stubbs S, Melero I. Advances in immunotherapy for hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 2021;18:525-43.
5. Hadjimichael C, Chanoumidou K, Papadopoulou N, Arampatzi P, Papamatheakis J, Kretsovali A. Common stemness regulators of embryonic and cancer stem cells. World J Stem Cells 2015;7:1150-84.
6. Garcia-Mayea Y, Mir C, Masson F, Paciucci R, LLeonart ME. Insights into new mechanisms and models of cancer stem cell multidrug resistance. Semin Cancer Biol 2020;60:166-80.
7. Huang T, Song X, Xu D, et al. Stem cell programs in cancer initiation, progression, and therapy resistance. Theranostics 2020;10:8721-43.
8. Zhou HM, Zhang JG, Zhang X, Li Q. Targeting cancer stem cells for reversing therapy resistance: mechanism, signaling, and prospective agents. Signal Transduct Target Ther 2021;6:62.
9. Lee TK, Guan XY, Ma S. Cancer stem cells in hepatocellular carcinoma - from origin to clinical implications. Nat Rev Gastroenterol Hepatol 2022;19:26-44.
10. Prasetyanti PR, Medema JP. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol Cancer 2017;16:41.
11. Zheng H, Pomyen Y, Hernandez MO, et al. Single-cell analysis reveals cancer stem cell heterogeneity in hepatocellular carcinoma. Hepatology 2018;68:127-40.
12. Wang XQ, Zhang W, Lui EL, et al. Notch1-Snail1-E-cadherin pathway in metastatic hepatocellular carcinoma. Int J Cancer 2012;131:E163-72.
13. Li MM, Yuan J, Guan XY, Ma NF, Liu M. Molecular subclassification of gastrointestinal cancers based on cancer stem cell traits. Exp Hematol Oncol 2021;10:53.
14. Silva-Diz V, Lorenzo-Sanz L, Bernat-Peguera A, Lopez-Cerda M, Muñoz P. Cancer cell plasticity: Impact on tumor progression and therapy response. Semin Cancer Biol 2018;53:48-58.
15. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 2009;9:265-73.
16. Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008;133:704-15.
17. Huang Z, Wu T, Liu AY, Ouyang G. Differentiation and transdifferentiation potentials of cancer stem cells. Oncotarget 2015;6:39550-63.
18. Gupta PB, Pastushenko I, Skibinski A, Blanpain C, Kuperwasser C. Phenotypic plasticity: driver of cancer initiation, progression, and therapy resistance. Cell Stem Cell 2019;24:65-78.
19. Liu M, Yan Q, Sun Y, et al. A hepatocyte differentiation model reveals two subtypes of liver cancer with different oncofetal properties and therapeutic targets. Proc Natl Acad Sci U S A 2020;117:6103-13.
20. Liu M, Chen L, Ma NF, et al. CHD1L promotes lineage reversion of hepatocellular carcinoma through opening chromatin for key developmental transcription factors. Hepatology 2016;63:1544-59.
21. Kong FE, Li GM, Tang YQ, et al. Targeting tumor lineage plasticity in hepatocellular carcinoma using an anti-CLDN6 antibody-drug conjugate. Sci Transl Med 2021;13:eabb6282.
22. Yang XD, Kong FE, Qi L, et al. PARP inhibitor Olaparib overcomes Sorafenib resistance through reshaping the pluripotent transcriptome in hepatocellular carcinoma. Mol Cancer 2021;20:20.
23. Cheng W, Li HL, Xi SY, et al. Growth differentiation factor 1-induced tumour plasticity provides a therapeutic window for immunotherapy in hepatocellular carcinoma. Nat Commun 2021;12:7142.
24. Paul R, Dorsey JF, Fan Y. Cell plasticity, senescence, and quiescence in cancer stem cells: Biological and therapeutic implications. Pharmacol Ther 2022;231:107985.
25. Boix L, López-Oliva JM, Rhodes AC, Bruix J. Restoring miR122 in human stem-like hepatocarcinoma cells, prompts tumor dormancy through Smad-independent TGF-β pathway. Oncotarget 2016;7:71309-29.
26. Fabregat I, Caballero-Díaz D. Transforming growth factor-β-induced cell plasticity in liver fibrosis and hepatocarcinogenesis. Front Oncol 2018;8:357.
27. Xia Y, Zhen L, Li H, et al. MIRLET7BHG promotes hepatocellular carcinoma progression by activating hepatic stellate cells through exosomal SMO to trigger hedgehog pathway. Cell Death Dis 2021;12:326.
28. Bayik D, Lathia JD. Cancer stem cell-immune cell crosstalk in tumour progression. Nat Rev Cancer 2021;21:526-36.
29. Shi GM, Xu Y, Fan J, et al. Identification of side population cells in human hepatocellular carcinoma cell lines with stepwise metastatic potentials. J Cancer Res Clin Oncol 2008;134:1155-63.
30. Hu C, Li H, Li J, et al. Analysis of ABCG2 expression and side population identifies intrinsic drug efflux in the HCC cell line MHCC-97L and its modulation by Akt signaling. Carcinogenesis 2008;29:2289-97.
31. Bhagyaraj E, Ahuja N, Kumar S, et al. TGF-β induced chemoresistance in liver cancer is modulated by xenobiotic nuclear receptor PXR. Cell Cycle 2019;18:3589-602.
32. Fung SW, Cheung PF, Yip CW, et al. The ATP-binding cassette transporter ABCF1 is a hepatic oncofetal protein that promotes chemoresistance, EMT and cancer stemness in hepatocellular carcinoma. Cancer Lett 2019;457:98-109.
33. Wang M, Wang Z, Zhi X, et al. SOX9 enhances sorafenib resistance through upregulating ABCG2 expression in hepatocellular carcinoma. Biomed Pharmacother 2020;129:110315.
34. Wei L, Wang X, Lv L, et al. The emerging role of microRNAs and long noncoding RNAs in drug resistance of hepatocellular carcinoma. Mol Cancer 2019;18:147.
35. Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther 2008;7:3129-40.
36. Hu B, Xu Y, Li YC, et al. CD13 promotes hepatocellular carcinogenesis and sorafenib resistance by activating HDAC5-LSD1-NF-κB oncogenic signaling. Clin Transl Med 2020;10:e233.
37. Lu S, Yao Y, Xu G, et al. CD24 regulates sorafenib resistance via activating autophagy in hepatocellular carcinoma. Cell Death Dis 2018;9:646.
38. Toh TB, Lim JJ, Hooi L, Rashid MBMA, Chow EK. Targeting Jak/Stat pathway as a therapeutic strategy against SP/CD44+ tumorigenic cells in Akt/β-catenin-driven hepatocellular carcinoma. J Hepatol 2020;72:104-18.
39. Cao W, Li M, Liu J, et al. LGR5 marks targetable tumor-initiating cells in mouse liver cancer. Nat Commun 2020;11:1961.
40. Gao Y, You M, Fu J, et al. Intratumoral stem-like CCR4+ regulatory T cells orchestrate the immunosuppressive microenvironment in HCC associated with hepatitis B. J Hepatol 2022;76:148-59.
41. Wu CX, Wang XQ, Chok SH, et al. Blocking CDK1/PDK1/β-Catenin signaling by CDK1 inhibitor RO3306 increased the efficacy of sorafenib treatment by targeting cancer stem cells in a preclinical model of hepatocellular carcinoma. Theranostics 2018;8:3737-50.
42. Wang S, Wang Y, Xun X, et al. Hedgehog signaling promotes sorafenib resistance in hepatocellular carcinoma patient-derived organoids. J Exp Clin Cancer Res 2020;39:22.
43. Leung HW, Lau EYT, Leung CON, et al. NRF2/SHH signaling cascade promotes tumor-initiating cell lineage and drug resistance in hepatocellular carcinoma. Cancer Lett 2020;476:48-56.
44. Leung HW, Leung CON, Lau EY, et al. EPHB2 activates β-catenin to enhance cancer stem cell properties and drive sorafenib resistance in hepatocellular carcinoma. Cancer Res 2021;81:3229-40.
45. Pollutri D, Patrizi C, Marinelli S, et al. The epigenetically regulated miR-494 associates with stem-cell phenotype and induces sorafenib resistance in hepatocellular carcinoma. Cell Death Dis 2018;9:4.
46. Xu J, Wan Z, Tang M, et al. N6-methyladenosine-modified CircRNA-SORE sustains sorafenib resistance in hepatocellular carcinoma by regulating β-catenin signaling. Mol Cancer 2020;19:163.
47. Li W, Dong X, He C, et al. LncRNA SNHG1 contributes to sorafenib resistance by activating the Akt pathway and is positively regulated by miR-21 in hepatocellular carcinoma cells. J Exp Clin Cancer Res 2019;38:183.
48. Song W, Zheng C, Liu M, et al. TRERNA1 upregulation mediated by HBx promotes sorafenib resistance and cell proliferation in HCC via targeting NRAS by sponging miR-22-3p. Mol Ther 2021;29:2601-16.
49. Zhou K, Nguyen R, Qiao L, George J. Single cell RNA-seq analysis identifies a noncoding RNA mediating resistance to sorafenib treatment in HCC. Mol Cancer 2022;21:6.
50. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet 2018;391:1163-73.
51. Ma XL, Hu B, Tang WG, et al. CD73 sustained cancer-stem-cell traits by promoting SOX9 expression and stability in hepatocellular carcinoma. J Hematol Oncol 2020;13:11.
52. Ma XL, Shen MN, Hu B, et al. CD73 promotes hepatocellular carcinoma progression and metastasis via activating PI3K/AKT signaling by inducing Rap1-mediated membrane localization of P110β and predicts poor prognosis. J Hematol Oncol 2019;12:37.
53. Talukdar S, Emdad L, Das SK, Fisher PB. EGFR: an essential receptor tyrosine kinase-regulator of cancer stem cells. Adv Cancer Res 2020;147:161-88.
54. Jin H, Shi Y, Lv Y, et al. EGFR activation limits the response of liver cancer to lenvatinib. Nature 2021;595:730-4.
55. He X, Hikiba Y, Suzuki Y, et al. EGFR inhibition reverses resistance to lenvatinib in hepatocellular carcinoma cells. Sci Rep 2022;12:8007.
56. Lu Y, Shen H, Huang W, et al. Genome-scale CRISPR-Cas9 knockout screening in hepatocellular carcinoma with lenvatinib resistance. Cell Death Discov 2021;7:359.
57. Hou W, Bridgeman B, Malnassy G, et al. Integrin subunit beta 8 contributes to lenvatinib resistance in HCC. Hepatol Commun 2022;6:1786-802.
58. Wang J, Zhao H, Zheng L, et al. FGF19/SOCE/NFATc2 signaling circuit facilitates the self-renewal of liver cancer stem cells. Theranostics 2021;11:5045-60.
59. Myojin Y, Kodama T, Maesaka K, et al. ST6GAL1 Is a Novel Serum Biomarker for Lenvatinib-Susceptible FGF19-Driven Hepatocellular Carcinoma. Clin Cancer Res 2021;27:1150-61.
60. Arechederra M, Bazai SK, Abdouni A, et al. ADAMTSL5 is an epigenetically activated gene underlying tumorigenesis and drug resistance in hepatocellular carcinoma. J Hepatol 2021;74:893-906.
61. Han T, Zheng H, Zhang J, et al. Downregulation of MUC15 by miR-183-5p.1 promotes liver tumor-initiating cells properties and tumorigenesis via regulating c-MET/PI3K/AKT/SOX2 axis. Cell Death Dis 2022;13:200.
62. Zhang P, Sun H, Wen P, Wang Y, Cui Y, Wu J. circRNA circMED27 acts as a prognostic factor and mediator to promote lenvatinib resistance of hepatocellular carcinoma. Mol Ther Nucleic Acids 2022;27:293-303.
63. Grothey A, Blay JY, Pavlakis N, Yoshino T, Bruix J. Evolving role of regorafenib for the treatment of advanced cancers. Cancer Treat Rev 2020;86:101993.
64. Liu R, Li Y, Tian L, et al. Gankyrin drives metabolic reprogramming to promote tumorigenesis, metastasis and drug resistance through activating β-catenin/c-Myc signaling in human hepatocellular carcinoma. Cancer Lett 2019;443:34-46.
65. Wang J, Zhang N, Han Q, et al. Pin1 inhibition reverses the acquired resistance of human hepatocellular carcinoma cells to Regorafenib via the Gli1/Snail/E-cadherin pathway. Cancer Lett 2019;444:82-93.
66. Suemura S, Kodama T, Myojin Y, et al. CRISPR Loss-of-Function Screen Identifies the Hippo Signaling Pathway as the Mediator of Regorafenib Efficacy in Hepatocellular Carcinoma. Cancers (Basel) 2019;11:1362.
67. Huang H, Hou J, Liu K, et al. RAB27A-dependent release of exosomes by liver cancer stem cells induces Nanog expression in their differentiated progenies and confers regorafenib resistance. J Gastroenterol Hepatol 2021;36:3429-37.
68. Karabicici M, Azbazdar Y, Ozhan G, Senturk S, Firtina Karagonlar Z, Erdal E. Changes in Wnt and TGF-β signaling mediate the development of Regorafenib resistance in hepatocellular carcinoma cell line HuH7. Front Cell Dev Biol 2021;9:639779.
69. Dai Z, Wang X, Peng R, Zhang B, Han Q, et al. Induction of IL-6Rα by ATF3 enhances IL-6 mediated sorafenib and regorafenib resistance in hepatocellular carcinoma. Cancer Lett 2022;524:161-71.
70. Dai X, Guo Y, Hu Y, et al. Immunotherapy for targeting cancer stem cells in hepatocellular carcinoma. Theranostics 2021;11:3489-501.
71. Pinyol R, Sia D, Llovet JM. Immune exclusion-Wnt/CTNNB1 class predicts resistance to immunotherapies in HCC. Clin Cancer Res 2019;25:2021-3.
72. Ruiz de Galarreta M, Bresnahan E, Molina-Sánchez P, et al. β-catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov 2019;9:1124-41.
73. Zhu GQ, Wang Y, Wang B, et al. Targeting HNRNPM inhibits cancer stemness and enhances antitumor immunity in wnt-activated hepatocellular carcinoma. Cell Mol Gastr Hep 2022;13:1413-47.
74. Wei CY, Zhu MX, Zhang PF, Huang XY, Wan JK, et al. PKCα/ZFP64/CSF1 axis resets the tumor microenvironment and fuels anti-PD1 resistance in hepatocellular carcinoma. J Hepatol 2022;77:163-176.
75. Huang XY, Zhang PF, Wei CY, et al. Circular RNA circMET drives immunosuppression and anti-PD1 therapy resistance in hepatocellular carcinoma via the miR-30-5p/snail/DPP4 axis. Mol Cancer 2020;19:92.
76. Kilgour E, Rothwell DG, Brady G, Dive C. Liquid biopsy-based biomarkers of treatment response and resistance. Cancer Cell 2020;37:485-95.
77. Parikh AR, Leshchiner I, Elagina L, et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat Med 2019;25:1415-21.
78. Ho DW, Tsui YM, Sze KM, et al. Single-cell transcriptomics reveals the landscape of intra-tumoral heterogeneity and stemness-related subpopulations in liver cancer. Cancer Lett 2019;459:176-85.
79. Zhou L, Yu KH, Wong TL, et al. Lineage tracing and single-cell analysis reveal proliferative Prom1+ tumour-propagating cells and their dynamic cellular transition during liver cancer progression. Gut 2022;71:1656-68.
80. Ma S, Lee TK, Zheng BJ, Chan KW, Guan XY. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 2008;27:1749-58.
81. Caruso S, Calatayud AL, Pilet J, et al. Analysis of liver cancer cell lines identifies agents with likely efficacy against hepatocellular carcinoma and markers of response. Gastroenterology 2019;157:760-76.
82. Wei L, Lee D, Law CT, et al. Genome-wide CRISPR/Cas9 library screening identified PHGDH as a critical driver for Sorafenib resistance in HCC. Nat Commun 2019;10:4681.
83. Samanta D, Park Y, Andrabi SA, Shelton LM, Gilkes DM, Semenza GL. PHGDH expression is required for mitochondrial Redox homeostasis, breast cancer stem cell maintenance, and lung metastasis. Cancer Res 2016;76:4430-42.
84. He S, Hu B, Li C, et al. PDXliver: a database of liver cancer patient derived xenograft mouse models. BMC Cancer 2018;18:550.
85. Broutier L, Mastrogiovanni G, Verstegen MM, et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat Med 2017;23:1424-35.
86. Nuciforo S, Fofana I, Matter MS, et al. Organoid models of human liver cancers derived from tumor needle biopsies. Cell Rep 2018;24:1363-76.
87. Cho K, Ro SW, Seo SH, et al. Genetically engineered mouse models for liver cancer. Cancers (Basel) 2019;12:14.
88. Wang N, Wang S, Li MY, et al. Cancer stem cells in hepatocellular carcinoma: an overview and promising therapeutic strategies. Ther Adv Med Oncol 2018;10:1758835918816287.
89. Liu YC, Yeh CT, Lin KH. Cancer stem cell functions in hepatocellular carcinoma and comprehensive therapeutic strategies. Cells 2020;9:1331.
90. Danan G, Teschke R. RUCAM in drug and herb induced liver injury: the update. Int J Mol Sci 2015;17:14.
91. Galle PR, Finn RS, Qin S, et al. Patient-reported outcomes with atezolizumab plus bevacizumab versus sorafenib in patients with unresectable hepatocellular carcinoma (IMbrave150): an open-label, randomised, phase 3 trial. Lancet Oncol 2021;22:991-1001.
92. Cheng AL, Qin S, Ikeda M, et al. Updated efficacy and safety data from IMbrave150: Atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J Hepatol 2022;76:862-73.
94. Akce M, El-Rayes BF, Bekaii-Saab TS. Frontline therapy for advanced hepatocellular carcinoma: an update. Therap Adv Gastroenterol 2022;15:17562848221086126.
95. Highlights of prescribing information for nexavar (sorafenib) tablets, for oral use. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/021923s020lbl.pdf [Last accessed on 22 Sep 2022].
96. NEXAVAR. Available from: https://www.rxlist.com/nexavar-drug.htm#indications [Last accessed on 22 Sep 2022].
97. Highlights of prescribing information for lenvima® (lenvatinib) capsules, for oral use. Available from: https://www.lenvima.com/-/media/Project/EISAI/Lenvima/PDF/prescribing-information.pdf [Last accessed on 22 Sep 2022].
98. LENVIMA. Available from: https://www.rxlist.com/lenvima-drug.htm#description [Last accessed on 22 Sep 2022].
99. Systemic anti cancer therapy protocol. Protocol ref: MPHAABHCGA (Version No: 1.0). 95. Available from: https://www.clatterbridgecc.nhs.uk/application/files/6016/1650/0037/Atezolizumab_Bevacizumab_Hepatocellular_Carcinoma_Protocol_V1.0.pdf [Last accessed on 22 Sep 2022].
100. NSW government. eviQ. Available from: https://www.eviq.org.au/medical-oncology/upper-gastrointestinal/hepatic/3881-hcc-advanced-or-metastatic-atezolizumab-and-b#side-effects [Last accessed on 22 Sep 2022].
101. STIVARGA. Available from: https://www.rxlist.com/stivarga-drug.htm#description [Last accessed on 22 Sep 2022].
102. CABOMETYX. Available from: https://www.rxlist.com/cabometyx-drug.htm#interaction [Last accessed on 22 Sep 2022].
103. CYRAMZA. Available from: https://www.rxlist.com/cyramza-drug.htm#dosage [Last accessed on 22 Sep 2022].
104. Highlights of prescribing information for KEYTRUDA® (pembrolizumab). Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/125514s012lbl.pdf [Last accessed on 22 Sep 2022].
105. PEMBROLIZUMAB. Available from: https://www.rxlist.com/consumer_pembrolizumab_keytruda/drugs-condition.htm#what_other_drugs_interact_with_pembrolizumab [Last accessed on 22 Sep 2022].
106. Nivolumab. Available from: https://www.pediatriconcall.com/drugs/nivolumab/29 [Last accessed on 22 Sep 2022].
107. Ipilimumab. Available from: https://www.pediatriconcall.com/drugs/ipilimumab/167 [Last accessed on 22 Sep 2022].
108. Fairfax BP, Pratap S, Roberts IS, et al. Fatal case of sorafenib-associated idiosyncratic hepatotoxicity in the adjuvant treatment of a patient with renal cell carcinoma. BMC Cancer 2012;12:590.
109. Shikdar S, Barry O, Choi E. A rare case of sorafenib induced liver injury in a patient with recurrent Hurthle cell carcinoma. J Case Rep Images Oncol 2018;4:100051Z10SS2018.
110. Douros A, Bronder E, Andersohn F, et al. Drug-induced liver injury: results from the hospital-based berlin case-control surveillance study. Br J Clin Pharmacol 2015;79:988-99.
111. Brooks AJ, Begg EJ, Chapman BA, Fitzharris BM. Two cases of severe liver injury possibly related to 5-fluorouracil and calcium folinate. Intern Med J 2007;37:344-5.
112. Kreitman K, Nair SP, Kothadia JP. Successful treatment of crizotinib-induced fulminant liver failure: a case report and review of literature. Case Reports Hepatol 2020;2020:8247960.
113. Cardenas V, Mankuzhy N, Mody R, McCaffery H, Fontana RJ, DiPaola F. Incidence and sequelae of liver injury among children treated for solid tumors: analysis of a large single-center prospective cohort. J Pediatr Gastroenterol Nutr 2020;71:197-202.
114. Tzadok R, Levy S, Aouizerate J, Shibolet O. Acute liver failure following a single dose of atezolizumab, as assessed for causality using the updated RUCAM. Case Rep Gastrointest Med 2022;2022:5090200.
115. Ahmed T, Pandey R, Shah B, Black J. Resolution of ipilimumab induced severe hepatotoxicity with triple immunosuppressants therapy. BMJ Case Rep 2015;2015:bcr2014208102.
116. Al-Nattah S, Lata Sharma K, Caldis M, Spengler E, Nicholas Rose W. Plasmapheresis for pembrolizumab-induced hepatitis in a patient with squamous cell carcinoma and prior orthotopic liver transplantation. Case Reports Hepatol 2022;2022:5908411.
117. Weber S, Benesic A, Rotter I, Gerbes AL. Early ALT response to corticosteroid treatment distinguishes idiosyncratic drug-induced liver injury from autoimmune hepatitis. Liver Int 2019;39:1906-17.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Li MM, He YT, Liang JK, Guan XY, Ma NF, Liu M. Cancer stem cell-mediated therapeutic resistance in hepatocellular carcinoma. Hepatoma Res 2022;8:36. http://dx.doi.org/10.20517/2394-5079.2022.43
AMA Style
Li MM, He YT, Liang JK, Guan XY, Ma NF, Liu M. Cancer stem cell-mediated therapeutic resistance in hepatocellular carcinoma. Hepatoma Research. 2022; 8: 36. http://dx.doi.org/10.20517/2394-5079.2022.43
Chicago/Turabian Style
Li, Mei-Mei, Yi-Ti He, Jie-Kai Liang, Xin-Yuan Guan, Ning-Fang Ma, Ming Liu. 2022. "Cancer stem cell-mediated therapeutic resistance in hepatocellular carcinoma" Hepatoma Research. 8: 36. http://dx.doi.org/10.20517/2394-5079.2022.43
ACS Style
Li, M.M.; He Y.T.; Liang J.K.; Guan X.Y.; Ma N.F.; Liu M. Cancer stem cell-mediated therapeutic resistance in hepatocellular carcinoma. Hepatoma. Res. 2022, 8, 36. http://dx.doi.org/10.20517/2394-5079.2022.43
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 21 clicks
Cite This Article 21 clicks

 Like This Article 2
likes
Like This Article 2
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.