
Molecular mechanisms of liver carcinogenesis related to metabolic syndrome
 0
0Abstract
Global prevalence of non-alcoholic fatty liver disease (NAFLD) and of NAFLD-hepatocellular carcinoma (HCC) is estimated to grow in the next years. The burden of NAFLD and the evidence that NAFLD-HCC arises also in non-cirrhotic patients, explain the urgent need of a better characterization of the molecular mechanisms involved in NAFLD progression. Obesity and diabetes cause a chronic inflammatory state which favors changes in serum cytokines and adipokines, an increase in oxidative stress, DNA damage, and the activation of multiple signaling pathways involved in cell proliferation. Moreover, a role in promoting NAFLD-HCC has been highlighted in the innate and adaptive immune system, dysbiosis, and alterations in bile acids metabolism. Several dietary, genetic, or combined mouse models have been used to study nonalcoholic steatohepatitis (NASH) development and its progression to HCC, but models that fully recapitulate the biological and prognostic features of human NASH are still lacking. In humans, four single nucleotide polymorphisms (PNPLA3, TM6SF2, GCKR, and MBOAT7) have been linked to the development of both NASH and HCC in cirrhotic and non-cirrhotic patients, whereas HSD17B13 polymorphism has a protective effect. In addition, higher rates of somatic ACVR2A mutations and a novel mutational signature have been recently discovered in NASH-HCC patients. The knowledge of the molecular pathogenesis of NAFLD-HCC will be helpful to personalized screening programs and allow for primary and secondary chemopreventive treatments for NAFLD patients who are more likely to progress to HCC.
Keywords
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is the liver manifestation of metabolic syndrome and encompasses a large spectrum of clinic-pathological entities including steatosis, nonalcoholic steatohepatitis (NASH), and cirrhosis and its complications[1]. Recently, the term metabolic dysfunction-associated fatty liver disease (MAFLD) has been proposed to better define the liver manifestations of metabolic disorder[2]. As opposed to NAFLD, which is a diagnosis of exclusion, MAFLD is defined by positive diagnostic criteria and do not exclude chronic alcohol intake[2]. NAFLD affects 25%-35% of the worldwide adult population and up to 50% of type 2 diabetes and up to 90% of morbidly obese patients[3]. It has been estimated that the prevalence of NAFLD will increase exponentially in the next few years and this will probably lead to an increase in the incidence of hepatocellular carcinoma (HCC) in both men and women[3]. NAFLD-related HCC incidence is extremely variable[4]. In patients with NAFLD-related cirrhosis, annual incidence of HCC ranges from 0.7% to 2.6% in USA and Europe and from 0.5% to 11.3% in Asia. Lower incidence values are reported in patients without cirrhosis (0.1-1.3 per 1000 patients-year in USA and Europe and 0.04%-0.6% in Asia)[4]. Interestingly, both in the Western and Eastern countries, the prevalence of HCC without cirrhosis is higher compared to other aetiologies [e.g., alcohol liver disease (ALD), hepatitis B virus, and/or hepatitis C virus chronic infection][4]. Due to the high prevalence of NAFLD without cirrhosis and the low incidence of HCC in this population, a systematic HCC screening targeting non-cirrhotic NAFLD patients is currently not considered cost-effective[5]. A risk stratification to guide targeted screening among these patients requires the characterization of the mechanisms underlying the development of HCC.
GENETIC PREDISPOSITION TO NAFLD-RELATED HCC
Numerous genetic and epigenetic determinants of NAFLD and NAFLD-related HCC have been discovered over the years and some common single nucleotide polymorphisms (SNP) have been robustly validated as associated with the risk of HCC development in NAFLD [Figure 1].
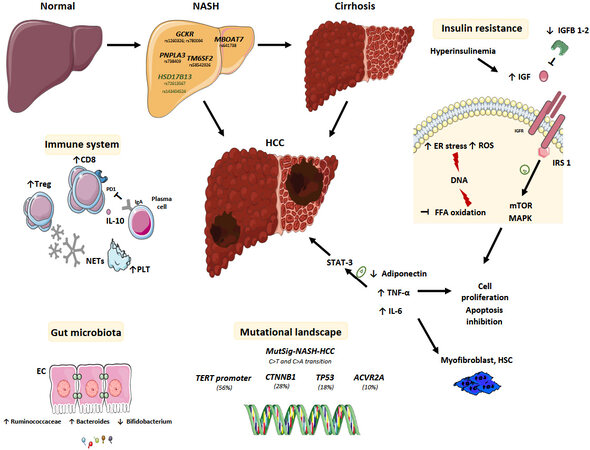
Figure 1. Multifactorial pathogenesis of NAFLD-related HCC. Single nucleotide polymorphism (GCKR, MBOAT7, PNPLA3, and TM6SF2) have been associated to the development of both NAFLD and HCC and the NASH-HCC mutational profiling (TERT promoter, CTNNB1, TP53, and ACVR2A) and signature (MutSig-NASH-HCC) have been recently described. Hyperinsulinemia and obesity favor chronic liver inflammation and DNA damage through ROS production, ER stress and inhibition of fatty free acid oxidation. The higher levels of IL-6 and TNF-α promote the activation of STAT-3, a transcription factor which is involved in HCC development. Hyperinsulinemia induced IGF1 expression that phosphorylates insulin receptor substrates 1 leading to the activation of mTOR and MAPK pathways and therefore to cell proliferation, inhibition of apoptosis and HCC development. A role for the immune system has also been described, involving the intratumor CD8+ PD1+ cells, platelet (PLT) and IgA plasma cells and the circulating NET. In addition, dysbiosis and gut hyperpermeability expose the liver to bacterial metabolites and MAMPs promoting HCC development. EC: Enterocyte cell; ER: endoplasmic reticulum; FFA: free fatty acid; GCKR: glucokinase regulator; HCC: hepatocellular carcinoma; HSC: hepatic stellate cell; HSD17B13: 17β-hydroxysteroid dehydrogenase 13; IgA: immunoglobulin A; IGF: insulin-like growth factor; IGFBP: insulin-like growth-factor binding protein; IGFR: insulin-like growth-factor receptor; IL: interleukin; IRS: insulin receptor substrate; MAMPS: microbe-associated molecular pattern; MAPK: mitogen-activated protein kinase; MBOAT7: membrane-bound O-acyltransferase domain containing 7; mTOR: mammalian target of rapamycin; NAFLD: non-alcoholic fatty liver disease; NET: neutrophil extracellular traps;
Patatin-like phospholipase domain-containing protein 3
Patatin-like phospholipase domain-containing protein 3 (PNPLA3), also known as adiponutrin, is a protein encoded by a gene located on chromosome 22. It is expressed in the endoplasmic reticulum and at the surface of lipid droplets both in hepatic stellate cells and hepatocytes[6]. PNPLA3 plays a role in hydrolysis of triglycerides and retinyl esters[7] and is regulated by nutritional status, being decreased during fasting and increased with carbohydrate-refeeding through the transcription induced by sterol regulatory element-binding protein-1c[8]. In a genome-wide association study, the rs738409 C>G SNP has been the first genetic variant found to be associated with NASH predisposition[9]. It is characterized by a loss of function due to the substitution of isoleucine in a methionine at position 148 (p.I148M) which restricts the access of triglycerides to the catalytic serine at residue 47[9]. Moreover, the mutated protein evades ubiquitination and inhibits the activity of adipose tissue triglyceride lipase (ATGL/PNPLA2)[10]. The rs738409 C>G SNP was strongly associated to the amount of hepatic fat regardless of diabetes, body mass index (BMI) values, and lipoprotein levels[9]. The rs738409 C>G (p.I148M) variant has been linked to fat accumulation[11], lower secretion of very low density lipoprotein (VLDL)[12], higher alanine aminotransferase (ALT) levels[13], and the full spectrum of NAFLD, both in children and adults[9,14-17]. PNPLA3 rs738409 C>G (p.I148M) has been also associated to HCC development regardless of the aetiology with a higher association in “fat-derived” HCC (i.e., NASH and ALD)[18,19]; its association with hepatitis C virus related HCC has been discussed in a meta-analysis[19]. Regarding NAFLD patients, Burza et al.[20] demonstrated the association between the rs738409 C>G variant and HCC development in obese patients. Furthermore, Liu et al.[21] demonstrated that the G allele was associated with an increased risk of HCC development independently of potential confounders including age, gender, BMI, presence of type 2 diabetes, and presence of advanced fibrosis or cirrhosis. These data have also been confirmed in a Japanese retrospective study which described an odds ratio (OR) of 6.36 for homozygous PNPLA3 p.I148M[22].
Transmembrane 6, superfamily member 2
Transmembrane 6, superfamily member 2 (TM6SF2) is a transmembrane Golgi protein encoded by a gene on chromosome 19p12 involved in lipid synthesis and in the enrichment of triglycerides to apolipoprotein B (ApoB) 100 in the VLDL secretion from hepatocytes[23,24]. The rs58542926 C>T variant causes the substitution of glutamate with lysine at residue 167 and leads to a 50% reduction in protein production compared with the wild type. It reduces triglyceride rich lipoproteins secretion and promotes the accumulation of lipid droplets within hepatocytes[23]. The rs58542926 C>T variant has been linked to fatty liver disease development and progression to cirrhosis, and to a lower risk of cardiovascular disease[25]. The TM6SF2 rs58542926 C>T variant has also been associated with the development of HCC both in ALD and NAFLD patients[26-28].
Membrane-bound O-acyltransferase domain containing 7
Membrane-bound O-acyltransferase domain containing 7 (MBOAT7) gene encodes for lysophosphatidyl-inositol acyltransferase 1, a membrane-anchored enzyme which preferentially incorporates arachidonic acid in phosphatidyl inositol (PI), a component of membrane phospholipids[29]. Dysregulation of its expression and/or function, may change the amount of free arachidonic acid that could trigger hepatic inflammation and fibrosis[30]. The rs641738 C>T variant leads to a reduction at both the mRNA and protein level, and it has been associated with the pathogenesis of alcohol-cirrhosis, fibrosis in hepatitis B virus and hepatitis C virus infection, and prognosis of patients with primary sclerosing cholangitis[31-33]. Moreover, the variant induces the increase in oleyl-PI and linoleyl-PI and a decrease in arachidonoyl-PI within hepatocytes fostering fat accumulation and the production of inflammatory mediators[34]. Teo et al.[35] have recently observed that the rs641738 C>T SNP is associated with high level of fat at computed tomography/magnetic resonance imaging scan, having NAFLD (OR = 1.17), having advanced fibrosis, and higher ALT levels in adult patients. An increased risk of evolution both to fibrosis and HCC development has also been described in an Italian cohort of 132 NAFLD-patients among whom the rs641738 C>T variant has been found to increase the risk of HCC by approximately 80%, especially in patients without advanced fibrosis[34].
17β-Hydroxysteroid dehydrogenase 13
17β-Hydroxysteroid dehydrogenase 13 (HSD17B13) gene is located in chromosome 4q22.1 and belongs to a family of 15 enzymes which catalyzes the conversion from the 17-keto to 17 hydroxysteroids. In 2019, Abul-Husn et al.[36] observed that the rs72613567 T to TA insertion variant, adjacent to the donor splice site downstream of exon 6, causes the production of a truncated and unstable protein with reduced enzymatic activity. This SNP was associated with a reduction in the risk of ALD and NAFLD. These data have been confirmed in a cohort of 356 patients with biopsy-proven metabolic disease; a protection against NAFLD, NASH, ballooning degeneration, lobular inflammation, and fibrosis has been recognized[37]. The HSD17B13 rs72613567 loss of function variant has been also identified to be protective of HCC development in patients with ALD, independent of the degree of liver fibrosis[27,38]. A role in protecting against the development of chronic liver disease has also been demonstrated for another variant of HSD17B13, characterized by a frameshift deletion at position 192 which causes the production of a truncated protein[39].
Glucokinase regulator
The role of glucokinase regulator (GCKR) in accumulation of fat in the liver and in a higher level of VLDL and triglycerides has been revealed in a large study enrolling 455 obese children and adolescent of different ethnicity[40]. The rs1260326 variant has been associated with the production of P446L protein which is unable to inhibit glucokinase in response to fructose-6-phosphate, resulting in an uncontrolled glucose uptake in the liver[40]. The sustained activation of glycolysis leads to a reduction in glycemia and to an increase in malonyl-COA levels which favor fat accumulation acting both as a substrate for lipogenesis and as an inhibitor of carnitine-palmitoyl transferase-a, an enzyme involved in fatty acid oxidation[40]. In 366 NAFLD patients, the rs780094 C>T variant has been associated with the severity of liver fibrosis and with higher serum triglyceride values[41], whereas the role of this SNP in determining the progression of NAFLD to HCC has not yet been clarified.
Other
The rs599839 A>G polymorphism, in the cluster of cadherin EGF LAG seven-pass G-type receptor 2 - proline and serine rich coiled-coil 1 - sortilin 1 (CELSR2-PSRC1-SORT), composed of genes involved in lipid handling and cell proliferation, has been linked to a lower risk of dyslipidemia and cardiovascular diseases and to an increased risk of HCC, regardless of fibrosis[42]. Truncating mutations in APOB, and consequently a dysfunctional ApoB 100 protein, have been related to the development of HCC in NAFLD patients, probably due to an increase in the oxidative stress and loss of the tumour suppressive activity of ApoB[43]. The same authors observed that variants in SQSTM1 gene lead to the cytoplasm accumulation of p62 favouring the hepatocyte transformation through the activation of oxidative stress and mammalian target of rapamicin (mTOR) pathways[43].
MOLECULAR MECHANISMS OF HCC DEVELOPED ON METABOLIC SYNDROME
Diabetes, insulin resistance, obesity, older age, male gender, ethnicity, and limited physical activity have been associated with the development of NAFLD-HCC regardless of the presence of cirrhosis[3]. The role of obesity as an independent risk factor in HCC development has been described in various cohort from Korea, the United States, and Sweden with a relative risk from 1.5 to 3.1[44]. Similar to obesity, hyperinsulinemia and diabetes are independent risk factors for HCC development ranging from 2 to 8 times in non-cirrhotic and cirrhotic patients respectively[45]. Overall, the risk is higher if other metabolic risk factors coexist and with increased length of exposure to these risk factors[3,45,46].
Insulin resistance and HCC development
Hyperinsulinemia and obesity lead to low-grade chronic systemic inflammation and may promote HCC development through various mechanisms. Lipid accumulation into hepatocytes, in particular free fatty acid, causes the generation of lipotoxic metabolites, reactive oxygen species and ER stress, dysregulation in gene transcription and signaling pathways, and mechanisms overall termed as lipotoxicity[6,44]. Hepatokines and adipokines expression is altered with reduced levels of the anti-inflammatory adiponectin and increased values of the proinflammatory leptin[6,44,47]. Consequently, the activation of the Janus kinase pathway is observed in NASH with signal transducer and activator of transcription (STAT)-1 activation which is involved in NASH development and STAT-3 in HCC occurrence[6,44,47,48]. Levels of tumor necrosis factor (TNF)-alpha and interleukin (IL)-6 are frequently increased in NAFLD-HCC patients[6,44,47]. These cytokines promote, together with the reduction of adiponectin, the development of insulin resistance. Insulin resistance inhibits free fatty acid oxidation and, as a consequence, leads to increased hepatocytes exposure to free fatty acid, leading to oxidative DNA damage[3]. Hyperinsulinemia favors HCC development through direct effects on transformed hepatocytes. Moreover, hyperinsulinemia plays an indirect role in carcinogenesis and lead to an increase in insulin-like growth factor 1 (IGF1) which levels are often reduced in patients with chronic liver diseases due to a reduced synthesis capacity of the cirrhotic liver[49]. In particular, hyperinsulinemia increases the expression of growth hormone receptor which controls the IGF1 production and secretion and reduced the synthesis of insulin-like growth factor-binding protein type 1 and 2, which negatively regulates the pathway[50]. IGF1 phosphorylates insulin receptor substrates 1 leading to the activation of mTOR and mitogen-activated protein kinase pathways and therefore to cell proliferation, inhibition of apoptosis and HCC development[6,44]. Finally, the uncontrolled cytokines production promotes the activation and proliferation of myofibroblasts, including hepatic stellate cells, leading to the deposition of fibrillar collagens and consequently to the development of cirrhosis and HCC occurrence[6,44].
The role of the immune system
The immune system contributes to NASH-driven liver damage and HCC[51]. Emerging data have suggested that CD8+ T cells play a key role in hepatocarcinogenesis in NASH patients[52]. In particular, Pfister et al.[52] have observed a high number of activated CD8+ T cells expressing the programmed cell death protein 1 (PD1) in a NASH-HCC mouse model, but despite the abundance of these cells, the anti-PD1 antibody did not reduce the number and/or size of tumours and moreover, an increase in liver fibrosis has been observed. The authors confirmed an enrichment of CD8+ PD1+ T cells with the same non-proliferative and tissue residency phenotype in NASH-HCC patients. They also conducted a meta-analysis of phase III randomized controlled trials showing that patients with HCC who had underlying NAFLD had significantly shorter overall survival after immunotherapy compared with other HCC aetiologies suggesting that NASH could promote resistance to immunotherapy[52]. T regulator cells are increased in NASH-HCC and play an important role both in the initiation and progression of the tumour. Recent evidence from mouse models, show that neutrophils extracellular traps favour the progression of NASH to HCC promoting the differentiation of naïve CD4+ to T regulator cells by increasing the expression of oxidative phosphorylation genes[53]. Moreover, elevated serum levels of neutrophils extracellular trap marker have been described in NASH patients who underwent liver resection, suggesting that the mechanism described in the preclinical model, could also promote hepatocarcinogenesis in humans[53]. Shalapour et al.[54] suggested a role for immunoglobulin A (IgA)+ plasma cells in the development of NAFLD-HCC suppressing CD8+ T cells via programmed cell death ligand 1 (PD-L1) and IL-10 expression, both in mice and humans. Preclinical models highlighted the possible role of platelets in NASH development and progression to HCC. Higher intrahepatic platelet levels have been found in the livers of mice fed with a choline deficient high-fat diet (CD-HFD) and not in mice fed with a lower calorie high fat diet, suggesting a specific link with NASH rather than with steatosis and/or insulin resistance. Moreover, glycoprotein Ib alpha (GPIba)-/- mice do not develop HCC, suggesting a possible role for antiplatelet and anti-GPIb alpha therapy into preventing NASH and NASH-HCC[55]. To date, robust clinical data about the role of antiplatelet therapy in prevention NASH-HCC development are not available even if some studies suggest a reduced HCC occurrence on cirrhosis or after a curative treatment for HCC in patients taking aspirin[56,57]. A role for myeloid cells in the progression of obesityrelated NAFLD to NASH has been also described, although the role of these cells in promoting HCC development has not been established yet[58].
The role of gut microbiota and bile metabolism
Gut microbiota and gut-liver axis influences the pathogenesis and the progression of various liver diseases, including NAFLD and HCC. A different microbial profile and alterations in intestinal permeability are usually found in NASH-cirrhosis, independent from the association with HCC, although specific differences in gut microbiota composition of NAFLD-HCC patients have been recently described[59]. Among these patients, an increase in Bacteroides and Ruminococcaceae and a decrease of Bifidobacterium have been reported. Additionally, increased levels of calprotectin have also been found[59]. The changes in gut microbiota composition have been linked to an increased production of secondary bile acids (BAs) (e.g., deoxycholic acid) which may reduce natural killer T cell recruitment and directly activate mTOR signaling fostering the HCC development[60]. Moreover, Behary et al.[61] have recently observed that the microbiota may modulate the immune peripheral response in HCC-NAFLD patients, promoting the expansion of T regulator cells, reduction of CD8+ T cells, and attenuation cytotoxic CD8+ T cells and total CD4+ T cells expansion. Pathogen-associated molecular patterns promote cytokine and chemokine production through the activation of toll-like receptors promoting oxidative stress and DNA damage, besides the activation of hepatic stellate cells which may promote hepatocyte proliferation[60].
Bile acids are involved in fat digestion and vitamin metabolism, and gene expression regulation targeting both nuclear farnesoid X receptor and membrane receptors G-protein-coupled bile acid receptor (TGR5). In NASH-HCC patients, an increased expression of BAs pool and steroidogenic acute regulatory protein 1 (STARD1), involved in the alternative mitochondrial pathway of BAs synthesis, has been observed, suggesting the role of BAs in HCC occurrence[62]. Moreover, in mice, STARD1 overexpression has been associated with multifocal carcinogenesis and the incubation of tumour-initiated stem-like cells or primary mouse hepatocytes with BAs seems to stimulate the expression of genes involved in stemness and inflammation[62]. The activation of farnesoid X receptor has been also linked to the increase in fibroblast growth factor (FGF) 19 blood levels[63]. FGF19 binds FGF receptor 4-b-Kloto complex expressed at hepatocyte surface inhibiting BA and reducing liver steatosis and insulin resistance[63]. These mechanisms represent the rationale of randomized clinical trial testing a humanized FGF19 analogue for NAFLD treatment (NCT03912532). Further research is needed to evaluate their role in NAFLD and NAFLD-HCC, considering that FGF19 is often overexpressed in these tumors[63].
Mutational landscape of NAFLD-related HCC
Pinyol et al.[64] have recently characterized the mutational landscape in 80 human NASH-HCCs, identifying TERT promoter (56%), CTNNB1 (28%), TP53 (18%), and ACVR2A (10%) mutations as the most frequently altered genes. A higher rate of ACVR2A mutations has been observed in NASH-HCC compared to HCC related to other etiologies (10% vs. 3%, respectively) regardless of the presence of cirrhosis[64]. ACVR2A modulates cell differentiation and proliferation, acting as a tumour suppressor in various malignancies, including HCC. In addition, a trend towards lower rates of TP53 mutations in NASH-HCC vs. non-NASH-HCC (18% vs. 32%, respectively) has also been observed[64]. They have also described a novel mutational signature (MutSig-NASH-HCC) characterised by a higher frequency of C>T and C>A transitions, almost exclusively found in NASH-HCC, predominantly in female patients[64]. Regarding molecular classes, NASH-HCCs have been found to be enriched in Wnt/TGF-beta class with a lower prevalence of CTNNB1 mutations. An increase in gene sets related to mTOR and mitochondria has also been observed, suggesting the involvement of bile acid and fatty acid metabolism as well as oxidative stress and inflammation in NASH-related HCC development[51]. Paradis et al.[65] reported that cullin 7, a E3 ligase that modulates cyclin D1 expression, is overexpressed in HCC arising from advanced fibrosis related to metabolic syndrome (60%). Cullin 7 was proposed to play an antiapoptotic effect through p53[65]. Among epigenetic alterations, an increased expression of lncRNA SNHG20[66] has been described in NAFLD-related patients whereas hypometilation of TRIM4, PRC1 and TUBA1B[67] has been observed in NASH-related HCC patients.
PRECLINICAL MODELS
In vitro and mouse models have improved the understanding of molecular pathways involved in liver carcinogenesis in NAFLD patients [Table 1].
Animal models of NAFLD-related HCC.
| Model | Obesity | Insulin resistance | NASH | Fibrosis | HCC |
| Dietary | |||||
| ALIOS[68-70] | + | + | + | + | 52 weeks |
| CDDA + HFD[52,71] | + | + | + | + | 24-30 weeks |
| Dietary and toxic | |||||
| STAM[72] | - | + | + | + | 16-20 weeks |
| CCl4 + WD[74] | + | + | + | + | 24 weeks |
| Dietary and genetic | |||||
| DIAMOND[75] | + | + | + | + | 52 weeks |
| MUP-uPA[76] | + | + | + | + | 40 weeks |
| TIRF-HFD[78] | - | NA | + | - | To be determined |
| Toxic and genetic | |||||
| Mc4rKODEN + WD[79] | + | + | + | + | 12 weeks |
Dietary models
Fructose consumption has been associated with NAFLD occurrence in humans[68]. Mice fed with HFD and fructose (40% fat, 0.2% of cholesterol, and a final fructose concentration of 42g/L), have increased body weight, increased body fat mass, and developed insulin resistance similar to HFD mice but with more liver fibrosis, inflammation, lipoapoptosis, and oxidative stress[69,70]. Moreover, Dowman et al.[71] have demonstrated that mice fed with a diet high in trans-fats and fructose corn syrup and who lead a sedentary lifestyle [American lifestyle-induced obesity syndrome (ALIOS)] develop features of NAFLD after 6 months and NASH after 12 months; six out of ten mice also developed hepatocellular neoplasm. These results were recently confirmed by Harris et al.[72] who state that the ALIOS diet in mice recapitulates many of the clinical features of NAFLD and, therefore, represents a robust and reproducible model for investigating the pathogenesis of NAFLD and its progression, showing an increased incidence of hepatic tumours after 52 weeks. The choline-deficient, L-amino acid-defined (CDAA) model develops steatohepatitis and liver fibrosis[6]. The combination of the CDAA diet with a HFD leads to obesity, NASH, and fibrosis; HCC occurs after 30 weeks in approximately 25% of mice[73]. These results have been recently confirmed by Wei et al.[74] which have observed that feeding C57Bl/6 mice with HFD-CDAA diet led to the development of NASH, cirrhosis, and liver cancer in 80% of cases within 24 weeks.
Dietary and toxic models
In the STAM (stelic animal model) model, a single dose of 200 micrograms of streptozotocin is given two days after birth to C57B16 mice to reduce insulin secretory capacity of damaging pancreatic beta-cells. Surviving mice are started on a HFD at 4 weeks of age and develop NASH, fibrosis, and HCC between 16 and 20 weeks of age[75]. Although histological features resemble human NASH, the mice lost weight. Moreover, a direct carcinogenic effect of streptozotocin cannot be ruled out[6]. The above data explain why this model is often considered as not adequate for the study of NASH and its progression to HCC[6,76]. A rapid progression to NASH with stage three fibrosis (12 weeks) and HCC development (24 weeks) has been observed in the mouse model developed by Tsuchida et al.[77] who administered to C57BL/6J mice a Western diet rich in fat, fructose, and cholesterol combined with a weekly dose of intraperitoneal carbon tetrachloride. This model mimics histological, immunological, and transcriptomic features of human NASH, and it could be used to study NASH and its progression to HCC[77]. Moreover, in a recent paper, Pinyol et al.[64] confirmed the association between Western diet + carbon tetrachloride murine model and the molecular features of both cirrhotic and non-cirrhotic human-NASH-HCC.
Dietary and genetic models
In genetic animal models of NASH, diet is used as a secondary trigger to mimic disease progression. The DIAMOND (diet induced animal model of NAFLD) model is based on an inbred isogenic strain of a C57Bl6/J and S129S1/svlmJ mice where approximately 60% of genes are from the C57Bl6/J background. When mice are fed with a high fat, high carbohydrate diet with 42% Kcal from fat and containing 0.1% cholesterol with ad libitum administration of glucose/fructose in drinking water, mice develop obesity, insulin resistance, and dyslipidemia. Moreover, after 16 weeks they develop steatohepatitis followed by stage 3-4 fibrosis (36 weeks) and HCC in 90% of cases (52 weeks)[78]. The DIAMOND model shows a strong concordance with the human NAFLD transcriptome throughout the various stages of disease development[78]. One of the mechanisms which fosters the HCC development in NAFLD patients is the ER stress consequent to the intracellular lipid accumulation. Mice which expressed high amounts of urokinase plasminogen activator in hepatocytes (MUP-uPA) has ER stress and the addition of HFD promotes expression of TNF and a spontaneous progression from NASH to HCC[79]. Moreover, in MUP-uPA mice fed with HFD, a strong p62 expression has been observed, and the p62 ablation (Sqstm1 Δhep/MUP), led to the reduction of both tumour number and size confirming the role of the autophagy adaptor p62 in NASH-HCC development. Similar results have been demonstrated also in STZ-HFD mice where p62 deletion suppresses HCC development without influencing fibrosis and steatosis development[80]. Recently, Bissig-Choisat et al.[81] generated a transgene-free Il2rg-/-/Rag2-/-/Fah-/- mouse populated with human hepatocytes and fed with HFD for 12 weeks. Interestingly, metabolic and histological changes of the human hepatocytes resemble those of NAFLD patients[81]. In particular, cholesterol metabolism changes are similar to NAFLD-patients[81]. Therefore, this model could be helpful in studying the malignant transformation on NAFLD[81].
Toxic and genetic models
Another more murine model has been recently proposed to assess drug efficacy in NASH-HCC. Two-week-old melanocortin-4-receptor knocked out mice receive a single intraperitoneal injection of diethylnitrosamine and are fed Western diet from 6 weeks of age up to 14 weeks. The advantage of this model is the fast development of HCC[82] with mice developing multiple liver tumours together with obesity, diabetes, and NASH in 3 months[82].
CLINICAL PERSPECTIVES AND CONCLUSION
Understanding the molecular pathogenesis of NAFLD-HCC patients could enable an early diagnosis and improve patient prognosis. It could also allow the development of HCC primary and secondary prevention strategies for patients with NASH, which to date has no approved treatment. HCC screening is recommended for cirrhotic patients, independent of the underlying aetiology[83]. As previously stated, the prevalence of NAFLD and the low incidence of HCC among patients without cirrhosis makes the surveillance in NAFLD non-cirrhotic patients not cost-effective. Moreover, sensitivity and specificity of ultrasonography may be compromised by obesity of cirrhotic NAFLD[5]. Whether non-cirrhotic NAFLD patients should undergo surveillance, which subgroup of patients should eventually be screened, and which test or combination of test should be used are questions that have still not been answered. Up to now various predictive factors have been assessed but none of these has been yet validated for clinical practice. A recent study by Younes et al.[84] evaluated non-invasive scoring systems, usually used to stratify fibrosis among patients with NAFLD [e.g., NAFLD fibrosis score-NFS, aspartate aminotransferase (AST) to platelet ratio index-APRI, Fibrosis 4-FIB-4, BMI, AST to ALT ratio, and BARD score] as predictors of long-term and extrahepatic events in a large cohort of patients. The authors identified a possible role of NFS in the prediction of HCC development (Harrell’s c index = 0.9)[84]. Moreover, the GALAD score (gender, age, lens culinaris-agglutinin-reactive fraction of AFP, AFP, and Des-g-carboxy-prothrombin) could be useful to detect HCC among NASH patients with or without cirrhosis[85]. Since no single SNP is capable of adequate HCC risk stratification due to their low odds ratio with HCC occurrence, polygenic risk scores based on the sum of risk alleles have been proposed[43,86,87]. Gellert-Kristensen et al.[87] demonstrated that a genetic risk score, based on the sum of three risk-increasing alleles (i.e., PNPLA3, TM6SF2, and HSD17B13), predicts the risk of developing cirrhosis and HCC in NAFLD-patients. In particular, a 29-fold higher risk of developing HCC has been observed in NAFLD patients with a score of 5 and 6[87]. A different genetic risk score for hepatic fat accumulation based on well characterized variants (PNPLA3, TM6SF2, GCKR, and MBOAT7) has been proposed, and a proportional impact of genomic variants on fibrosis and hepatic fat has been demonstrated[86]. This score and its adjusted version for HSD17B13 (PRS-5) was able to predict HCC development in NAFLD and obese patients regardless of the degree of fibrosis[88]. Thresholds (≥ 0.532 for PRS-HFC and 0.495 for PRS-5) to recognise NAFLD and dysmetabolism patients with a high risk of HCC development with a good specificity have been proposed[88]. Moreover, circulating tumour DNA, circulating tumour cells, and extracellular vesicles are potential candidate biomarkers that need to be explored in NAFLD-HCC patients. Further research should be deepening their role in the surveillance among non-cirrhotic NAFLD patients. Finally, more data are required in preclinical model but also in real life practice in order to confirm the link between NASH and resistance to immunotherapy for advanced HCC.
DECLARATIONS
Authors’ contributionsConceptualization: Campani C, Nault JC
Writing - original draft preparation: Campani C
Writing - review and editing: Nault JC
Supervision: Nault JC
Availability of data and materialsNot applicable.
Financial support and sponsorshipNault JC received research grant from Bayer and Ipsen.
Conflicts of interestNault JC received research grants from bayer and Ipsen. Campani C declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2022.
REFERENCES
1. Younossi ZM, Corey KE, Lim JK. AGA clinical practice update on lifestyle modification using diet and exercise to achieve weight loss in the management of nonalcoholic fatty liver disease: expert review. Gastroenterology 2021;160:912-8.
2. Eslam M, Newsome PN, Sarin SK, et al. A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J Hepatol 2020;73:202-9.
3. Younossi ZM, Henry L. Epidemiology of non-alcoholic fatty liver disease and hepatocellular carcinoma. JHEP Rep 2021;3:100305.
4. Huang DQ, El-Serag HB, Loomba R. Global epidemiology of NAFLD-related HCC: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 2021;18:223-38.
5. Loomba R, Lim JK, Patton H, El-Serag HB. AGA clinical practice update on screening and surveillance for hepatocellular carcinoma in patients with nonalcoholic fatty liver disease: expert review. Gastroenterology 2020;158:1822-30.
6. Nahon P, Allaire M, Nault JC, Paradis V. Characterizing the mechanism behind the progression of NAFLD to hepatocellular carcinoma. Hepat Oncol 2020;7:HEP36.
7. Eslam M, Valenti L, Romeo S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J Hepatol 2018;68:268-79.
8. Huang Y, Cohen JC, Hobbs HH. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J Biol Chem 2011;286:37085-93.
9. Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461-5.
10. BasuRay S, Wang Y, Smagris E, Cohen JC, Hobbs HH. Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc Natl Acad Sci U S A 2019;116:9521-6.
11. He S, McPhaul C, Li JZ, et al. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J Biol Chem 2010;285:6706-15.
12. Pirazzi C, Adiels M, Burza MA, et al. Patatin-like phospholipase domain-containing 3 (PNPLA3) I148M (rs738409) affects hepatic VLDL secretion in humans and in vitro. J Hepatol 2012;57:1276-82.
13. Yuan X, Waterworth D, Perry JR, et al. Population-based genome-wide association studies reveal six loci influencing plasma levels of liver enzymes. Am J Hum Genet 2008;83:520-8.
14. Speliotes EK, Butler JL, Palmer CD, Voight BF, Hirschhorn JN. GIANT Consortium, MIGen Consortium, NASH CRN. PNPLA3 variants specifically confer increased risk for histologic nonalcoholic fatty liver disease but not metabolic disease. Hepatology 2010;52:904-12.
15. Rotman Y, Koh C, Zmuda JM, Kleiner DE, Liang TJ. NASH CRN. The association of genetic variability in patatin-like phospholipase domain-containing protein 3 (PNPLA3) with histological severity of nonalcoholic fatty liver disease. Hepatology 2010;52:894-903.
16. Sookoian S, Pirola CJ. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 2011;53:1883-94.
17. Salameh H, Hanayneh MA, Masadeh M, et al. PNPLA3 as a genetic determinant of risk for and severity of non-alcoholic fatty liver disease spectrum. J Clin Transl Hepatol 2016;4:175-91.
18. Falleti E, Fabris C, Cmet S, et al. PNPLA3 rs738409C/G polymorphism in cirrhosis: relationship with the aetiology of liver disease and hepatocellular carcinoma occurrence. Liver Int 2011;31:1137-43.
19. Trépo E, Nahon P, Bontempi G, et al. Association between the PNPLA3 (rs738409 C>G) variant and hepatocellular carcinoma: Evidence from a meta-analysis of individual participant data. Hepatology 2014;59:2170-7.
20. Burza MA, Pirazzi C, Maglio C, et al. PNPLA3 I148M (rs738409) genetic variant is associated with hepatocellular carcinoma in obese individuals. Dig Liver Dis 2012;44:1037-41.
21. Liu YL, Patman GL, Leathart JB, et al. Carriage of the PNPLA3 rs738409 C >G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J Hepatol 2014;61:75-81.
22. Seko Y, Sumida Y, Tanaka S, et al. Development of hepatocellular carcinoma in Japanese patients with biopsy-proven non-alcoholic fatty liver disease: Association between PNPLA3 genotype and hepatocarcinogenesis/fibrosis progression. Hepatol Res 2017;47:1083-92.
23. Kozlitina J, Smagris E, Stender S, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2014;46:352-6.
24. Prill S, Caddeo A, Baselli G, et al. The TM6SF2 E167K genetic variant induces lipid biosynthesis and reduces apolipoprotein B secretion in human hepatic 3D spheroids. Sci Rep 2019;9:11585.
25. Dongiovanni P, Petta S, Maglio C, et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 2015;61:506-14.
26. Newberry EP, Hall Z, Xie Y, et al. Liver-specific deletion of mouse Tm6sf2 promotes steatosis, fibrosis, and hepatocellular cancer. Hepatology 2021;74:1203-19.
27. Yang J, Trépo E, Nahon P, et al. PNPLA3 and TM6SF2 variants as risk factors of hepatocellular carcinoma across various etiologies and severity of underlying liver diseases. Int J Cancer 2019;144:533-44.
28. Liu YL, Reeves HL, Burt AD, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat Commun 2014;5:4309.
29. Tanaka Y, Shimanaka Y, Caddeo A, et al. LPIAT1/MBOAT7 depletion increases triglyceride synthesis fueled by high phosphatidylinositol turnover. Gut 2021;70:180-93.
30. Meroni M, Longo M, Fracanzani AL, Dongiovanni P. MBOAT7 down-regulation by genetic and environmental factors predisposes to MAFLD. EBioMedicine 2020;57:102866.
31. Buch S, Stickel F, Trépo E, et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat Genet 2015;47:1443-8.
32. Freund C, Wahlers A, Begli NH, et al. The MBOAT7 rs641738 variant is associated with an improved outcome in primary sclerosing cholangitis. Clin Res Hepatol Gastroenterol 2020;44:646-52.
33. Thabet K, Chan HLY, Petta S, et al. The membrane-bound O-acyltransferase domain-containing 7 variant rs641738 increases inflammation and fibrosis in chronic hepatitis B. Hepatology 2017;65:1840-50.
34. Donati B, Dongiovanni P, Romeo S, et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci Rep 2017;7:4492.
35. Teo K, Abeysekera KWM, Adams L, et al. EU-PNAFLD Investigators, GOLD Consortium. rs641738C>T near MBOAT7 is associated with liver fat, ALT and fibrosis in NAFLD: a meta-analysis. J Hepatol 2021;74:20-30.
36. Abul-Husn NS, Cheng X, Li AH, et al. A protein-truncating HSD17B13 variant and protection from chronic liver disease. N Engl J Med 2018;378:1096-106.
37. Pirola CJ, Garaycoechea M, Flichman D, et al. Splice variant rs72613567 prevents worst histologic outcomes in patients with nonalcoholic fatty liver disease. J Lipid Res 2019;60:176-85.
38. Stickel F, Lutz P, Buch S, et al. Genetic variation in HSD17B13 reduces the risk of developing cirrhosis and hepatocellular carcinoma in alcohol misusers. Hepatology 2020;72:88-102.
39. Kozlitina J, Stender S, Hobbs HH, Cohen JC. HSD17B13 and chronic liver disease in blacks and hispanics. N Engl J Med 2018;379:1876-7.
40. Santoro N, Zhang CK, Zhao H, et al. Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology 2012;55:781-9.
41. Petta S, Miele L, Bugianesi E, et al. Glucokinase regulatory protein gene polymorphism affects liver fibrosis in non-alcoholic fatty liver disease. PLoS One 2014;9:e87523.
42. Meroni M, Longo M, Paolini E, et al. The rs599839 A>G variant disentangles cardiovascular risk and hepatocellular carcinoma in NAFLD patients. Cancers (Basel) 2021;13:1783.
43. Pelusi S, Baselli G, Pietrelli A, et al. Rare pathogenic variants predispose to hepatocellular carcinoma in nonalcoholic fatty liver disease. Sci Rep 2019;9:3682.
44. Margini C, Dufour JF. The story of HCC in NAFLD: from epidemiology, across pathogenesis, to prevention and treatment. Liver Int 2016;36:317-24.
45. Kanwal F, Kramer JR, Li L, et al. Effect of metabolic traits on the risk of cirrhosis and hepatocellular cancer in nonalcoholic fatty liver disease. Hepatology 2020;71:808-19.
46. Simon TG, King LY, Chong DQ, et al. Diabetes, metabolic comorbidities, and risk of hepatocellular carcinoma: results from two prospective cohort studies. Hepatology 2018;67:1797-806.
47. Kucukoglu O, Sowa JP, Mazzolini GD, Syn WK, Canbay A. Hepatokines and adipokines in NASH-related hepatocellular carcinoma. J Hepatol 2021;74:442-57.
48. Grohmann M, Wiede F, Dodd GT, et al. Obesity drives STAT-1-dependent NASH and STAT-3-dependent HCC. Cell 2018;175:1289-1306.e20.
50. Allaire M, Nault JC. Type 2 diabetes-associated hepatocellular carcinoma: a molecular profile. Clin Liver Dis (Hoboken) 2016;8:53-8.
51. Sutti S, Albano E. Adaptive immunity: an emerging player in the progression of NAFLD. Nat Rev Gastroenterol Hepatol 2020;17:81-92.
52. Pfister D, Núñez NG, Pinyol R, et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021;592:450-6.
53. van der Windt DJ, Sud V, Zhang H, et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology 2018;68:1347-60.
54. Shalapour S, Lin X, Bastian IN, et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 2017;551:340-5.
55. Malehmir M, Pfister D, Gallage S, et al. Platelet GPIbα is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat Med 2019;25:641-55.
56. Simon TG, Ma Y, Ludvigsson JF, et al. Association between aspirin use and risk of hepatocellular carcinoma. JAMA Oncol 2018;4:1683-90.
57. Simon TG, Duberg AS, Aleman S, Chung RT, Chan AT, Ludvigsson JF. Association of aspirin with hepatocellular carcinoma and liver-related mortality. N Engl J Med 2020;382:1018-28.
58. Krenkel O, Hundertmark J, Abdallah AT, et al. Myeloid cells in liver and bone marrow acquire a functionally distinct inflammatory phenotype during obesity-related steatohepatitis. Gut 2020;69:551-63.
59. Ponziani FR, Bhoori S, Castelli C, et al. Hepatocellular carcinoma is associated with gut microbiota profile and inflammation in nonalcoholic fatty liver disease. Hepatology 2019;69:107-20.
60. Zhou J, Tripathi M, Sinha RA, Singh BK, Yen PM. Gut microbiota and their metabolites in the progression of non-alcoholic fatty liver disease. Hepatoma Res 2021;7:11.
61. Behary J, Amorim N, Jiang XT, et al. Gut microbiota impact on the peripheral immune response in non-alcoholic fatty liver disease related hepatocellular carcinoma. Nat Commun 2021;12:187.
62. Conde de la Rosa L, Garcia-Ruiz C, Vallejo C, et al. STARD1 promotes NASH-driven HCC by sustaining the generation of bile acids through the alternative mitochondrial pathway. J Hepatol 2021;74:1429-41.
63. Marra F, Svegliati-Baroni G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J Hepatol 2018;68:280-95.
64. Pinyol R, Torrecilla S, Wang H, et al. Molecular characterisation of hepatocellular carcinoma in patients with non-alcoholic steatohepatitis. J Hepatol 2021;75:865-78.
65. Paradis V, Albuquerque M, Mebarki M, et al. Cullin7: a new gene involved in liver carcinogenesis related to metabolic syndrome. Gut 2013;62:911-9.
66. Wang B, Li X, Hu W, Zhou Y, Din Y. Silencing of lncRNA SNHG20 delays the progression of nonalcoholic fatty liver disease to hepatocellular carcinoma via regulating liver Kupffer cells polarization. IUBMB Life 2019;71:1952-61.
67. Tian Y, Arai E, Makiuchi S, et al. Aberrant DNA methylation results in altered gene expression in non-alcoholic steatohepatitis-related hepatocellular carcinomas. J Cancer Res Clin Oncol 2020;146:2461-77.
68. Rietman A, Sluik D, Feskens EJM, Kok FJ, Mensink M. Associations between dietary factors and markers of NAFLD in a general Dutch adult population. Eur J Clin Nutr 2018;72:117-23.
69. Kohli R, Kirby M, Xanthakos SA, et al. High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology 2010;52:934-44.
70. Charlton M, Krishnan A, Viker K, et al. Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am J Physiol Gastrointest Liver Physiol 2011;301:G825-34.
71. Dowman JK, Hopkins LJ, Reynolds GM, et al. Development of hepatocellular carcinoma in a murine model of nonalcoholic steatohepatitis induced by use of a high-fat/fructose diet and sedentary lifestyle. Am J Pathol 2014;184:1550-61.
72. Harris SE, Poolman TM, Arvaniti A, Cox RD, Gathercole LL, Tomlinson JW. The American lifestyle-induced obesity syndrome diet in male and female rodents recapitulates the clinical and transcriptomic features of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol 2020;319:G345-60.
73. Wolf MJ, Adili A, Piotrowitz K, et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 2014;26:549-64.
74. Wei G, An P, Vaid KA, et al. Comparison of murine steatohepatitis models identifies a dietary intervention with robust fibrosis, ductular reaction, and rapid progression to cirrhosis and cancer. Am J Physiol Gastrointest Liver Physiol 2020;318:G174-88.
75. Fujii M, Shibazaki Y, Wakamatsu K, et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med Mol Morphol 2013;46:141-52.
76. Santhekadur PK, Kumar DP, Sanyal AJ. Preclinical models of non-alcoholic fatty liver disease. J Hepatol 2018;68:230-7.
77. Tsuchida T, Lee YA, Fujiwara N, et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J Hepatol 2018;69:385-95.
78. Asgharpour A, Cazanave SC, Pacana T, et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J Hepatol 2016;65:579-88.
79. Nakagawa H, Umemura A, Taniguchi K, et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014;26:331-43.
80. Umemura A, He F, Taniguchi K, et al. p62, Upregulated during preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed HCC-initiating cells. Cancer Cell 2016;29:935-48.
81. Bissig-Choisat B, Alves-Bezerra M, Zorman B, et al. A human liver chimeric mouse model for non-alcoholic fatty liver disease. JHEP Rep 2021;3:100281.
82. Yoshioka N, Tanaka M, Ochi K, et al. The sodium-glucose cotransporter-2 inhibitor Tofogliflozin prevents the progression of nonalcoholic steatohepatitis-associated liver tumors in a novel murine model. Biomed Pharmacother 2021;140:111738.
83. Association for the Study of the Liver. Electronic address: easloffice@easloffice.eu, European Association for the Study of the Liver. EASL Clinical Practice Guidelines: management of hepatocellular carcinoma. J Hepatol 2018;69:182-236.
84. Younes R, Caviglia GP, Govaere O, et al. Long-term outcomes and predictive ability of non-invasive scoring systems in patients with non-alcoholic fatty liver disease. J Hepatol 2021;75:786-94.
85. Best J, Bechmann LP, Sowa JP, et al. GALAD score detects early hepatocellular carcinoma in an international cohort of patients with nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol 2020;18:728-35.e4.
86. Dongiovanni P, Stender S, Pietrelli A, et al. Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. J Intern Med 2018;283:356-70.
87. Gellert-Kristensen H, Richardson TG, Davey Smith G, Nordestgaard BG, Tybjaerg-Hansen A, Stender S. Combined effect of PNPLA3, TM6SF2, and HSD17B13 variants on risk of cirrhosis and hepatocellular carcinoma in the general population. Hepatology 2020;72:845-56.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Campani C, Nault JC. Molecular mechanisms of liver carcinogenesis related to metabolic syndrome. Hepatoma Res 2022;8:3. http://dx.doi.org/10.20517/2394-5079.2021.126
AMA Style
Campani C, Nault JC. Molecular mechanisms of liver carcinogenesis related to metabolic syndrome. Hepatoma Research. 2022; 8: 3. http://dx.doi.org/10.20517/2394-5079.2021.126
Chicago/Turabian Style
Campani, Claudia, Jean-Charles Nault. 2022. "Molecular mechanisms of liver carcinogenesis related to metabolic syndrome" Hepatoma Research. 8: 3. http://dx.doi.org/10.20517/2394-5079.2021.126
ACS Style
Campani, C.; Nault J.C. Molecular mechanisms of liver carcinogenesis related to metabolic syndrome. Hepatoma. Res. 2022, 8, 3. http://dx.doi.org/10.20517/2394-5079.2021.126
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 9 clicks
Cite This Article 9 clicks

 Like This Article 19
likes
Like This Article 19
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.