
Treatment of advanced biliary tract cancers: from chemotherapy to targeted agents
 0
0Abstract
Biliary tract cancers (BTCs) are usually diagnosed at an advanced stage and have a dismal prognosis. The treatment of advanced disease is mainly based on systemic chemotherapy, which is demonstrated to improve survival in the first- and second-line setting. Following the results of phase III clinical trials, the combination of cisplatin and gemcitabine is the regimen of choice in the frontline, while 5-fluorouracil plus oxaliplatin is considered the standard after first-line progression in unselected patients. Recent advances in molecular biology have unravelled the molecular heterogeneity of BTCs and identified patient subgroups harbouring unique molecular aberrations such as isocitrate dehydrogenase (IDH) mutations and fibroblast growth factor receptor (FGFR) fusions that can be targeted by specific agents. This knowledge has opened the way to personalised medicine in BTCs. Molecules targeting IDH and FGFR are currently approved for the treatment of advanced, refractory, intrahepatic cholangiocarcinoma. Beyond targeted therapies, novel combinatorial approaches that target the immune microenvironment and the crosstalk between cancer and stroma are being explored based on strong preclinical rationale. This review discusses the current therapeutic opportunities for the management of patients with advanced BTCs and provides an overview of the promising new strategies on the horizon with a particular focus on ongoing clinical trials.
Keywords
INTRODUCTION
Advanced stage is the most common clinical presentation of biliary tract cancers (BTCs). Given the paucity of specific early symptoms and the lack of early-detection programs, up to 80% of patients are diagnosed with unresectable tumours[1,2]. In addition, recurrence rate after surgery ranges between 50% and 79%, even for those patients diagnosed with early-stage cancer[3-5]. Advanced disease at presentation or relapse after local treatment, along with the scarce availability of active treatments and the significant chemo-resistance, make BTCs one of the deadliest solid tumours with a global five-year survival rate of 10%[6]. In this scenario, untreated patients have an overall survival (OS) of 3-4 months, while those receiving active treatment can reach up to 18-20 months with two therapeutic lines[7-10]. Systemic approach represents the standard of care of advanced biliary tract cancers (aBTCs) and aims at increasing survival, delaying disease progression, relieving symptoms, and improving/maintaining the quality of life. Historically, cytotoxic chemotherapy was the only available option and was associated with marginal improvements in oncological outcomes. More recently, emerging compounds targeting isocitrate dehydrogenase (IDH) 1 mutations and fibroblast growth factor receptor (FGFR) 2 fusions, as well as immune checkpoint inhibitors (ICIs), showed promising results in molecularly selected patients[11]. Thanks to advances in molecular biology, many other therapeutic vulnerabilities have recently been identified both in the tumour and in the surrounding microenvironment, providing a plethora of novel potential strategies to be further investigated for the treatment of BTCs[12].
In this paragraph, we aim to offer an overview of the current therapeutic approaches for the management of patients with aBTCs, and to discuss the novel opportunities on the near horizon.
UNSELECTED TREATMENT APPROACH: PAST, PRESENT AND FUTURE
The history of the therapeutic development in BTCs has been characterised by “all-comers” design of clinical trials exploring the efficacy of cytotoxic agents and targeted therapies. This “one size fits all” model has shown clinical benefit from chemotherapy, allowing the identification of current standard first and second-line regimens[7,8]. Chemotherapeutic drugs combination is demonstrated to increase efficacy by using different mechanisms of cytotoxic action. The two chemotherapy combinations regimens approved following positive results of phase III clinical trials include cisplatin and gemcitabine (CisGem) for frontline treatment and 5-fluorouracil plus oxaliplatin (FOLFOX) after first-line progression. More recently, two combinatorial regimens showed promising results in phase II studies for the first-line (cisplatin, gemcitabine, and nab-paclitaxel)[13] and second-line (5-fluorouracil and liposomal irinotecan; Nal-Iri) setting[14]. Clinical studies investigating targeted therapies and ICIs have been widely unsuccessful in unselected patients instead[15-24].
First-line chemotherapy
Limited data supported effective management of aBTCs before 2010. A pooled analysis of 104 small and nonrandomised trials, including 2810 patients conducted in 2007, showed combination chemotherapy with CisGem or oxaliplatin being the most active strategy[11]. The first evidence of superior regimen from randomised phase II study was reported in 2007, when CisGem demonstrated better time to progression compared to gemcitabine alone (8 months vs. 4 months, respectively) and improved disease control rate (DCR)[25]. This association became the standard first-line treatment for patients with metastatic BTCs since 2010, when the Advanced Biliary Tract Cancer (ABC-02) phase III trial demonstrated the superiority of CisGem vs. gemcitabine alone[7]. The median overall survival (mOS) resulted superior for the experimental arm compared to gemcitabine [11.7 months vs. 8.1 months, respectively; hazard ratio (HR) = 0.64; 95% confidence interval (CI): 0.52-0.8; P < 0.001]. The combination also demonstrated a better rate of partial response (PR) compared to the gemcitabine (26% vs. 16%, respectively)[7]. Similar results were reported in the Japanese BT22 randomised phase II trial that investigated the antitumour activity of the same agents (mOS 11.2 months vs. 7.7 months; HR = 0.69; 95%CI: 0.42-1.13; P = 0.139)[26]. The meta-analysis of the two trials confirmed significant improvement in progression-free survival (PFS) (HR = 0.64; 95%CI: 0.53-0.76; P < 0.001) and OS (HR = 0.65; 95%CI: 0.54-0.78; P < 0.001) of CisGem over gemcitabine for intra-and extra-hepatic cholangiocarcinomas (iCCA and eCCA) and gallbladder cancer, with most marked efficacy among patients with good performance status (PS 0-1) (HR for PFS = 0.61; 95%CI: 0.51-0.74; P < 0.001; HR for OS = 0.64; 95%CI: 0.53-0.77; P < 0.001)[27]. Table 1 shows an overview of these three studies.
First-line chemotherapy in Biliary tract cancers: an overview of pivotal studies
| Authors | Year | Country | Study design | Regimen | n | Median PFS (months) | HR | Median OS (months) | HR |
| Advanced biliary tract cancer (ABC)-02 | 2010 | United Kingdom | Randomized phase 3 | CisGem | 204 | 8.0 | 0.63; 95%CI: 0.51-0.77, P < 0.001 | 11.7 | 0.64; 95%CI: 0.52-0.80, P < 0.001 |
| GEM | 206 | 5.0 | 8.1 | ||||||
| Biliary tract (BT) 22 study | 2010 | Japan | Randomized phase 2 | CisGem | 41 | 5.8 | 0.66; 95%CI: 0.41-1.05, P = 0.077 | 11.2 | 0.69; 95%CI: 0.42-1.13, P = 0.139 |
| GEM | 42 | 3.7 | 7.7 | ||||||
| Cisplatin and gemcitabine for advanced biliary tract cancer: a meta-analysis of two randomised trials | 2014 | United Kingdom and Japan | Meta-analysis | CisGem | - | 8.8 | 0.64; 95%CI: 0.53-0.76, P < 0.001 | 11.6 | 0.65; 95%CI: 0.54-0.78, P < 0.001 |
| GEM | - | 6.7 | 8 |
Subsequent studies explored the role of other compounds such as oxaliplatin and fluoropyrimidines. Oxaliplatin showed a more favourable toxicity profile than cisplatin, with mOS reaching 12.4 months and overall response rate (ORR) up to 50%[21,22], but its activity has never been tested in a head-to-head comparison with CisGem. Nonetheless, this agent may be adopted when cisplatin is contraindicated. Fluoropyrimidines have also been shown to be active in aBTCs studies, but their precise role is still controversial as a direct comparison with gemcitabine-based regimens is lacking[28,29].
Another treatment option in the first-line setting is represented by gemcitabine plus S-1 (a fluoropyrimidine derivative), as suggested by the FUGA-BT phase III trial showing non-inferiority of this regimen compared with CisGem in the Japanese population[30].
Whether intensifying chemotherapy could improve oncological outcomes by maintaining a good quality of life is still an unanswered question and is currently under clinical investigation. In the Japanese population, the association of cisplatin, gemcitabine, and S1 could represent a treatment option on the basis of a phase III trial, demonstrating an improved OS of the triplet compared with CisGem (13.5 months vs. 12.6 months; HR = 0.791; 90%CI: 0.620-0.996; P = 0.046)[31].
Results from a phase II trial evaluating cisplatin, gemcitabine hydrochloride (salt of an analogue of the antimetabolite nucleoside deoxycytidine with antineoplastic activity), and nab-paclitaxel demonstrated median PFS (mPFS) of 11.8 months, mOS of 19.2 months, 45% response rate (RR) and 84% DCR with this regimen[13]. The ongoing phase III SWOG-1815 clinical trial will establish whether the triplet is more efficacious than the standard CisGem (NCT03768414). Likewise, a phase III study comparing CisGem vs. FOLFIRINOX (oxaliplatin, irinotecan, and 5-fluorouracil) has recently completed recruitment (NCT02591030) and will define if non-gemcitabine regimes have a place in first-line. Furthermore, a phase II study is exploring the activity of Nal-Iri in combination with 5-fluorouracil (5-FU)/leucovorin (LV) vs. CisGem (NCT03044587)[12].
Second-line chemotherapy
Following a failure to first-line chemotherapy, 25%-40% of BTC patients are still fit to receive second-line chemotherapy[32]. The choice of second-line treatment is extremely limited, particularly in unselected patients. In daily practice, 5-FU-based chemotherapy has been the most widely adopted regimen, despite the absence of robust evidence and the poor clinical efficacy (mPFS of 2.5-5.5 months and a mOS of 7.5-13.5 months in published series)[33].
Only recently, the phase III ABC-06 study demonstrated the superiority of FOLFOX vs. active symptom control (ASC, i.e., proactive detection of biliary obstruction, sepsis, and symptom management) in patients progressing to CisGem[8]. This study enrolled 162 patients with aBTC, including 72% of CCA following progression on standard first-line CisGem. Patients were randomised to ASC (81 patients) or ASC plus FOLFOX (81 patients). The ABC-06 trial demonstrated a benefit from second-line chemotherapy in terms of OS (HR = 0.69; 95%CI: 0.50-0.97; P = 0.031). Even though absolute differences in mOS were modest (5.3 months for ASC arm vs. 6.2 months for ASC + FOLFOX arm), differences in the survival rate at 6 months (35.5% for ASC arm vs. 50.6% for ASC + FOLFOX arm) and 12 months (11.4% for ASC arm vs. 25.9% for ASC + FOLFOX arm) were clinically meaningful[8]. Based on this evidence, FOLFOX is currently considered the standard of care second-line chemotherapy for patients with aBTCs without driver mutations who remains fit following CisGem progression.
The efficacy and safety of FOLFIRINOX with two different dosages (standard and modified) was investigated in a recent phase II trial enrolling 40 patients who had disease progression or unacceptable toxicity after ≥ 3 cycles of CisGem. The mPFS and mOS were 6.2 and 10.7 months, and the toxicity profile was acceptable[34]. Therefore, in patients with aBTCs who progress after or are intolerant to CisGem, FOLFIRINOX could be considered as an option for salvage treatment, even though borderline fitness of BTC patients in second-line may limit its applications[12].
Recent data from the NIFTY randomised, phase IIb study presented at ASCO 2021 showed that Nal-IRI plus 5-FU/LV significantly improved PFS and OS compared to 5-FU/LV in BTC patients who progressed on prior GemCis[14]. mPFS per investigator review in Nal-IRI plus 5-FU/LV group and 5-FU/LV group was 3.9 months and 1.6 months respectively (HR = 0.48; CI: 0.34-0.69; P < 0.0001); mOS was 8.6 months and 5.5 months respectively (HR = 0.68; CI: 0.48-0.98; P = 0.0349); ORR was 14.8% and 19.3% and 2.3%, respectively (P = 0.0002)[14].
One of the main challenges for the completion of adequately powered studies is that, given the aggressive clinical behaviour of CCA, < 40% of patients are considered eligible for second-line treatment while many others rapidly drop out of clinical trials due to clinical deterioration[35-39]. Amongst the subtypes of BTC, iCCA is usually associated with better performance status, with increased access to personalised approaches based on the tumour molecular profile, which will be discussed in the next chapter.
Prognostication
Reliable prognostic/predictive biomarkers to assist the therapeutic decision making in patients with aBTCs are still lacking. To date, only clinical prognostic factors independently associated with OS have been suggested, including Eastern Cooperative Oncology Group (ECOG) performance status (PS), blood tests (white blood cells and neutrophil count, haemoglobin and bilirubin levels), disease status, and gender, from the post-hoc analysis of CisGem pivotal trials[7,26,40]; and ECOG PS, prior resection, tumour grading, baseline carcinoembryonic antigen and carbohydrate antigen 19.9 (CA19-9), from the Italian Group of Cholangiocarcinoma, G.I.Co[36].
Recently, a prognostic score was derived by baseline neutrophil count, lymphocytes-monocytes ratio, neutrophil-lymphocytes ratio and albumin: the A.L.A.N. score. This prognostication has been shown to correlate with OS in a cohort of 123 aBTCs patients undergoing first-line chemotherapy. A.L.A.N. score was able to identify three classes of patients with significantly different OS (high-risk: median OS, 5 months; intermediate-risk: median OS, 12 months and low-risk: median OS, 22 months; P < 0.001)[14]. Many other clinical prognostic scores have been proposed, however, no standard currently exists[41-45].
The post-hoc analysis of ABC-01/02/03 trials of CisGem has also indicated a more favourable prognosis of iCCA and iCCA with liver-only disease. iCCA had a longer OS compared with other non-iCCA biliary tract cancers (HR = 0.58; 95%CI: 0.35-0.95; P = 0.03); liver-only iCCA patients also showed longer OS even though findings did not reach statistical significance (HR = 0.65; 95%CI: 0.36-1.19; P = 0.16)[40].
Despite being informative, evidence is yet to be confirmed in large, prospective, clinical studies, but until then, the optimal prognostication to adopt in the therapeutic decision of aBTCs remains uncertain.
To summarise, CisGem is the regimen of choice in the frontline, while FOLFOX is the standard after first-line progression in unselected patients [Figure 1]. Other options (i.e., single agents) may be carefully considered when the standard treatment is contraindicated or due to patient’s preferences. Several novel combinations are currently under clinical investigation. A selection of the most relevant ongoing clinical trials based on cytotoxic chemotherapy are shown in Table 2.
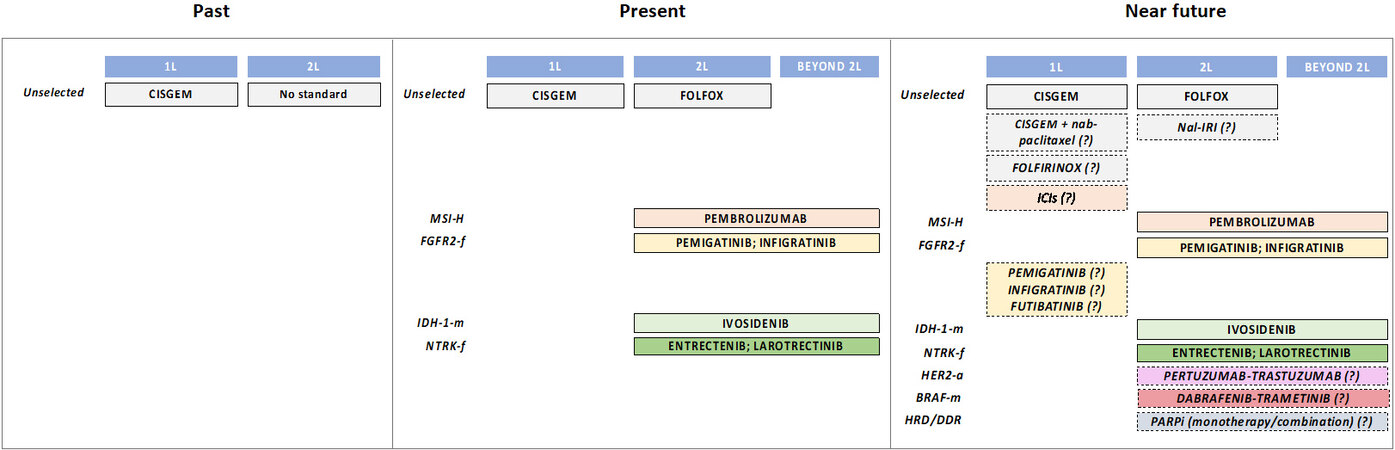
Figure 1. Evolution of systemic treatments of aBTCs. aBTCs: Advanced biliary tract cancers; L: line; ICIs: immune checkpoint inhibitors; m: mutations; f: fusions; a: amplifications; italic: under current clinical investigation; CISGEM: cisplatin + gemcitabine; FOLFOX: 5-fluorouracil + folinic acid + oxaliplatin; Nal-IRI: nanoliposomal irinotecan; FOLFIRINOX: 5-fluorouracil + folinic acid + oxaliplatin + irinotecan; PARPi: poly-ADP ribose polymerase inhibitors.
Cytotoxic chemotherapy: selection of ongoing clinical trials
| Trial number | Phase | Approach | Drug(s) | Line of treatment |
| NCT04163900 | 3 | Unselected | NUC-1031 + cisplatin | I |
| NCT03768414 | 3 | Unselected | Gemcitabine hydrochloride + cisplatin + nab-Paclitaxel | I |
| NCT04066491 | 2/3 | Unselected | GEM/CIS + bintrafusp alfa (M7824) | I |
| NCT04005339 | 2 | Unselected | Fluorouracil + leucovorin + NALIRI | II |
| NCT03044587 | 2 | Unselected | Fluorouracil + leucovorin + NALIRI | I |
| NCT03043547 | 2 | Unselected | NALIRI + 5-FU | II |
| NCT04111380 | 2 | Unselected | Nab-paclitaxel + cisplatin | II |
| NCT04692051 | 2 | Unselected | Nab-paclitaxel + cisplatin | I |
| NCT04203160 | 2 | Unselected | GEM/CIS + devimistat (CPI-613) | I |
| NCT04076761 | 2 | Unselected | Trifluridine/tipiracil | II |
| NCT03943043 | 1/2 | Unselected | GEMOX + Nab-paclitaxel | II |
| NCT03368963 | 1/2 | Unselected | Trifluridine/tipiracil + NALIRI | II |
| NCT04491942 | 1 | Unselected | Cisplatin or GEM/CIS + elimusertib (BAY 1895344) | II |
The allocation of locoregional strategies, complementary to chemotherapy, should be considered in the context of clinical studies.
Reliable prognostic and predictive biomarkers are still lacking and desirable, particularly in the second-line setting.
PRECISION MEDICINE: BIOLOGICAL RATIONALE AND THERAPEUTIC STRATEGIES
Advances in Next-Generation Sequencing technologies and large-scale initiatives from cooperative groups such as The Cancer Genome Atlas, the International Cancer Genome Consortium and others have allowed the characterisation of the molecular landscape of BTCs over the last decade, resulting in the identification of key actionable oncogenic drivers and signalling networks[46-54]. Firstly, biological differences according to anatomic location (iCCAs vs. eCCA), geographical location (Eastern vs. Western), and etiology (Hepatitis B, fluke, other causes) have been identified. Secondly, somatic - potentially targetable - alterations have been described in up half of patients with BTC[55-58].
This evidence has paved the way for the precision medicine revolution in this rare group of malignancies.
The molecular landscape of BTCs: single genetic alterations, molecular networks and subgroups
Data from published and non-published studies available in the cancer genomic dataset c-bioportal[59] indicate TP53, ARID1A, IDH1, BAP1, PBRM1, KRAS, SMAD4 and ATM as the most frequent molecular alterations in BTCs. As already mentioned, different patterns of mutations according to the specific anatomic subtype have been described, highlighting the high molecular heterogeneity - and possibly diverse etiopathogenesis - of histologically similar cancers [Figure 2]. The predominant molecular changes in BTCs are associated with epigenetic processes, particularly for iCCA, characterised by a higher frequency of mutations in IDH1, relevant for the cell metabolism processes (found in 10%-20% of cases) and FGFR2 rearrangements (found in up to 23% of patients). The latter consists in the fusion of the intact kinase domain of FGFR2 with several genes (BICC1, AHCYL1, TACC3, MGEA5 and PPHLN1), which constitutively activates the FGFR2 fusion protein and the downstream pathways, thus promoting tumour growth[60-66]. In contrast, alterations in other genes such as BAP1, TP53, KRAS, ERBB2, SMAD4, ARID1B, ELF, PBRM1, and STK11 are more common in perihilar, eCCA, and gallbladder cancer[46,56,67,68].
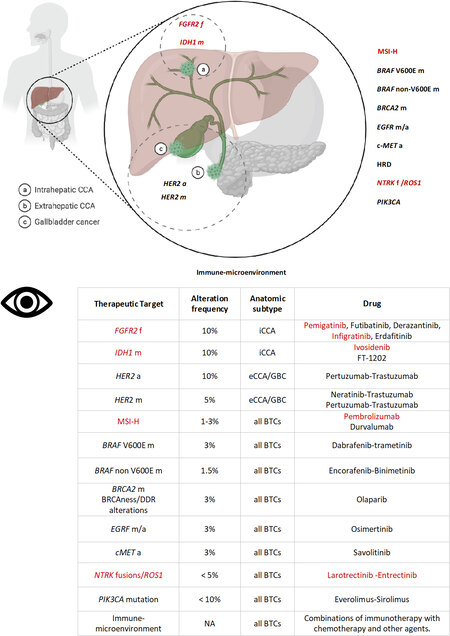
Figure 2. BTCs subtypes and therapeutic targets. Figure 2 shows the recurrent molecular aberrations in biliary tract cancers (BTCs) with a focus on the prevalence variations according to the anatomic subtypes. Fibroblast growth factor receptor 2 (FGFR2) fusions (f) and isocitrate dehydrogenase 1 (IDH-1) mutations (m) are more common in intrahepatic cholangiocarcinoma (iCCA), while human epidermal growth factor receptor 2 (HER2) mutations and amplifications (a) are more common in extrahepatic cholangiocarcinoma (eCCA) and gallbladder cancer (GCB). Aberrations in other genes and pathways (including immune microenvironment characteristics) are independent of the anatomic location (indicated outside the circle). In red: established targets and approved drugs; in black: targeted therapies under clinical investigation. CCA: Cholangiocarcinoma; DDR: DNA damage repair. HRD: homologous recombination deficiency; MSI-H: microsatellite instability-high; NA: not available.
Integrative genomics studies have also unravelled the genomic characterisation of the most prevalent oncogenic signalling pathways, including those involved in cell survival, angiogenesis, signal transduction and transcriptional control[69,70]. Genetic and epigenetic changes in BTCs cooperate in complex oncogenic networks, including NADPH metabolism, SWI-SNF complex, de-ubiquitination, and histone
In this context, distinct recurring molecular subgroups with different prognoses and responses to therapy have been identified. Sia et al.[69] described two subtypes of iCCA with different activated pathways and prognosis: the inflammation class (38% of cases) associated with the activation of inflammatory pathways, overexpression of cytokines, and STAT3 activation; and the proliferation class (62% of cases) associated with KRAS and BRAF mutations and oncogenic pathways activation (i.e., RAS, MAPK, and MET).
Through integrated genomic analysis of iCCA (whole-exome sequencing, targeted exome sequencing) and epigenomic data from 496 patients, Nepal et al.[71] described unique mutational signatures, structural variants and epigenomic alterations and proposed four different molecular subgroups with specific drug-response profiles: IDH mutant (drug: metabolic modulators), KRAS mutant (drug: microtubule modulators), TP53 mutant (drug: topoisomerase inhibitors), and “undetermined” (drug: mTOR inhibitors).
Another interesting classification that considers the tumour microenvironment (TME) characteristics of iCCA has identified 4 TME-based subtypes related to distinct immune escape mechanisms and patient outcomes[72]. About 45% of iCCA displayed an immune desert phenotype, while the 11% presented a massive T lymphocyte infiltration, activation of inflammatory and immune checkpoint pathways, and was associated with the longest patient survival (inflamed subtype). The other two subtypes differed in nature (lymphoid, myeloid, mesenchymal) and abundance of tumour-infiltrating cells[72]. The inflamed subgroup constitutes the most clinically relevant as it may potentially identify patients who are more likely to benefit from ICIs.
Personalised therapeutic approaches in aBTCs
The above data indicate the potential for oncogenic network characterisation to enable efficient stratification and therapy optimisation for patients with biliary cancers. A deeper understanding of the molecular biology underlying biliary tract malignancies and the high genomic/anatomical heterogeneity identified across histologically similar tumours may also partially explain the limited clinical advances achieved in the past decades. The lack of therapeutic targets, along with the “all-comers” design of clinical trials investigating targeted therapies in unselected patients (widely unsuccessful) may be an additional explanation for disappointing results achieved in past years[15-24].
On the other hand, promising results of personalised medicine for aBTCs have been demonstrated in molecularly driven studies. Among 43 pre-treated patients with aBTCs included in the prospective clinical trial MOSCATO-01, successful biopsy with multiple high-throughput molecular analysis was obtained in 34 patients. Orientation to an appropriate early clinical trial or administration of molecular-targeted agents was possible for 23 of these patients (68%), and 18 (53%) have received matched treatment. Among them, the ORR was 33%, and the DCR was 88%, while a PFS ≥ 6 months was observed in 37%. These patients had a lower risk for death as compared to the 20 patients not orientated to a matched treatment (mOS 17 months vs. 5 months; HR = 0.29; 95%CI: 0.11-0.76; P = 0.008)[73].
This analysis highlighted the relevance of histology-agnostic personalised medicine in the identification of patient subgroups to be matched with compounds targeting the specific molecular alteration and paved the way for the next-generation personalised medicine programs in BTCs.
Several compounds targeting single gene alterations have shown clinical activity and efficacy in advanced BTCs [Figure 2], others are currently under clinical investigation [Table 3], and many others are on the horizon [Figure 2].
Targeted therapies: ongoing clinical trials
| Trial number | Phase | Pathways targeted | Drug(s) | Line of treatment |
| NCT03656536 | 3 | FGFR | Pemigatinib | I |
| NCT03773302 | 3 | FGFR | Infigratinib (BGJ398) | I |
| NCT04093362 | 3 | FGFR | Futibatinib (TAS-120) | I |
| NCT04256980 | 2 | FGFR | Pemigatinib | II |
| NCT03873532 | 2/3 | MAPK | Surufatinib | II |
| NCT03230318 | 2 | FGFR | Derazantinib | II |
| NCT02150967 | 2 | FGFR | Infigratinib | II |
| NCT04238715 | 2 | FGFR2 | E7090 | II |
| NCT02699606 | 2 | FGRF | Erdafitinib | II |
| NCT03212274 | 2 | PARP | Olaparib | II |
| NCT03878095 | 2 | PARP | Olaparib Ceralasertib (AZD6738) | II |
| NCT04298021 | 2 | PARP | Ceralasertib (AZD6738) + durvalumab or AZD6738 + Olaparib | II |
| NCT04042831 | 2 | PARP | Olaparib | II |
| NCT03521219 | 2 | VEGFR2 | Apatinib | II |
| NCT03427242 | 2 | VEGFR2 | Apatinib | II |
| NCT02520141 | 2 | VEGF | Ramucirumab | II |
| NCT03377179 | 2 | SK2 | Opaganib/HCQ | II |
| NCT03185988 | 2 | HER2 | Trastuzumab | II |
| NCT04466891 | 2 | HER2 | ZW25 (Zanidatamab) | II |
| NCT03833661 | 2 | PD-L1; TGF-β Trap fusion protein | M7824 | II |
| NCT02576431 | 2 | NTRK | Larotrectinib | II |
| NCT02568267 | 2 | NTRK | Entrectinib | II |
| NCT03207347 | 2 | PARP | Niraparib | II |
| NCT02052778 | 1/2 | FGFR | Futibatinib (TAS-120) | II |
| NCT03684811 | 1/2 | IDH1 | FT-2102 | II |
| NCT04209465 | 1/2 | EGFR/ErbB | BDTX-189 | II |
| NCT03422679 | 1/2 | NOTCH | CB-103 | II |
| NCT03149549 | 1/2 | TAA CD166 | CX-2009 | II |
| NCT02908451 | 1 | TAA AG7 | AbGn-107 | II |
Currently, the most promising and clinically relevant target therapies are IDH inhibitors for patients carrying IDH-mutations and compounds targeting FGFR2 gene fusions[48]. Several IDH inhibitors are being explored for the treatment of advanced iCCA: inhibitors of IDH1 (ivosidenib), IDH2 (AG221), and pan-IDH1/2 (AG881). Ivosidenib showed encouraging results in a phase I dose escalation and expansion basket study. A total of 77 IDH1 mutant patients with refractory iCCA received the IDH1 inhibitor with stable disease and partial response achieved in 40 and 4 patients, respectively[74]. The positive results were subsequently confirmed in the ClarIDHy phase III trial, where 185 patients with IDH1-mutant CCA were randomised to receive the IDH1 inhibitor ivosidenib or placebo after 1-2 lines of unsuccessful systemic therapy. Ivosidenib significantly improved PFS (the primary endpoint). mPFS was 2.7 months in the experimental group and 1.4 months in the placebo group (HR = 0.37; 95%CI: 0.25-0.54; P < 0.001), with a 12 months PFS rate of 22% vs. 0%[10]. The intention to treat analysis showed a mOS of 10.3 months in patients treated with ivosidenib vs. 7.5 months in the placebo group (HR = 0.79; 95%CI: 0.56-1.12; P = 0.093)[75]. However, the design of the study allowed crossover to ivosidenib in the placebo arm at progression resulting in 35 patients receiving the experimental drug in the control group. A rank preserving structural failure time-adjusted analysis taking into consideration the effects of the crossover showed a more clinically meaningful result with a 4-months increase in median OS (10.3 months vs. 5.1 months; P = 0.0008)[75]. On the basis of this trial, ivosidenib has been recently approved by the Food and Drug Administration (FDA) for adult patients with previously treated, locally advanced or metastatic IDH1-mutated iCCA, which represent around 13% of all CCA[76].
Multiple inhibitors of FGFR isoforms 1-3 have shown activity in phase I/II clinical trials of advanced, pre-treated, CCA with FGFR2 translocations, including several ATP-competitive, reversible inhibitors (erdafitinib, infigratinib, pemigatinib, and derazantinib) and non-ATP competitive, covalent inhibitor (futibatinib)[24]. The pan FGFR inhibitors BGJ398/infigratinib and ARQ087/Derazantinib showed meaningful clinical benefits in chemorefractory iCCA patients carrying FGFR2 fusions in a phase II trial, with tumour stabilisation or regression achieved in the majority of patients. The ORR was 18.8% and 20.7%, respectively, and DCR 83.3% and 82.8%[77,78]. Infigratinib has recently received FDA approval for the treatment of chemoresistant FGFR2 rearranged CCA. The FIGHT-202 study investigated the FGFR1-3 inhibitor pemigatinib in 107 pre-treated CCA patients with FGFR2 fusions, obtaining an impressive 37% ORR, with a median duration of response of 8.1 months, PFS of 7.0 months and mOS of 17.5 months[14,79]. No activity was recorded for pemigatinib in tumours harbouring mutations of FGFR2, making this approach specifically for the 10% of CCA with FGFR2 genomic rearrangements. Despite the exciting result of the FIGHT-202 trial, the enthusiasm is slightly hampered by the single-arm design of the trial, which lacks randomisation. Nonetheless, FDA and the European Medicines Evaluation Agency have approved pemigatinib in the second-line setting of CCA with FGFR2 fusions based on the comparison with historical data. Indeed, mOS for aBTC patients treated with ASC or chemotherapy in second-line is 6 months, as highlighted in the ABC-06 and ClarIDHy trial. Even considering the better prognosis of iCCA patients[8], according to the subgroup analysis of the ABC trials, this is expected to be around 16 months from diagnosis, leaving an expected survival of < 8 months from the starting of second-line treatment providing a mPFS of 8 months for first-line CisGem [Figure 3]. FGFR2 fusions are also known to be associated with a better prognosis with a higher number of patients being diagnosed at an early stage which justifies the difference in OS between patients with and without FGFR2 rearranged CCA in a retrospective multi-institutional American cohort. In this analysis of 377 patients, mOS from diagnosis was 37 months vs.
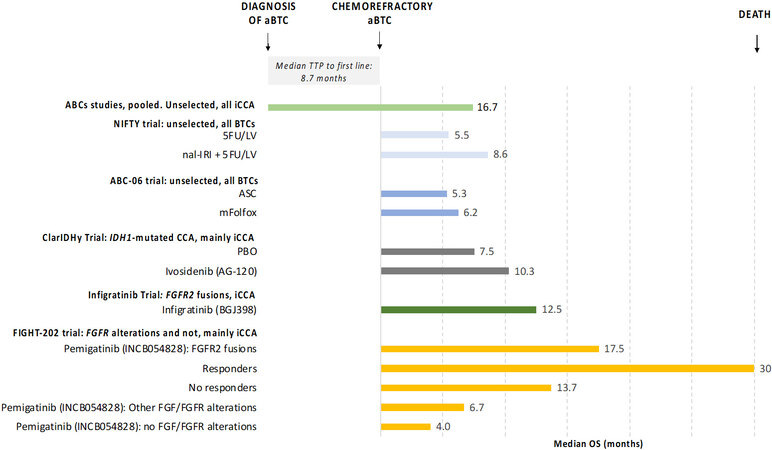
Figure 3. Overall survival of chemorefractory aBTC. Figure 3 shows overall survival (OS) of chemorefractory aBTCs from pooled analysis of ABCs studies and second line clinical trials. ABCs studies include ABC-01, -02 and -03 clinical trials which explored the role of first-line systemic chemotherapy in advanced BTCs [cisplatin and gemcitabine vs. gemcitabine (ABC-01 and ABC-02) and cisplatin-gemcitabine-cediranib vs. cisplatin-gemcitabine-placebo (ABC-03)]. ASC: Active symptom control; PBO: placebo; FGF: fibroblast growth factor; FGFR: fibroblast growth factor receptor; IDH-1: isocitrate dehydrogenase 1; CCA: cholangiocarcinoma; BTCs: biliary tract cancers; aBTCs: advanced biliary tract cancers; 5FU/LV: 5-fluorouracil plus leucovorin; nal-IRI: fluorouracil and liposomial irinotecan; mFOLFOX: 5-fluorouracil plus oxaliplatin.
On the basis of the data described above, FGFR2 inhibitors have now become the standard of care for chemorefractory CCA with FGFR2 genomic rearrangements. As their use increase in clinic, we are called to face other challenges. First, it is a common observation to notice secondary resistance with FGFR2 inhibitors. According to data by Goyal et al.[83], resistance seems to be associated with de-novo FGFR2 mutations. The development of the second generation of FGFR2 inhibitors is ongoing to address this issue. Futibatinib, a highly selective irreversible FGFR1-4 inhibitor, seemed to be effective to overcome resistance, likely due to an effective target engagement due to its covalent binding to the FGFR2 kinase[84]. Futibatinib is currently tested in the phase II FOENIX-CCA2 trial in chemoresistant FGFR2 fused CCA and showed promising preliminary results (ORR = 34%, DCR = 76%, median duration of response 6.2 months)[84]. The next steps would be to increase the specificity of drugs to the FGFR2 isoform to prevent side effects such as hyperphosphatemia and identify biomarkers for patients’ selection. Sub-analysis of the FIGHT-202 showed that co-occurrence of TP53 is associated with lack of response (ORR 0% vs. 45% in TP53-mutated vs. TP53-wild type tumours), raising the question of whether FISH for identification of FGFR2 fusion is too limited for an optimal selection of patients[85].
Currently, results from randomised controlled phase III trials recruiting patients with FGFR2-fusions/rearrangements that compare the standard of care (CisGem) with different FGFR inhibitors in first-line setting are awaited (pemigatinib, NCT03656536; infigratinib, NCT03773302; futibatinib, NCT04093362)[9].
The whole plethora of FGFR-targeted therapies, characterised by different mechanisms of action, offers a unique opportunity for a paradigm shift in the treatment of a subgroup of patients with advanced BTCs, providing the potential for personalised treatment with active compounds and a sequential approach to be investigated in the next future. The challenge of primary and secondary resistance limits the efficacy of this class of drugs[83-85]. Exploring the potential of combining FGFR inhibitors with other treatments able to target signalling shared by wild-type and resistant tumour cells may help prevent the emergence of resistance mutations. This strategy stands on the biological rationale that blocking a driver kinase is more efficacious when downstream pathway components are targeted as well[86,87].
Expanding precision medicine in aBTCs: challenges and opportunities
Despite the opportunity to improve treatment and outcomes in a personalised manner, a major challenge, consisting in the design (and conduction) of adequately powered clinical trials in small patient subgroups, limits a rapid and effective drug development process based on traditional clinical trials design. However, the advent of precision medicine has been followed by novel clinical trial models to identify biomarker-matched subgroups of patients likely to benefit from specific targeted therapies, independently from histology. The overall objective of this innovation is to answer multiple questions at the same time and more efficiently in order to accelerate clinical drug testing and approval. In this light, novel strategies such as platform studies with adaptive designs which consider innovative endpoints may represent a potential opportunity to advance the precision drug development process in rare tumours such as BTCs. Recently, basket trials have been developed to investigate the efficacy of molecular-targeted therapy for similar oncogene-defined subgroups across different tumour types[88].
The activity of the BRAF V600 kinase inhibitor vemurafenib has been explored in a basket trial enrolling 120 patients with refractory BRAF mutated cancers. In the BTCs patient cohort, vemurafenib monotherapy showed 62% of DCR[89].
Another phase II basket trial of the BRAF inhibitor dabrafenib in combination with the MEK inhibitor trametinib in multiple refractory tumour types with BRAFV600E mutation showed ORR in 51% (22/43) of patients with BTCs[90]. Common treatment-related adverse events included pyrexia, rash, and nausea. The most common grade 3 or worse adverse event was increased γ-glutamyltransferase in five (12%) patients. No treatment-related deaths were reported. Further investigation in larger trials is warranted to confirm these early promising results.
A similar experience included 251 patients with 35 different advanced refractory tumour types with alterations in HER-2, EGFR, BRAF, and Hedgehog pathway, who were treated with pertuzumab plus trastuzumab, erlotinib, vemurafenib, or vismodegib, respectively[91]. In the small BTCs cohort (n = 7),
In another basket trial of patients with HER-2 or HER-3 mutations treated with neratinib, 2/9 patients of the BTCs cohort experienced partial response[90].
Basket trials have also demonstrated the clinical activity of tropomyosin receptor kinase (TRK) inhibitors in TRK fusion-positive cancers. Entrectinib and Larotrectinib are potent, highly selective inhibitors of tropomyosin TRK A, B, and C, which has been shown promising results with RR up to 75% in NTRK gene fusion-positive advanced solid tumours, including BTCs, in early-phase trials[90]. The long duration of response (10 months for entrectinib and not reached for larotrectinib), and the good tolerability, led to expedite approval from the FDA in 2018 and 2019 for larotrectinib and entrectinib for patients with histology-agnostic solid tumours harbouring NTRK fusions. Although present in a small percentage of BTCs patients (3%-5%), NTRK fusion constitutes a promising therapeutic target to further investigate in this disease. NTRK inhibitors are currently being explored in clinical studies, which include advanced BTCs (NCT02576431, NCT02568267).
The above-mentioned studies demonstrate that histology-independent, biomarker-driven trials are feasible and represent a potential tool for developing targeted therapies for small - molecularly selected - patient subgroups of refractory cancers, thus constituting a unique opportunity to advance the paradigm shift from unselected to genome-driven oncology. However, major efforts are necessary to expand biomarker-directed clinical trials that are adequately powered for small groups of patients with a wide range of potentially actionable genetic aberrations.
In this context, several studies based on tumour sequencings such as TAPUR (NCT02693535) and MATCH (NCT02465060) trials are investigating the clinical activity of a series of compounds in patients with advanced solid tumours, including BTCs. The rationale is that patients with genetic abnormalities (such as mutations, amplifications, or translocations) may benefit from treatments matching the genetic profile that are either in clinical trials or already approved for the treatment of other cancer types.
Immunotherapy
Immunotherapy based on monoclonal antibodies targeting the immune checkpoint regulators such as cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and programmed death 1 (PD-1) or programmed death ligand 1 (PD-L1), has emerged as a promising therapeutic strategy for several tumour types, including melanoma, non-small cell lung cancer, urothelial carcinoma, renal-cell carcinoma, head and neck cancer, and hepatocellular carcinoma[92]. In contrast, this strategy was associated with modest efficacy in unselected patients with aBTCs[93,94]. The only FDA-approved immunotherapy in BTCs is pembrolizumab, an
The safety and activity of Pembrolizumab in patients with aBTCs have been investigated in the phase 1b multicohort study KEYNOTE-028 (NCT02054806) and the phase 2 multicohort study KEYNOTE-158 (NCT02628067), respectively[96]. PD-L1 positive tumours were required for eligibility in KEYNOTE-028 only. These two studies demonstrated that pembrolizumab monotherapy provides durable antitumor activity in 6% to 13% of patients with advanced BTC, regardless of PD-L1 expression, and has a manageable toxicity profile. In KEYNOTE-028, ORR was 13% (3/23), mOS and mPFS were 5.7 and 1.8 months. In KEYNOTE-158, ORR was 5.8% (6/104), mOS and mPFS were 7.4 and 2 months. ORR was 6.6% (4/61) and 2.9% (1/34) among PD-L1-expressers (n = 61) defined by using a threshold of ≥ 1% of tumours cells or infiltrating lymphocytes expressing PD-L1, and PD-L1-nonexpressers (n = 34), respectively[96]. Whether this improved ORR translates into improved longer overall survival in patients with PD-L1-negative tumours remains unclear.
The overall disappointing results achieved with ICIs monotherapy in BTCs compared to other tumour types can be explained by their biological features. BTCs are characterised by “cold” TME that lacks dendritic cells and effector T cells, essential for an effective immunomodulatory activity[97,98]. A different TME/immune cell infiltrate may also explain the higher sensitivity to ICIs observed in patients with hepatocellular carcinoma[99].
The only subgroup of patients with higher sensitivity to ICIs is that with dMMR/MSI high, as already mentioned. This genomic alteration represents the only potential biomarker that can predict the activity of this class of drugs identified to date. In the BTC cohort of 22 patients enrolled in the KEYNOTE-158 phase II study evaluating pembrolizumab in previously-treated patients with advanced non-colorectal dMMR/MSI high cancer, the ORR was 40.9% (including complete response in 2 patients), mPFS 4.2 months, and mOS 24.3 months[100]. The biological explanation of these results is that cancers with dMMR/MSI high (either due to inherited germline variants or epigenetic inactivation of MMR genes) are associated with a high degree of somatic mutations and have peculiar biological characteristics such as increased expression of neoantigens, abundant tumour-infiltrating lymphocyte (TIL), and high tumour mutational burden (TMB) that stimulate the immune checkpoint axis. Beyond dMMR/MSI-H, TMB is emerging as an additional predictive biomarker for PD-1/PD-L1 immune checkpoint inhibitors. A whole-exome-sequencing study of paired tumour/normal BTCs showed that 6% (14/239) were defined as high TMB using a threshold of > 11.13 Δ/Mb, which corresponded to a median of 641 non-silent mutations in this subset of hypermutated tumours. Hypermutation was associated with concurrent MMR deficiency and/or MSI in about 36% of cases[57].
Given to poor results achieved with immune checkpoint inhibitors monotherapy, combinatorial strategies to enhance the efficacy of this class of drugs in patients with BTCs are currently under clinical investigation. One promising approach is represented by immunotherapy-chemotherapy combination. The anti-PD-1 nivolumab has been associated with CisGem chemotherapy in phase II clinical trials. The association resulted in 37% ORR and mPFS and mOS of 4.2 and 15.4 months[101]. Phase III studies aimed at assessing the clinical efficacy of this therapeutic opportunity are currently ongoing, including the TOPAZ-1 and KEYNOTE-966, in which patients are randomised towards receiving CisGem alone or in combination with the anti-PD-L1 durvalumab or and PD-1 pembrolizumab, respectively. A phase II trial is investigating the activity of the triplet combination of durvalumab, tremelimumab (anti-CTLA-4), and paclitaxel (NCT03704480).
The inhibition of the transforming growth factor β (TGF-β) pathway has shown promising preliminary results. Bintrafusp alfa (M7824) is a first-in-class bifunctional fusion protein composed of the extracellular domain of the transforming growth factor (TGF)βRII receptor (a TGF-β trap) fused to a human IgG1 monoclonal antibody blocking PD-L1. It has been tested in an expansion cohort from a phase 1 study (NCT02699515), including 30 patients with advanced refractory BTCs. The ORR by central assessment was 20%, mPFS was 2.6 months and mOS 12.7 months[99]. However, despite initial enthusiasm, results from recent phase II clinical trials showed disappointing results in first- and second-line settings. Single-agent bintrafusp alfa demonstrated efficacy, durability, and an acceptable toxicity profile when used in patients with locally advanced or metastatic BTC who failed or were intolerant to frontline chemotherapy platinum-based chemotherapy, according to recent data from the phase 2 INTR@PID BTC 047 trial (NCT03833661). After more than 9 months of follow-up, the independent review committee adjudicated an ORR of 10.1% (95%CI: 5.9%-15.8%). Despite the benefit showcased, the trial did not meet the predefined threshold that would have allowed for regulatory filing for the agent’s use in the second-line treatment of BTC[102]. Bintrafusp alfa was also under investigation in combination with CisGem in the frontline treatment of patients with BTC as part of the phase 2/3 INTR@PID BTC 055 trial (NCT04066491). However, due to disappointing preliminary results, the trial has been recently discontinued[103].
Immune checkpoint inhibitors have also been tested in association with anti-angiogenic drugs. However, results in BTCs were not satisfactory, in contrast with high efficacy observed in hepatocellular carcinoma[99]. Pembrolizumab plus ramucirumab, anti-vascular-endothelial growth factor, showed limited efficacy in patients with previously treated aBTC, with a mOS of 6.4 months and mPFS of 1.6 months[104]. Data from the ongoing IMbrave-151 phase II trial are awaited to assess the role of the combination of CisGem chemotherapy with Atezolizumab plus/minus Bevacizumab in the first-line setting (NCT04677504).
In conclusion, the efficacy and safety of immunotherapies for BTC are still unclear. ICIs monotherapy has limited efficacy in unselected patients, while the combinations of ICIs and chemotherapies/targeted therapies seem to achieve better results. However, further clinical trials are necessary to find the optimal combination strategy and identify the predictive biomarkers for improved treatment selection. Table 4 shows a selection of ongoing clinical trials exploring ICIs.
Immunotherapy combinations: ongoing clinical trials
| Trial number | Phase | Pathways targeted | Drug(s) | Line of treatment |
| NCT03478488 | 3 | PDL-1, chemotherapy | KN035/GEMOX | I |
| NCT04003636 | 3 | PD-1, chemotherapy | Pembrolizumab/GEM/CIS | I |
| NCT03875235 | 3 | PDL-1, chemotherapy | Durvalumab or Placebo and GEM/CIS | I |
| NCT04066491 | 2/3 | PD-L1; TGF-β Trap fusion protein, chemotherapy | GEM/CIS with or without Bintrafusp alfa (M7824) | I |
| NCT04677504 | 2 | PD-L1, chemotherapy, VEGF | GEM/CIS/Atezolizumab with or without Bevacizumab | I |
| NCT04027764 | 2 | PD-1, chemotherapy | Toripalimab/S1/Albumin Paclitaxel | I |
| NCT03796429 | 2 | PD-1, chemotherapy | Toripalimab/Gemcitabine-S1 | I |
| NCT04191343 | 2 | PD-1, chemotherapy | Toripalimab/GEMOX | I |
| NCT03486678 | 2 | PD-1, chemotherapy | SHR-1210/GEMOX | I |
| NCT03111732 | 2 | PD-1, chemotherapy | Pembrolizumab/capecitabine/oxaliplatin | II |
| NCT03785873 | 2 | PD-L1, chemotherapy | Nivolumab/NALIRI/5-FU, leucovorin | II |
| NCT03486678 | 2 | PD-1, chemotherapy | SHR-1210/GEMOX | I |
| NCT03046862 | 2 | PDL-1, CTLA-4, chemotherapy | Durvalumab/Tremelimumab/GEM/CIS | I |
| NCT02834013 | 2 | PD-1, CTLA-4 | Nivolumab/Ipilimumab | II |
| NCT03092895 | 2 | PD-1, VEGF, chemotherapy | SHR-1210/Apatinib, GEMOX or FOLFOX | I/II |
| NCT04211168 | 2 | PD-1, VEGF | Toripalimab/Lenvatinib | II |
| NCT03797326 | 2 | PD-1, VEGF | Pembrolizumab/Lenvatinib | II |
| NCT03895970 | 2 | PD-1, VEGF | Pembrolizumab/Lenvatinib | II |
| NCT04057365 | 2 | PD-1, DKK1 | Nivolumab/DKN-01 | II |
| NCT03250273 | 2 | PD-1, Histone deacetylase inhibitor | Nivolumab/Entinostat | II |
| NCT03639935 | 2 | PD-1, PARP | Nivolumab/Rucaparib | II |
| NCT04004234 | 1 /2 | PD-1, chemotherapy | Manganese/anti-PD-1/Nabpaclitaxel-gemcitabine | I/II |
| NCT03937895 | 1/ 2 | PD-1, SMT-NK | Pembrolizumab/Allogeneic NK cell | II |
| NCT03475953 | 1 /2 | PD-1, MAPK | Avelumab/Regorafenib | I/II |
| NCT03475953 | 1/ 2 | PD-1, MAPK | Avelumab/Regorafenib | I/II |
| NCT03849469 | 1 | PD-1, CTLA-4, LAG-3 | XmAb®22841/Pembrolizumab | II |
| NCT03257761 | 1 | PDL-1, DNMT inhibitor | Durvalumab/Guadecitabine (SGI-110) | II |
Targeting the TME
For many tumour types, targeting the TME is an attractive therapeutic strategy in BTCs. High heterogeneity of TME both at the molecular and the cellular level has been demonstrated through multi-omics studies, and different clinically relevant subtypes of BTCs have been suggested based on TME specific characteristics[72,105,106]. This increasing knowledge has led to the identification of possible therapeutic strategies targeting several cell types and/or signalling pathways of the TME. In addition to treatments targeting the immune cell infiltrate (already discussed above), many others addressed towards different components of the TME have been recently proposed. In particular, their association with cytotoxic chemotherapy and tumour cells targeted therapies represents a promising opportunity[107].
BTCs are characterised by significant desmoplastic reactions orchestrated by stromal cells, and different types of immune cells infiltrate. The complex TME ecosystem is populated with a wide variety of cells, including cancer cells, cancer-associated fibroblasts (CAFs), myeloid-derived suppressor cells, tumour-associated macrophages (TAMs), TILs, tumour-associated neutrophils, natural killer cells, and many others[108,109]. TME has a significant role in shaping a chemoresistant phenotype. On the one hand, it constitutes a barrier that obstacles drug delivery; on the other, TME cells interact in dynamic crosstalk with components of the extracellular matrix (ECM), soluble factors - including cytokines, chemokines, growth factors - and biliary tumour cells to modulate chemo-resistance and drive tumour progression[110-112]. From a therapeutic point, the most interesting cells are CAFs and TAMs.
CAFs are a heterogeneous group of cells characterised by the expression of alpha-smooth muscle actin and platelet-derived growth factor receptor beta. They are the key protagonists in the TME of BTCs and have a pivotal role in ECM composition and crosstalk with cancer cells through the secretion of TGF-β, stromal cell‐derived factor 1, hepatocyte growth factor, connective tissue growth factor, epidermal growth factor and platelet-derived growth factor[108]. CAFs are first recruited by CCA tumour cells which subsequently exert positive feedback and amplify their activity. Once activated, CAFs promote CCA progression.
TAMs and exerts a key role in cancer-related inflammation by promoting tumour-cell proliferation, angiogenesis, matrix turnover and suppression of the adaptive immune response[113]. TAMs create a favourable niche by interacting with biliary cancer cells and other stromal cells via releasing multiple protumour factors (including IL-4, IL-6 and IL-10, CCL17, CCL18, and MMP9) that ultimately sustain CCA growth. In addition, TAMs exert immunosuppressive and chemoresistant effects.
Several therapeutic strategies targeting TME and associated cell types showed promising preclinical activity. Navitoclax (Bcl-2 inhibitor) was shown to induce apoptosis in CAFs leading to decreased ECM and stroma deposition and tumour size in vivo using rat xenograft models transplanted with rat CCA cell line BDEneu cells[114]. Similarly, 1D11, a monoclonal antibody targeting TGF-β, demonstrated the ability to inhibit TGF-β, decrease fibrogenesis, ECM deposition, and CCA development in rats[115].
WNT pathway is important for CCA progression and growth, and its activation is maintained by inflammatory macrophages in the tumour stroma in vivo. Mouse and rat CCA models demonstrated that depletion of TAMs or inhibition of WNT signalling with one of two small molecules significantly reduces tumour proliferation and increased apoptosis, resulting in tumour regression[115].
TWEAK/Fn14 pathway is also increasingly expressed in CCA. It not only supports tumour proliferation but also modulates migration and polarisation of TME cells, including macrophages and CAFs. The pharmacological inhibition of TWEAK/Fn14 and of its downstream signals significantly reduced CCA xenograft growth, inflammation and fibrosis, while TWEAK overexpression drove cancer-associated fibroblast proliferation and collagen deposition in the tumour niche in preclinical models[115].
These preliminary studies indicate that targeting TME is a promising therapeutic strategy to further investigate in clinical trials.
Other potential therapeutic targets on the horizon
In addition to those discussed in the previous sections, many other potential targets include DNA damage response (DDR), MET aberrations, PI3K/AKT/mTOR signalling pathway, and chromatin remodelling gene alterations (ARID1, BAP1 and PBRM1).
DDR can be found in up to 60% of BTCs and may offer several therapeutic advantages with PARP inhibitors (PARPi)[116,117]. Inactivation of BRCA1 and BRCA2 genes is the most frequent event associated with DDR[118]. In a retrospective multicentric study, Golan et al.[119] demonstrated that 4/18 CCA patients with BRCA pathogenic variants (somatic or germline) treated with PARPi achieved a favourable response in first or subsequent lines, of whom one experienced sustained disease response with a PFS of 42.6 months. The 44% of patients (8/18) had a previous personal or a family history of BRCA-associated cancer (breast, ovarian, prostate and pancreatic cancer).
Several PARPi are currently being investigated in BTCs patients, including olaparib, rucaparib, and niraparib, as single agents or associated with other strategies [Table 3]. In particular, the combination of PARPi and IDH1 inhibitors or ICIs is based on the preclinical rationale of potential synergistic activity. Mutations in IDH gene are shown to significantly decrease homologous recombination repair activity by inhibiting the αKG-dependent dioxygenases, therefore enhancing the sensitivity to PARPi[120]. Ongoing clinical trials are exploring the efficacy of PARPi in IDH1/2 mutant iCAA in order to assess their synthetic lethality and to target IDH1/2-related dependencies (NCT03212274, NCT03878095).
Treatment with PARPi has also been associated with increased genomic instability, immune pathway activation, and PD-L1 expression on cancer cells, which might promote responsiveness to ICIs[121].
High c-Met expression and MET amplifications are described in up to 16% and 7% of BTCs, respectively[122]. Cabozantinib is a multi-kinase inhibitor of MET, already approved for the treatment of advanced renal cell and thyroid carcinoma[123]. It showed limited activity and significant toxicity in a study of 19 unselected, previously treated, BTCs patients with a mPFS and mOS of 1.8 (95%CI: 1.6-5.4) and 5.2 (95%CI: 2.7-10.5) months, respectively[124]. However, the administration of this agent-based on molecular selection may be more effective.
Mutations in the PI3K/AKT/mTOR signalling pathway have been described in 40% of eCCA and 25% of iCCA (25%)[125,126]. Preclinical studies have shown activity of PI3K/AKT/mTOR inhibition in BTCs, suggesting that this therapeutic strategy may have clinical utility for a subset of BTCs harbouring aberrations in this pathway[125]. However, despite initial enthusiasm from clinical studies with rapamycin analogues, showing interesting activity with an acceptable toxicity profile, novel strategies exploring AKT and PI3K inhibitors have raised serious safety concerns, highlighting the need for improved patient selection and increased target specificity for the clinical development of these agents[126]. Several ongoing trials are currently investigating the combination (or sequential approach) of AKT-mTOR inhibitors or PI3K-mTOR inhibitors plus chemotherapy (NCT02465060, NCT02836847, NCT02631590).
Lastly, numerous chromatin-modifying enzymes are frequently mutated or altered by copy number aberrations in BTCs. Blocking these targets when found dysregulated represent a compelling strategy to explore in BTCs[54,127,128]
CONCLUSION
Overall, BTCs constitute a neglected disease for which novel therapeutic strategies are urgently needed. After years of “one-size fits all” approach, characterised by a series of failures in improving patient outcomes with only marginal progress made thanks to the allocation of first-line chemotherapy, we are now witnessing the dawn of a new era based on molecularly driven decision making. The current standard therapeutic portfolio includes chemotherapy in first-line (CisGem) and second-line for unselected patients (FOLFOX); patients with molecular aberrations (IDH mutations, FGFR fusions, MSI high) can be treated with targeted therapies or pembrolizumab following progression to frontline chemotherapy [Figure 1].
Despite currently being limited to small patient subgroups, “precision medicine” for BTCs is possible - and desirable - given the numerosity of ongoing biomarker-based/enriched clinical trials and potential therapeutic vulnerabilities recently identified, both in the tumour and in the immune-microenvironment. To reach this ambitious goal, a revolution in patient management is much needed. Large-scale efforts to implement tumour’s molecular characterisation at diagnoses with a rapid translation of information to clinical research, in addition to expanding clinical trials (and trial enrolment), should be a health priority. Implementing liquid biopsy studies will be a crucial step to facilitate sample collection for biomarker discovery, treatment monitoring, and disease surveillance in BTCs. In parallel, enhanced preclinical research will be essential to the identification of predictive biomarkers and to the elucidation of mechanisms of primary and acquired resistance. Multi-omics studies and international collaborations are also fundamental to accelerate the precision therapeutic development in this aggressive disease.
DECLARATIONS
Authors’ contributionsConception and design of the study: Braconi C, Casolino R
Drafting the manuscript: Casolino R
Review of the manuscript: Braconi C
Approval of the final version of the manuscript: Casolino R, Braconi C
Availability of data and materialsNot applicable.
Financial support and sponsorshipBraconi C is a recipient of the Lord Kelvin Adam Smith Readership from the University of Glasgow.
Conflicts of interestBraconi C (or spouse) received honoraria from Incyte, Bayer, Eli-Lilly, Pfizer, Merck-Serono.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Valle JW, Borbath I, Khan SA, Huguet F, Gruenberger T, Arnold D. ESMO Guidelines Committee. Biliary cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2016;27:v28-37.
3. Spolverato G, Kim Y, Ejaz A, et al. Conditional probability of long-term survival after liver resection for intrahepatic cholangiocarcinoma: a multi-institutional analysis of 535 patients. JAMA Surg 2015;150:538-45.
4. Hyder O, Hatzaras I, Sotiropoulos GC, et al. Recurrence after operative management of intrahepatic cholangiocarcinoma. Surgery 2013;153:811-8.
5. Spolverato G, Yakoob MY, Kim Y, et al. The impact of surgical margin status on long-term outcome after resection for intrahepatic cholangiocarcinoma. Ann Surg Oncol 2015;22:4020-8.
7. Valle J, Wasan H, Palmer DH, et al. ABC-02 Trial Investigators. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med 2010;362:1273-81.
8. Lamarca A, Palmer DH, Wasan HS, et al. Second-line FOLFOX chemotherapy versus active symptom control for advanced biliary tract cancer (ABC-06): a phase 3, open-label, randomised, controlled trial. Lancet Oncol 2021;22:690-701.
9. Javle MM, Borbath I, Clarke SJ, et al. Infigratinib versus gemcitabine plus cisplatin multicenter, open-label, randomized, phase 3 study in patients with advanced cholangiocarcinoma with FGFR2 gene fusions/translocations: the PROOF trial. J Clin Oncol 2019;37:TPS4155.
10. Abou-alfa GK, Macarulla T, Javle MM, et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): a multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol 2020;21:796-807.
11. Lamarca A, Barriuso J, McNamara MG, Valle JW. Molecular targeted therapies: Ready for “prime time” in biliary tract cancer. J Hepatol 2020;73:170-85.
12. Marin JJG, Prete MG, Lamarca A, et al. working group 6 of the COST-action 18122 (Euro-Cholangio-NET) as part of the European Network for the study of Cholangiocarcinoma (ENSCCA). Current and novel therapeutic opportunities for systemic therapy in biliary cancer. Br J Cancer 2020;123:1047-59.
13. Shroff RT, Javle MM, Xiao L, et al. Gemcitabine, cisplatin, and nab-paclitaxel for the treatment of advanced biliary tract cancers: a phase 2 clinical trial. JAMA Oncol 2019;5:824-30.
14. Yoo C, Kim K, Kim I, et al. Liposomal irinotecan (nal-IRI) in combination with fluorouracil (5-FU) and leucovorin (LV) for patients with metastatic biliary tract cancer (BTC) after progression on gemcitabine plus cisplatin (GemCis): Multicenter comparative randomized phase 2b study (NIFTY). J Clin Oncol 2021;39:4006-4006.
15. Gruenberger B, Schueller J, Heubrandtner U, et al. Cetuximab, gemcitabine, and oxaliplatin in patients with unresectable advanced or metastatic biliary tract cancer: a phase 2 study. Lancet Oncol 2010;11:1142-8.
16. Chen JS, Hsu C, Chiang NJ, et al. Taiwan Cooperative Oncology Group. A KRAS mutation status-stratified randomized phase II trial of gemcitabine and oxaliplatin alone or in combination with cetuximab in advanced biliary tract cancer. Ann Oncol 2015;26:943-9.
17. Peck J, Wei L, Zalupski M, O'Neil B, Villalona Calero M, Bekaii-Saab T. HER2/neu may not be an interesting target in biliary cancers: results of an early phase II study with lapatinib. Oncology 2012;82:175-9.
18. Ahn DH, Li J, Wei L, et al. Results of an abbreviated phase-II study with the akt inhibitor MK-2206 in patients with advanced biliary cancer. Sci Rep 2015;5:12122.
19. Bridgewater J, Lopes A, Beare S, et al. A phase 1b study of Selumetinib in combination with Cisplatin and Gemcitabine in advanced or metastatic biliary tract cancer: the ABC-04 study. BMC Cancer 2016;16:153.
20. Goyal L, Yurgelun MB, Abrams TA, et al. A phase II trial of cabozantinib (XL-184) in patients with advanced cholangiocarcinoma. J Clin Oncol 2015;33:800-800.
21. Lee J, Park SH, Chang H, et al. Gemcitabine and oxaliplatin with or without erlotinib in advanced biliary-tract cancer: a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 2012;13:181-8.
22. Malka D, Cervera P, Foulon S, et al. Gemcitabine and oxaliplatin with or without cetuximab in advanced biliary-tract cancer (BINGO): a randomised, open-label, non-comparative phase 2 trial. Lancet Oncol 2014;15:819-28.
23. Zhu AX, Meyerhardt JA, Blaszkowsky LS, et al. Efficacy and safety of gemcitabine, oxaliplatin, and bevacizumab in advanced biliary-tract cancers and correlation of changes in 18-fluorodeoxyglucose PET with clinical outcome: a phase 2 study. Lancet Oncol 2010;11:48-54.
24. Demols A, Borbath I, Van den Eynde M, et al. Regorafenib after failure of gemcitabine and platinum-based chemotherapy for locally advanced/metastatic biliary tumors: REACHIN, a randomized, double-blind, phase II trial. Ann Oncol 2020;31:1169-77.
25. Valle JW, Wasan H, Johnson P, et al. Gemcitabine alone or in combination with cisplatin in patients with advanced or metastatic cholangiocarcinomas or other biliary tract tumours: a multicentre randomised phase II study - The UK ABC-01 Study. Br J Cancer 2009;101:621-7.
26. Okusaka T, Nakachi K, Fukutomi A, et al. Gemcitabine alone or in combination with cisplatin in patients with biliary tract cancer: a comparative multicentre study in Japan. Br J Cancer 2010;103:469-74.
27. Valle JW, Furuse J, Jitlal M, et al. Cisplatin and gemcitabine for advanced biliary tract cancer: a meta-analysis of two randomised trials. Ann Oncol 2014;25:391-8.
28. Sasaki T, Isayama H, Nakai Y, et al. Multicenter, phase II study of gemcitabine and S-1 combination chemotherapy in patients with advanced biliary tract cancer. Cancer Chemother Pharmacol 2010;65:1101-7.
29. Schinzari G, Rossi E, Mambella G, et al. First-line treatment of advanced biliary ducts carcinoma: a randomized phase II study evaluating 5-FU/LV plus oxaliplatin (Folfox 4) versus 5-FU/LV (de Gramont Regimen). Anticancer Res 2017;37:5193-7.
30. Morizane C, Okusaka T, Mizusawa J, et al. members of the Hepatobiliary and Pancreatic Oncology Group of the Japan Clinical Oncology Group (JCOG-HBPOG). Combination gemcitabine plus S-1 versus gemcitabine plus cisplatin for advanced/recurrent biliary tract cancer: the FUGA-BT (JCOG1113) randomized phase III clinical trial. Ann Oncol 2019;30:1950-8.
31. Sakai D, Kanai M, Kobayashi S, et al. Randomized phase III study of gemcitabine, cisplatin plus S-1 (GCS) versus gemcitabine, cisplatin (GC) for advanced biliary tract cancer (KHBO1401-MITSUBA). Ann Oncol 2018;29:viii205.
32. Tella SH, Kommalapati A, Borad MJ, Mahipal A. Second-line therapies in advanced biliary tract cancers. Lancet Oncol 2020;21:e29-41.
33. Lamarca A, Hubner RA, David Ryder W, Valle JW. Second-line chemotherapy in advanced biliary cancer: a systematic review. Ann Oncol 2014;25:2328-38.
34. Belkouz A, de Vos-Geelen J, Mathôt RAA, et al. Efficacy and safety of FOLFIRINOX as salvage treatment in advanced biliary tract cancer: an open-label, single arm, phase 2 trial. Br J Cancer 2020;122:634-9.
35. Bridgewater J, Palmer D, Cunningham D, et al. Outcome of second-line chemotherapy for biliary tract cancer. Eur J Cancer 2013;49:1511.
36. Leone F, Filippi R, Palloni A, et al. Prognostic factors in unresectable biliary tract cancer: a GICO (Gruppo Italiano COlangiocarcinoma) retrospective analysis. Ann Oncol 2017;28:vi48.
37. Brieau B, Dahan L, De Rycke Y, et al. Second-line chemotherapy for advanced biliary tract cancer after failure of the gemcitabine-platinum combination: a large multicenter study by the Association des Gastro-Entérologues Oncologues. Cancer 2015;121:3290-7.
38. Schweitzer N, Kirstein MM, Kratzel AM, et al. Second-line chemotherapy in biliary tract cancer: outcome and prognostic factors. Liver Int 2019;39:914-23.
39. Takahara N, Nakai Y, Isayama H, et al. Second-line chemotherapy in patients with advanced or recurrent biliary tract cancer: a single center, retrospective analysis of 294 cases. Invest New Drugs 2018;36:1093-102.
40. Lamarca A, Ross P, Wasan HS, et al. Advanced intrahepatic cholangiocarcinoma: post hoc analysis of the ABC-01, -02, and -03 clinical trials. J Natl Cancer Inst 2020;112:200-10.
41. Salati M, Filippi R, Vivaldi C, et al. The prognostic nutritional index predicts survival and response to first-line chemotherapy in advanced biliary cancer. Liver Int 2020;40:704-11.
42. Filippi R, Leone F, Fornaro L, et al. Clinical insights and prognostic factors from an advanced biliary tract cancer case series: a real-world analysis. J Chemother 2021:1-10.
43. Filippi R, Montagnani F, Lombardi P, et al. A prognostic model in patients with advanced biliary tract cancer receiving first-line chemotherapy. Acta Oncol 2021;60:1317-24.
44. Casadei-Gardini A, Filippi R, Rimini M, et al. Effects of metformin and vitamin D on clinical outcome in cholangiocarcinoma patients. Oncology 2021;99:292-9.
45. Rovesti G, Leone F, Brandi G, et al. Prognostic role of a new index tested in European and Korean advanced biliary tract cancer patients: the PECS index. J Gastrointest Cancer 2021; doi: 10.1007/s12029-021-00596-z.
46. Jusakul A, Cutcutache I, Yong CH, et al. Whole-genome and epigenomic landscapes of etiologically distinct subtypes of cholangiocarcinoma. Cancer Discov 2017;7:1116-35.
47. Lowery MA, Ptashkin R, Jordan E, et al. Comprehensive molecular profiling of intrahepatic and extrahepatic cholangiocarcinomas: potential targets for intervention. Clin Cancer Res 2018;24:4154-61.
48. Chan-On W, Nairismägi ML, Ong CK, et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat Genet 2013;45:1474-8.
49. Welcome to the Pan-Cancer Atlas. Available from: https://www.cell.com/pb-assets/consortium/pancanceratlas/pancani3/index.html [Last accessed on 16 Nov 2021].
50. Ong CK, Subimerb C, Pairojkul C, et al. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nat Genet 2012;44:690-3.
51. Li M, Zhang Z, Li X, et al. Whole-exome and targeted gene sequencing of gallbladder carcinoma identifies recurrent mutations in the ErbB pathway. Nat Genet 2014;46:872-6.
52. Narayan RR, Creasy JM, Goldman DA, et al. Regional differences in gallbladder cancer pathogenesis: Insights from a multi-institutional comparison of tumor mutations. Cancer 2019;125:575-85.
53. Zou S, Li J, Zhou H, et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat Commun 2014;5:5696.
54. Jiao Y, Pawlik TM, Anders RA, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat Genet 2013;45:1470-3.
55. Bogenberger JM, DeLeon TT, Arora M, Ahn DH, Borad MJ. Emerging role of precision medicine in biliary tract cancers. NPJ Precis Oncol 2018;2:21.
56. Wardell CP, Fujita M, Yamada T, et al. Genomic characterization of biliary tract cancers identifies driver genes and predisposing mutations. J Hepatol 2018;68:959-69.
57. Nakamura H, Arai Y, Totoki Y, et al. Genomic spectra of biliary tract cancer. Nat Genet 2015;47:1003-10.
58. Chaisaingmongkol J, Budhu A, Dang H, et al. TIGER-LC Consortium. Common molecular subtypes among Asian hepatocellular carcinoma and cholangiocarcinoma. Cancer Cell 2017;32:57-70.e3.
59. cBioPortal for Cancer Genomics. Available from: https://www.cbioportal.org/study/summary?id=chol_nccs_2013 [Last accessed on 16 Nov 2021].
60. Arai Y, Totoki Y, Hosoda F, et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014;59:1427-34.
61. Graham RP, Barr Fritcher EG, Pestova E, et al. Fibroblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma. Hum Pathol 2014;45:1630-8.
62. Ross JS, Wang K, Gay L, et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist 2014;19:235-42.
63. Sia D, Losic B, Moeini A, et al. Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat Commun 2015;6:6087.
64. Voss JS, Holtegaard LM, Kerr SE, et al. Molecular profiling of cholangiocarcinoma shows potential for targeted therapy treatment decisions. Hum Pathol 2013;44:1216-22.
65. Wu YM, Su F, Kalyana-Sundaram S, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov 2013;3:636-47.
66. Borad MJ, Champion MD, Egan JB, et al. Integrated genomic characterization reveals novel, therapeutically relevant drug targets in FGFR and EGFR pathways in sporadic intrahepatic cholangiocarcinoma. PLoS Genet 2014;10:e1004135.
67. Putra J, de Abreu FB, Peterson JD, et al. Molecular profiling of intrahepatic and extrahepatic cholangiocarcinoma using next generation sequencing. Exp Mol Pathol 2015;99:240-4.
68. Zhu AX, Borger DR, Kim Y, et al. Genomic profiling of intrahepatic cholangiocarcinoma: refining prognosis and identifying therapeutic targets. Ann Surg Oncol 2014;21:3827-34.
69. Sia D, Hoshida Y, Villanueva A, et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 2013;144:829-40.
70. Farshidfar F, Zheng S, Gingras MC, et al. Cancer Genome Atlas Network. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep 2017;18:2780-94.
71. Nepal C, O'Rourke CJ, Oliveira DVNP, et al. Genomic perturbations reveal distinct regulatory networks in intrahepatic cholangiocarcinoma. Hepatology 2018;68:949-63.
72. Job S, Rapoud D, Dos Santos A, et al. Identification of four immune subtypes characterized by distinct composition and functions of tumor microenvironment in intrahepatic cholangiocarcinoma. Hepatology 2020;72:965-81.
73. Verlingue L, Malka D, Allorant A, et al. Precision medicine for patients with advanced biliary tract cancers: An effective strategy within the prospective MOSCATO-01 trial. Eur J Cancer 2017;87:122-30.
74. Lowery MA, Abou-alfa GK, Burris HA, et al. Phase I study of AG-120, an IDH1 mutant enzyme inhibitor: results from the cholangiocarcinoma dose escalation and expansion cohorts. J Clin Oncol 2017;35:4015-4015.
75. Zhu AX, Macarulla T, Javle MM, et al. Final overall survival efficacy results of ivosidenib for patients with advanced cholangiocarcinoma with IDH1 mutation: the phase 3 randomized clinical ClarIDHy trial. JAMA Oncol 2021; doi: 10.1001/jamaoncol.2021.3836.
76. FDA approves ivosidenib for advanced or metastatic cholangiocarcinoma Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-ivosidenib-advanced-or-metastatic-cholangiocarcinoma [Last accessed on 16 Nov 2021].
77. Javle M, Lowery M, Shroff RT, et al. Phase II Study of BGJ398 in Patients With FGFR-Altered Advanced Cholangiocarcinoma. J Clin Oncol 2018;36:276-82.
78. Mazzaferro V, El-Rayes BF, Droz Dit Busset M, et al. Derazantinib (ARQ 087) in advanced or inoperable FGFR2 gene fusion-positive intrahepatic cholangiocarcinoma. Br J Cancer 2019;120:165-71.
79. Vogel A, Sahai V, Hollebecque A, et al. FIGHT-202: a phase II study of pemigatinib in patients (pts) with previously treated locally advanced or metastatic cholangiocarcinoma (CCA). Ann Oncol 2019;30:v876.
80. Jain A, Borad MJ, Kelley RK, et al. Cholangiocarcinoma with FGFR genetic aberrations: a unique clinical phenotype. JCO Precision Oncology 2018; doi: 10.1200/po.17.00080.
81. Javle MM, Sadeghi S, El-khoueiry AB, et al. A retrospective analysis of post second-line chemotherapy treatment outcomes for patients with advanced or metastatic cholangiocarcinoma and FGFR2 fusions. J Clin Oncol 2020;38:4591.
82. Abou-alfa GK, Sahai V, Hollebecque A, et al. Pemigatinib for previously treated locally advanced/metastatic cholangiocarcinoma (CCA): update of FIGHT-202. J Clin Oncol 2021;39:4086.
83. Goyal L, Saha SK, Liu LY, et al. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov 2017;7:252-63.
84. Goyal L, Shi L, Liu LY, et al. TAS-120 overcomes resistance to ATP-competitive FGFR inhibitors in patients with FGFR2 fusion-positive intrahepatic cholangiocarcinoma. Cancer Discov 2019;9:1064-79.
85. Silverman IM, Murugesan K, Lihou CF, et al. Comprehensive genomic profiling in FIGHT-202 reveals the landscape of actionable alterations in advanced cholangiocarcinoma. J Clin Oncol 2019;37:4080.
86. Cocco E, Schram AM, Kulick A, et al. Resistance to TRK inhibition mediated by convergent MAPK pathway activation. Nat Med 2019;25:1422-7.
87. Misale S, Bozic I, Tong J, et al. Vertical suppression of the EGFR pathway prevents onset of resistance in colorectal cancers. Nat Commun 2015;6:8305.
88. Park JJH, Hsu G, Siden EG, Thorlund K, Mills EJ. An overview of precision oncology basket and umbrella trials for clinicians. CA Cancer J Clin 2020;70:125-37.
89. Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373:726-36.
90. Subbiah V, Lassen U, Élez E, et al. Dabrafenib plus trametinib in patients with BRAFV600E-mutated biliary tract cancer (ROAR): a phase 2, open-label, single-arm, multicentre basket trial. Lancet Oncol 2020;21:1234-43.
91. Hainsworth JD, Meric-Bernstam F, Swanton C, et al. Targeted therapy for advanced solid tumors on the basis of molecular profiles: results from MyPathway, an open-label, phase IIa multiple basket study. J Clin Oncol 2018;36:536-42.
93. Kim RD, Chung V, Alese OB, et al. A phase 2 multi-institutional study of nivolumab for patients with advanced refractory biliary tract cancer. JAMA Oncol 2020;6:888-94.
94. Shen T, Zheng S, Geng L, et al. Experience with anti-PD-1 antibody, camrelizumab, monotherapy for biliary tract cancer patients and literature review. Technol Cancer Res Treat 2020;19:1533033820979703.
95. Marcus L, Lemery SJ, Keegan P, Pazdur R. FDA approval summary: pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clin Cancer Res 2019;25:3753-8.
96. Piha-Paul SA, Oh DY, Ueno M, et al. Efficacy and safety of pembrolizumab for the treatment of advanced biliary cancer: results from the KEYNOTE-158 and KEYNOTE-028 studies. Int J Cancer 2020;147:2190-8.
97. Gani F, Nagarajan N, Kim Y, et al. Program death 1 immune checkpoint and tumor microenvironment: implications for patients with intrahepatic cholangiocarcinoma. Ann Surg Oncol 2016;23:2610-7.
98. Kitano Y, Yamashita YI, Nakao Y, et al. Clinical significance of PD-L1 expression in both cancer and stroma cells of cholangiocarcinoma patients. Ann Surg Oncol 2020;27:599-607.
99. Pinato DJ, Guerra N, Fessas P, et al. Immune-based therapies for hepatocellular carcinoma. Oncogene 2020;39:3620-37.
100. Marabelle A, Le DT, Ascierto PA, et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: results from the phase II KEYNOTE-158 study. J Clin Oncol 2020;38:1-10.
101. Ueno M, Ikeda M, Morizane C, et al. Nivolumab alone or in combination with cisplatin plus gemcitabine in Japanese patients with unresectable or recurrent biliary tract cancer: a non-randomised, multicentre, open-label, phase 1 study. Lancet Gastroenterol Hepatol 2019;4:611-21.
102. Merck reports topline data for Bintrafusp alfa as second-line monotherapy treatment in biliary tract cancer. Available from: https://www.merckgroup.com/en/news/bintrafusp-topline-data-biliary-tract-cancer-16-03-2021.html [Last accessed on 16 Nov 2021].
103. Merck statement on phase II study of Bintrafusp alfa in first-line treatment of biliary tract cancer. Available from: https://www.merckgroup.com/en/news/bintrafusp-alfa-update-23-08-2021.html [Last accessed on 16 Nov 2021].
104. Arkenau HT, Martin-Liberal J, Calvo E, et al. Ramucirumab plus pembrolizumab in patients with previously treated advanced or metastatic biliary tract cancer: nonrandomized, open-label, phase I trial (JVDF). Oncologist 2018;23:1407-e136.
105. Sulpice L, Rayar M, Desille M, et al. Molecular profiling of stroma identifies osteopontin as an independent predictor of poor prognosis in intrahepatic cholangiocarcinoma. Hepatology 2013;58:1992-2000.
106. Sulpice L, Desille M, Turlin B, et al. Gene expression profiling of the tumor microenvironment in human intrahepatic cholangiocarcinoma. Genom Data 2016;7:229-32.
107. Louis C, Edeline J, Coulouarn C. Targeting the tumor microenvironment in cholangiocarcinoma: implications for therapy. Expert Opin Ther Targets 2021;25:153-62.
108. Brivio S, Cadamuro M, Strazzabosco M, Fabris L. Tumor reactive stroma in cholangiocarcinoma: The fuel behind cancer aggressiveness. World J Hepatol 2017;9:455-68.
109. Martín-Sierra C, Martins R, Laranjeira P, et al. Functional and phenotypic characterization of tumor-infiltrating leukocyte subsets and their contribution to the pathogenesis of hepatocellular carcinoma and cholangiocarcinoma. Transl Oncol 2019;12:1468-79.
110. Sirica AE, Gores GJ. Desmoplastic stroma and cholangiocarcinoma: clinical implications and therapeutic targeting. Hepatology 2014;59:2397-402.
111. Zhou G, Sprengers D, Mancham S, et al. Reduction of immunosuppressive tumor microenvironment in cholangiocarcinoma by ex vivo targeting immune checkpoint molecules. J Hepatol 2019;71:753-62.
112. Chen Z, Guo P, Xie X, Yu H, Wang Y, Chen G. The role of tumour microenvironment: a new vision for cholangiocarcinoma. J Cell Mol Med 2019;23:59-69.
113. Raggi C, Correnti M, Sica A, et al. Cholangiocarcinoma stem-like subset shapes tumor-initiating niche by educating associated macrophages. J Hepatol 2017;66:102-15.
114. Mertens JC, Fingas CD, Christensen JD, et al. Therapeutic effects of deleting cancer-associated fibroblasts in cholangiocarcinoma. Cancer Res 2013;73:897-907.
115. Ling H, Roux E, Hempel D, et al. Transforming growth factor β neutralization ameliorates pre-existing hepatic fibrosis and reduces cholangiocarcinoma in thioacetamide-treated rats. PLoS One 2013;8:e54499.
116. Mehta R, Wood AC, Yu J, Kim R. Investigational PARP inhibitors for the treatment of biliary tract cancer: spotlight on preclinical and clinical studies. Expert Opin Investig Drugs 2021;30:451-61.
117. Ahn DH, Bekaii-Saab T. Biliary tract cancer and genomic alterations in homologous recombinant deficiency: exploiting synthetic lethality with PARP inhibitors. Chin Clin Oncol 2020;9:6.
119. Golan T, Raitses-Gurevich M, Kelley RK, et al. Overall survival and clinical characteristics of BRCA-associated cholangiocarcinoma: a multicenter retrospective study. Oncologist 2017;22:804-10.
120. Sulkowski PL, Corso CD, Robinson ND, et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med 2017;9:eaal2463.
121. Peyraud F, Italiano A. Combined PARP inhibition and immune checkpoint therapy in solid tumors. Cancers (Basel) 2020;12:1502.
122. Miyamoto M, Ojima H, Iwasaki M, et al. Prognostic significance of overexpression of c-Met oncoprotein in cholangiocarcinoma. Br J Cancer 2011;105:131-8.
123. Lyseng-Williamson KA. Cabozantinib as first-line treatment in advanced renal cell carcinoma: a profile of its use. Drugs Ther Perspect 2018;34:457-65.
124. Goyal L, Zheng H, Yurgelun MB, et al. A phase 2 and biomarker study of cabozantinib in patients with advanced cholangiocarcinoma. Cancer 2017;123:1979-88.
125. Zuo M, Rashid A, Churi C, et al. Novel therapeutic strategy targeting the Hedgehog signalling and mTOR pathways in biliary tract cancer. Br J Cancer 2015;112:1042-51.
126. Corti F, Nichetti F, Raimondi A, et al. Targeting the PI3K/AKT/mTOR pathway in biliary tract cancers: a review of current evidences and future perspectives. Cancer Treat Rev 2019;72:45-55.
127. Goeppert B, Konermann C, Schmidt CR, et al. Global alterations of DNA methylation in cholangiocarcinoma target the Wnt signaling pathway. Hepatology 2014;59:544-54.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Casolino R, Braconi C. Treatment of advanced biliary tract cancers: from chemotherapy to targeted agents. Hepatoma Res 2021;7:76. http://dx.doi.org/10.20517/2394-5079.2021.79
AMA Style
Casolino R, Braconi C. Treatment of advanced biliary tract cancers: from chemotherapy to targeted agents. Hepatoma Research. 2021; 7: 76. http://dx.doi.org/10.20517/2394-5079.2021.79
Chicago/Turabian Style
Casolino, Raffaella, Chiara Braconi. 2021. "Treatment of advanced biliary tract cancers: from chemotherapy to targeted agents" Hepatoma Research. 7: 76. http://dx.doi.org/10.20517/2394-5079.2021.79
ACS Style
Casolino, R.; Braconi C. Treatment of advanced biliary tract cancers: from chemotherapy to targeted agents. Hepatoma. Res. 2021, 7, 76. http://dx.doi.org/10.20517/2394-5079.2021.79
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 11 clicks
Cite This Article 11 clicks

 Like This Article 1
likes
Like This Article 1
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.