
Pathology and molecular pathology of cholangiocarcinoma

Abstract
Biliary tract cancers are a wide group of heterogeneous neoplasms of the biliary tree, composed of intrahepatic cholangiocarcinoma perihilar bile duct cancer and distal bile duct cancer, according to location. The variability in location reflects the different morphologies and molecular alterations. In particular, intrahepatic peripheral mass forming cholangiocarcinoma is represented by the “small duct type” cholangiocarcinoma, which is different from the “large duct type” cholangiocarcinoma that, although intrahepatic, behaves similar to extrahepatic bile duct cancers, perihilar and distal ones. Recently, molecular targetable alterations, mainly FGFR2 fusions and IDH1 mutations, have been described, mostly in the intrahepatic “small duct type” subgroup and have opened the way, together with rarer targetable alterations, for personalisation of therapy also in these aggressive neoplasms.
Keywords
BACKGROUND
Biliary tract cancer is a heterogeneous and very aggressive group of neoplasms that includes intrahepatic cholangiocarcinoma and extrahepatic bile duct cancer. It arises in different clinical contexts and often in non-cirrhotic livers, without clear aetiology. It can be found on surgical specimens associated with preneoplastic lesions that at least partially resemble the pancreatic counterpart. Recently, the morphological classification of this neoplasm has been improved and finds confirmation in the molecular profile. In fact, important targetable molecular alterations have recently been described, regarding around 15%-20% of the intrahepatic cholangiocarcinoma, whilst the extrahepatic bile duct cancer is rarely a suitable subject for targeted therapy. These aberrations are FGFR2 fusions, IDH1/2 mutations, more rarely BRAF V600E and BRCA1/2 alterations, neurotrophic receptor tyrosine kinase (NTRK) fusions and microsatellite instability (MSI). These represent an important advancement in the understanding of the disease, although further investigation is mandatory for our patients.
HISTOPATHOLOGY OF BILIARY TRACT CANCER
Cholangiocarcinoma is a malignant epithelial neoplasm, generally an adenocarcinoma, arising from the biliary tree and includes a wide range of malignant tumours, commonly in the context of a non-cirrhotic liver. The term “cholangiocarcinoma” should be used for intrahepatic malignancies (iCCA), whilst the extrahepatic ones should be called “of the extrahepatic bile ducts” (eCCA) (sec. WHO 2019[1]). This classification is supposed to follow the probable cell of origin of these neoplasms and agree with the TNM system. In fact, the TNM staging system subdivides iCCA as located proximal to the second order bile ducts, from the perihilar eCCA, developing in the right and left hepatic duct or at the confluence between the two, and the distal eCCA that involves the common bile duct. The eCCA are divided from the insertion of the cystic duct. iCCA can arise from the bile ductules until the second-order bile ducts or segmental bile ducts, and the WHO 2019 subdivides between “small duct type” and “large duct type” iCCA[1]. The “small duct type” is generally peripheral, mass forming, typically made of small tubules and ductules, not producing mucus [Figure 1] and can arise in the context of cirrhosis[2,3]. The “large duct type” more commonly arises in the intrahepatic perihilar region and generally presents as a highly desmoplastic lesion with poorly defined margins, typically with periductal infiltrating pattern of growth. It generally shows all the typical aspects of the extrahepatic bile duct adenocarcinoma in terms of aggressiveness, with large vessels and nerves infiltration [Figure 2].
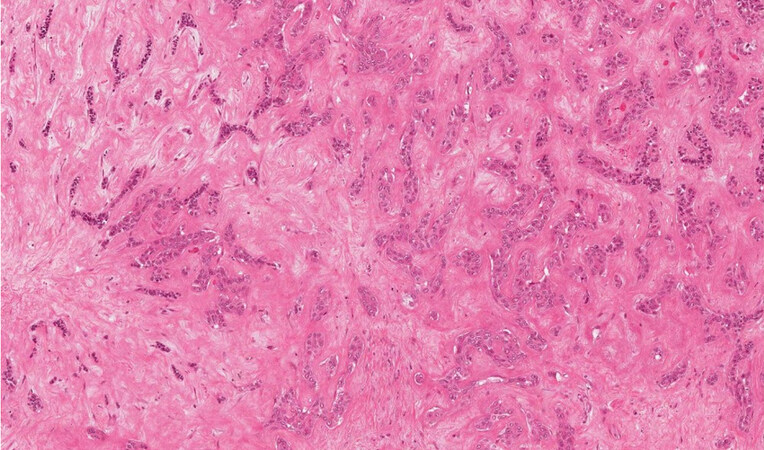
Figure 1. iCCA small duct type with mass forming type pattern of growth (haematoxylin-eosin staining, 20×).
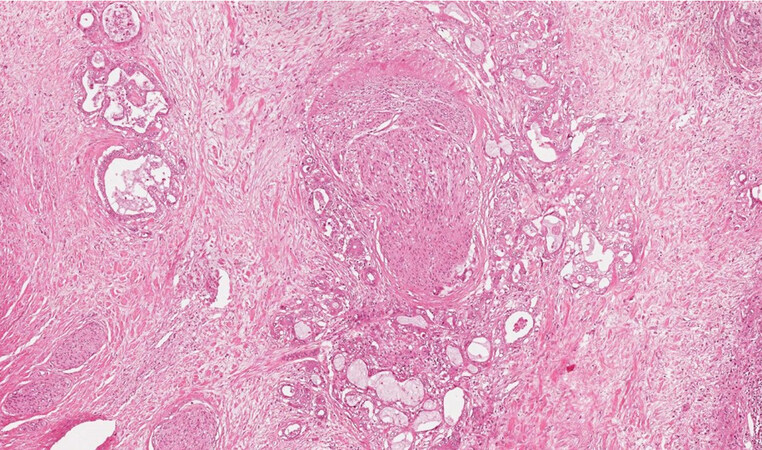
Figure 2. iCCA large duct type with extensive perineural infiltration (haematoxylin-eosin staining, 20×).
The “small duct type” iCCA can arise in the context of a ductal plate malformation or a biliary adenofibroma, as occasionally reported in the literature[4,5]. Biliary adenofibroma is a recently introduced category in the WHO 2019[1], representing a mass forming lesion made of glands and stroma, with cystic dilatation, covered by biliary cuboidal epithelium without significant atypia not communicating with the biliary tree[6], and in fact after extensive sampling of the tumour it is possible to appreciate aspects resembling an underlying biliary adenofibroma [Figure 3A and B]. The “large duct type” iCCA is usually associated with preinvasive neoplasms such as biliary intraepithelial neoplasms (BilIN) or intraductal papillary neoplasms of the bile ducts (IPNB)[1]. BilIN presents with flat or micropapillary proliferation of the biliary epithelium not radiologically visible, now subdivided between low grade and high grade depending on architecture and cytological alterations[1] [Figure 4]. IPNB is a grossly visible lesion, radiologically detectable and generally associated with dilatation of the bile ducts. Both the preinvasive lesions of the biliary tree have their pancreatic counterpart in the pancreatic intraepithelial neoplasms and intraductal mucinous papillary neoplasms (IPMN), although with some morphological and phenotypical differences[7], IPNB protrudes as a polyp in the lumen of a dilated bile duct and can also develop in the context of the peribiliary glands[8].
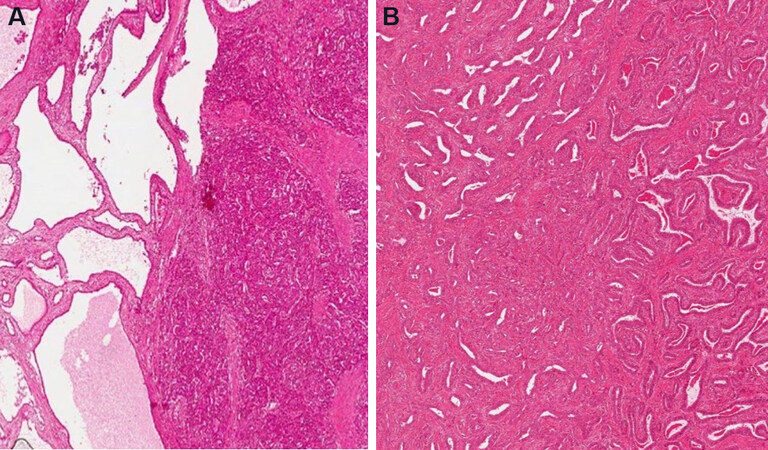
Figure 3. (A, B) iCCA with aspects of biliary adenofibroma (haematoxylin-eosin staining, 20×).
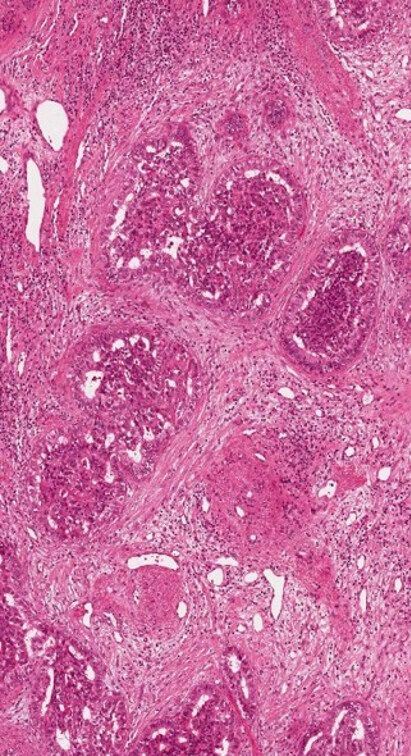
Figure 4. BilIN (haematoxylin-eosin staining, 20×).
The IPNB are subdivided between types I and II. Type I IPNB mainly arises in the intrahepatic bile ducts, phenotypically gastric or intestinal [Figure 5A] and less aggressive than the type II IPNB, which is more typical of the extrahepatic bile ducts, has an irregular architecture and shows intestinal or pancreaticobiliary phenotype[1] [Figure 5B].
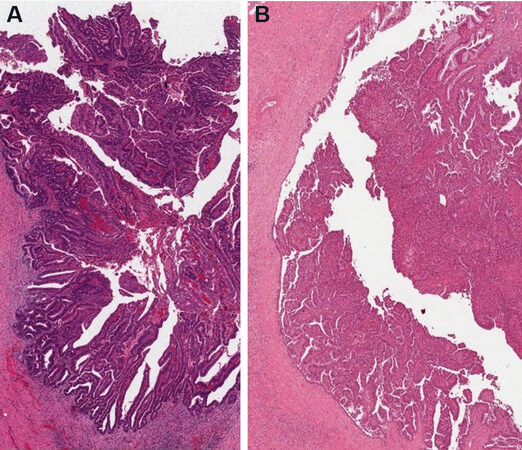
Figure 5. Intestinal type (A) and pancreatico-biliary type (B) IPNB (haematoxylin-eosin staining, 20×).
Another recently described entity, with a well-established counterpart in the pancreas and radiologically identifiable, is the intraductal tubulo-papillary neoplasm of the bile ducts, which has a more compact architecture and is mainly made of small tubules, with small or no mucin production[9] [Figure 6].
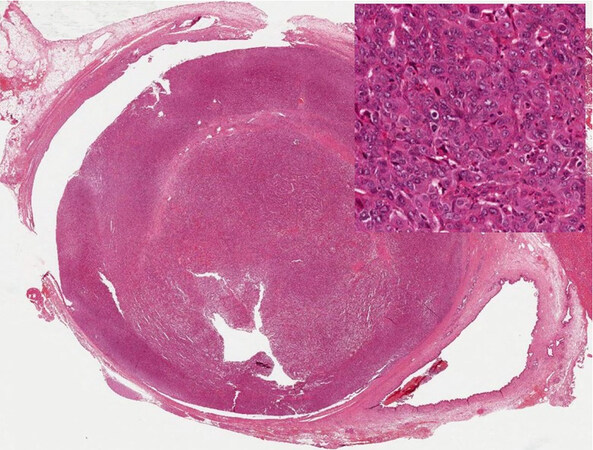
Figure 6. Intraductal tubulopapillary neoplasm of the bile duct (with high-power view) (haematoxylin-eosin staining, 4×).
The mucinous cystic neoplasm of the liver is a neoplastic lesion characterised in general by a mucinous epithelium with underlying ovarian-type stroma [Figure 7]. This lesion is considered a possible precursor of iCCA, because the cuboidal or mucinous columnar epithelium covering the cyst can become dysplastic and then undergo malignant degeneration. The amount of mucous produced by this lesion is very variable[10], sometimes making the diagnosis difficult especially if the cyst undergoes fenestration. Moreover, the ovarian-type stroma can also be patchy and loose[10], sometimes difficult to be recognised in intraoperative consultation. It is mandatory to resect the whole cyst once the diagnosis is performed because of the neoplastic nature of the lesion.
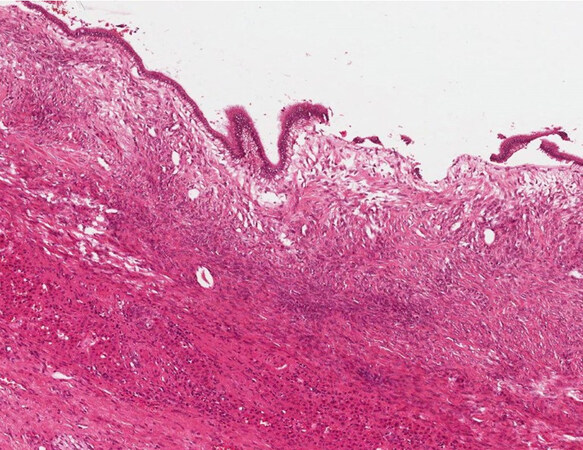
Figure 7. Mucinous cystic neoplasm of the liver (haematoxylin-eosin staining, 40×) without dysplasia of the epithelium.
In cystoadenocarcinoma [Figure 8], it is often difficult to find the ovarian type stroma, and it is not clear if they represent a mucinous cystic neoplasm that has undergone regression of the stroma or a different entity. The suggested term in the WHO 2019[1] is mucinous cystic neoplasm with associated invasive carcinoma, and it is important to recognise it because its behaviour depends on the complete or incomplete resection of the cyst and the amount of parenchymal invasion.
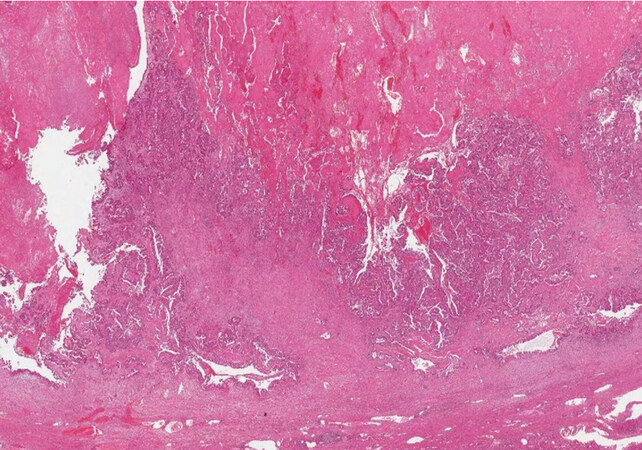
Figure 8. Cystoadenocarcinoma of the liver (haematoxylin-eosin staining, 20×).
The combined hepatocellular-cholangiocarcinoma (cHCC-CCA) [Figure 9] is now a category itself inside WHO 2019[1] with some simplification compared with the previous WHO 2010[11]. This entity is very heterogeneous and rare, thus controlled studies to understand its biology and potential treatment are needed. The diagnosis should rely on morphology first, and immunohistochemistry should be used for confirmation. Although the real risk factors for cHCC-CCA are largely unknown, they are more commonly found in cirrhotic liver compared to iCCA, reported to be around 40% in a large series[12]. Some reports found that it behaves more as an HCC[13], but actually their treatment is still without defined guidelines[14]. The so-called “cholangiolocarcinoma” can be a part of cHCC-CCA, but it is more commonly found of part of iCCA and is now considered a subtype of iCCA [Figure 10][1]. Differently, the presence of distinct HCC and iCCA in the same liver has been reported in fewer than 40 cases in the literature[15], mainly in cirrhotic liver explants.
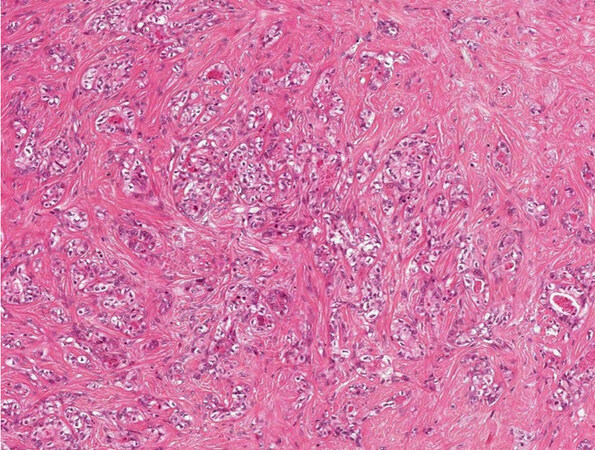
Figure 9. Combined hepatocellular-cholangiocarcinoma (haematoxylin-eosin staining, 20×).
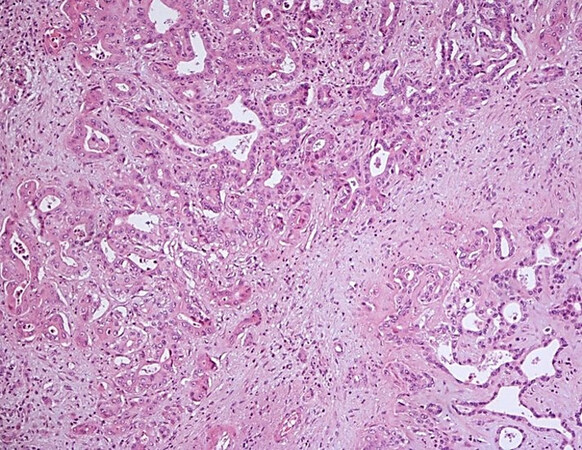
Figure 10. Cholangiolocarcinoma together with small duct type iCCA (haematoxylin-eosin staining, 20×).
Carcinosarcoma and the undifferentiated carcinoma of the biliary tree do exist, although they are very rare, the first showing both components [Figure 11], the sarcoma differentiation lacking the epithelial differentiation and the second showing only the epithelial differentiation[1].
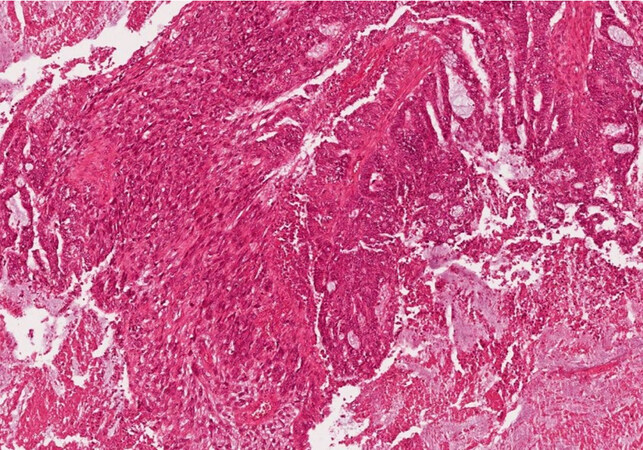
Figure 11. Carcinosarcoma of the liver (haematoxylin-eosin staining, 20×).
IMMUNOHISTOCHEMISTRY
Although the distinction between iCCA and HCC based only on morphology can be difficult in some cases, especially in poorly differentiated ones, some immunohistochemical markers are helpful for suggesting hepatocellular differentiation such as HepPar‐1 (hepatocyte in paraffin 1) or hepatocyte specific antigen, arginase‐1, alpha‐fetoprotein, bile salt export pump and glutamine synthetase. CD10 and pCEA have to show canalicular pattern of expression. There are no markers specific for biliary differentiation until now, but CK7 and CK19, although not specific, are usually variably expressed by the biliary tumours. Be careful with CK19 in poorly differentiated tumours because different subtype can show expression[16]. Concerning the usefulness of immunohistochemistry in the diagnosis of cHCC-iCCA, some markers were suggested in the previous WHO 2010[11] such as CD56n and KIT in the case of the so-called stem cell subtype (this term is no longer recommended by the current WHO[1]), both of them mainly staining the intermediate cell subtype[17]. Both CD56n and KIT are reported to be expressed by canals of Hering and hepatic progenitors[18].
Mucins, in particular MUC1, MUC2, MUC5AC and MUC6, can be of some importance as they are for IPMN[1] for distinguishing among immunophenotypes of IPNB, in particular MUC2 for intestinal subtype, MUC1 for pancreatico-biliary and MUC5AC and MUC6 which suggest gastric differentiation[7].
MOLECULAR CHARACTERIZATION OF BILIARY TRACT CANCER
Since 2014[19], the biliary tract cancers have been discovered to be targetable because of distinctive molecular alterations that can be found in around 50% of cases in large series[20], allowing patients to have more chances to be treated also when relapsing and opening the way to personalisation of therapies (see Table 1 for a summary concerning targetable alterations and detection methods, modified from the very useful one from Valle et al.[21]). iCCAs has targetable alterations definitely more commonly than eCCAs, but this is true for the mass forming “small duct type” iCCAs, whilst the “large duct type” behave mostly as an eCCA, showing mutations in oncogenes and tumour suppressor genes[19]. The targetable alterations mainly regard FGFR2 and IDH1[20], representing around 20% and 15% of cases, respectively[22].
Summary table of molecular targets with methods for detection
| Molecular target | Frequency of alteration | Methods for detection |
| FGFR alterations | 20% of intrahepatic cholangiocarcinoma cases | FISH testing for FGFR2 translocation, if available also next-generation DNA sequencing including FGFR2 intronic region, targeted RNAseq |
| IDH1/IDH2 | 15% of intrahepatic cholangiocarcinoma cases | DNA sequencing for hotspot mutations in coding region of IDH1 (immunohistochemistry not applicable) |
| MSI-high or MMR deficiency | 5% of biliary tract cancer cases | Immunohistochemistry, PCR or tumour next-generation DNA sequencing |
| BRCAness | 3.60% | Next generation DNA sequencing |
| ERBB2 (HER2) | 1.4%-2.4% | Immunohistochemistry and FISH for validation/interpretation of immunohistochemistry to identify eventual amplification, tumour next-generation DNA sequencing for mutations |
| BRAF | 1% of intrahepatic cholangiocarcinoma cases | Immunohistochemistry, DNA sequencing for hotspot mutations in coding region of BRAF |
| NTRK | 0.75% | Immunohistochemistry with Pan-TRK antibodies, next-generation DNA sequencing including NTRK intronic region or targeted RNAseq, or FISH testing for NTRK translocation |
FGFR2 rearrangements seems to occur mainly in young adults and possibly confer a better prognosis[23]. FGFR2 aberrations can present as hotspot mutations, deletion-in-frame and fusions, which generally regard the breakpoint within intron 17 or exon 18 and more frequently involve the bicaudal family RNA binding protein 1 (BICC1) in 27.9% of cases[24]. Interestingly, 32.9% of patients have a peculiar unique rearrangement and/or the aberration involves an intronic region[24]. Fusions are a part of the rearrangements and induce the constitutive activation of FGFR2[25], happening between the N-terminus containing exons 1-19 of FGFR2 with a functional tyrosine kinase domain and the C-terminus that contains the partner dimerisation domain. Recently, deletion-in-frame alteration has also been described in 2.8% of cases in a large series of 178 patients, also finding that this kind of alteration is targetable with FGFR inhibitors[26]. For this reason, although the first analysis should consider fluorescence in situ hybridisation (FISH) when looking for translocation and fusions, it is recommended to also search for FGFR2 aberrations through next-generation DNA sequencing including analyses of the intronic region[21].
IDH1 alterations are missense mutations that generally involve a single residue in the active site of the enzyme. They generally regard “small duct type” iCCA with poor grade of differentiation[27-29]. The most frequent hot spot mutations identified are R132C (70%) and less frequently R132L (15%) or R132G
Fusion events can involve the NTRK kinase (NTRK 1-3 genes) with different upstream partners, leading to the overexpression of chimeric protein and constitutively active, ligand-independent downstream signalling. NTRK fusions are rare in the routine practice for biliary tract cancer. In the recent study by Demols et al.[31], the cases were screened by immunohistochemistry and confirmed by NGS, and NTRK was found to be altered in 0.75%. Moreover, we have to remember that NTRK alterations are definitely important for the treatment of different malignancies[32]. In the metastatic settings, the guidelines recommend to also directly apply NGS for detecting NTRK fusions[33].
MSI is found in around 5% of iCCAs, sometimes occurring in Lynch syndrome[34,35]. The MSI status does not directly correlate with the site of tumour, unless the distal bile duct cancer seems to always be stable[35] and was found mainly in those with mucinous or papillary morphology[35]. MSI status does have an impact on survival[35] and is the only predictive marker so far for sensitivity to immune checkpoint inhibitors[36]. In fact, until now, the immunoexpression of PD-L1 has not been proven to be relevant since it is usually found in macrophages and not in cancer cells[37]. More studies are needed to investigate this important aim.
BRCA mutations are found in around 3.6% of iCCAs[38] without differences in terms of site, providing rationale for targeted therapies. BRCA mutant biliary tract cancers are more frequently associated with MSI and high mutational burden[39], but not with PD-L1 expression. Nevertheless, the clinical significance of BRCA status in biliary tract cancer also needs further investigation.
Another rare mutation (1%) regards BRAFV600E[40], which can be detectable with immunohistochemistry and/or directly investigated with DNA-based methods[41].
Rarely, HER2 can be found overexpressed by immunohistochemistry in biliary tract cancer (around 1.4% in a large study by Albrecht et al.[42]), more commonly in distal bile duct cancer[42], and its detection requires screening with immunohistochemistry and confirmation with FISH, mainly because of the rare detections and absence of guidelines for evaluation of immunostaining. In fact, the quantitative and qualitative reporting of HER2 immunohistochemical expression still relies on the same system applied for gastric cancer[43].
Concerning eCCAs, as mentioned above, they rarely show targetable alterations tending to be similar to pancreatic adenocarcinoma, with mutations regarding TP53, KRAS and SMAD4[44]. ERBB2 amplification is found only in around 1.3% of cases, mainly characterised by papillary architecture[44], which regards a small percentage of whole eCCAs in the routine practice, since most of them are generally perihilar adenocarcinomas with periductal pattern of growth. IDH1/2 mutations are found in 4.7% of cases of eCCA, whilst other eventual targets to be evaluated for the future are found in other around 20% of cases[44]. In the important study by Montal et al.[44], 11.5% of cases were “inflamed” and overexpressing PD-L1, thus being good candidates for immune checkpoint inhibitors therapy. Nevertheless, eCCAs is still a great orphan concerning molecular targets and the available therapies for this poorly responsive cancer are scant[45].
Since patients present already in advanced stage of disease, another important issue regards the possible application of liquid biopsy technology in biliary tract cancer. Importantly, a large recent study[20] has assessed the possibility to identify actionable alterations in primary, metastasis and liquid biopsy.
CONCLUSIONS
Biliary tract cancer is a large group of neoplasms including intrahepatic and extrahepatic biliary neoplasms with a wide spectrum of morphological characteristics and recently discovered important molecular targets for personalisation of therapies [Figure 12].
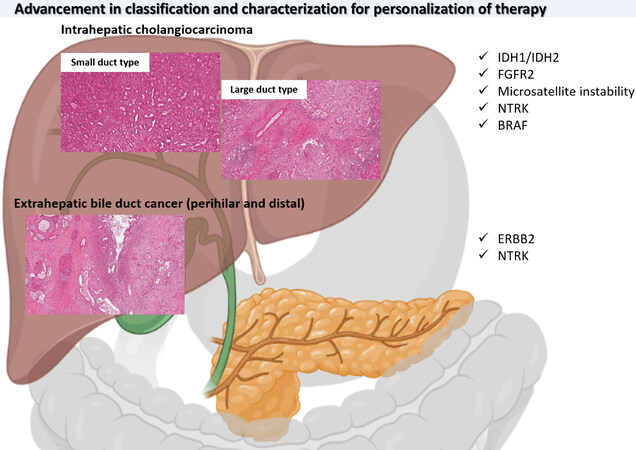
Figure 12. Graphic abstract summarizing morphological and molecular advancement in biliary tract cancer.
Future developments are urgently needed because, although the new molecular targets have allowed important advancements in clinical practice, these chances regard only a part of our patients, variable from 15%-20%, as described in the literature, to around 40%-50% of the patients tested in our routine practice[46].
Moreover, another important issue regards the possibility to perform an early diagnosis or identify an early relapse with new biomarkers, such as plasma N-glycoproteins with specific N-glycan compositions[47], or change therapy according to molecular profiling performed through liquid biopsy as it happens for lung cancer[48].
Another important issue regards the need for a better understanding of the disease progression through the study of tumour microenvironment, to find solutions for preventing advanced stage[49]. For example, it has recently been discovered that α-1,2-mannosylated N-glycans are expressed on the proteins constituting the membrane of the cancer cell and may be targeted for preventing metastasis[50].
Although all these important new topics and the incessant investigation of morpho-molecular characterisation of biliary tract cancer are quickly changing the landscape of opportunities for the treatment of our patients, strong support from the scientific community is necessary to bring them to application in clinical practice in the near future.
DECLARATIONS
AcknowledgmentsWe are grateful and honoured to work with our large team in the Pathology Unit and in particular we thank our Head of the Pathology Unit, Professor Claudio Doglioni, our Associate Director of the Molecular Diagnostic Lab, Dr.ssa Maria Giulia Cangi and our Chief of the Oncological Cytogenetics section, Dr.ssa Lorenza Pecciarini.
Authors’ contributionsDrafting the manuscript, design of the work, image collection, critical revision of the article, final approval of the version to be published: Pedica F
Critical revision of the article, in particular the molecular pathology section and final approval of the version to be published: Grassini G
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestBoth authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Nagtegaal ID, Odze RD, Klimstra D, et al. WHO Classification of Tumours Editorial Board. The 2019 WHO classification of tumours of the digestive system. Histopathology 2020;76:182-8.
2. Cardinale V, Bragazzi MC, Carpino G, et al. Intrahepatic cholangiocarcinoma: review and update. Hepatoma Res 2018;4:20.
3. Komuta M, Govaere O, Vandecaveye V, et al. Histological diversity in cholangiocellular carcinoma reflects the different cholangiocyte phenotypes. Hepatology 2012;55:1876-88.
4. Nakanuma Y, Sato Y, Ikeda H, et al. Intrahepatic cholangiocarcinoma with predominant "ductal plate malformation" pattern: a new subtype. Am J Surg Pathol 2012;36:1629-35.
5. Chua D, Chiow AKH, Ang TL, Wang LM. Malignant transformation arising within unusual and rare hepatic lesions: fibropolycystic disease form of ductal plate malformation and biliary adenofibroma. Int J Surg Pathol 2018;26:542-50.
6. Kaminsky P, Preiss J, Sasatomi E, Gerber DA. Biliary adenofibroma: a rare hepatic lesion with malignant features. Hepatology 2017;65:380-3.
7. Nakanuma Y, Uesaka K, Kakuda Y, et al. Intraductal papillary neoplasm of bile duct: updated clinicopathological characteristics and molecular and genetic alterations. J Clin Med 2020;9:E3991.
8. Nakanishi Y, Nakanuma Y, Ohara M, et al. Intraductal papillary neoplasm arising from peribiliary glands connecting with the inferior branch of the bile duct of the anterior segment of the liver. Pathol Int 2011;61:773-7.
9. Schlitter AM, Jang KT, Klöppel G, et al. Intraductal tubulopapillary neoplasms of the bile ducts: clinicopathologic, immunohistochemical, and molecular analysis of 20 cases. Mod Pathol 2015;28:1249-64.
10. Quigley B, Reid MD, Pehlivanoglu B, et al. Hepatobiliary mucinous cystic neoplasms with ovarian type stroma (so-called "hepatobiliary cystadenoma/cystadenocarcinoma"): clinicopathologic analysis of 36 cases illustrates rarity of carcinomatous change. Am J Surg Pathol 2018;42:95-102.
11. Bosman FT, Carneiro F, Hruban RH, Theise ND. . WHO classification of tumours of the digestive system. 4th ed. Geneva: World Health Organization; 2010.
12. Taguchi J, Nakashima O, Tanaka M, Hisaka T, Takazawa T, Kojiro M. A clinicopathological study on combined hepatocellular and cholangiocarcinoma. J Gastroenterol Hepatol 1996;11:758-64.
13. Joseph NM, Tsokos CG, Umetsu SE, et al. Genomic profiling of combined hepatocellular-cholangiocarcinoma reveals similar genetics to hepatocellular carcinoma. J Pathol 2019;248:164-78.
14. Beaufrère A, Calderaro J, Paradis V. Combined hepatocellular-cholangiocarcinoma: an update. J Hepatol 2021;74:1212-24.
15. Geramizadeh B, Gity R, Bahraini A, Malek-Hosseini SA. Synchronous hepatocellular carcinoma and cholangiocarcinoma in a patient transplanted for cryptogenic cirrhosis. Int J Org Transplant Med 2014;5:125-8.
16. Jain R, Fischer S, Serra S, Chetty R. The use of Cytokeratin 19 (CK19) immunohistochemistry in lesions of the pancreas, gastrointestinal tract, and liver. Appl Immunohistochem Mol Morphol 2010;18:9-15.
17. Akiba J, Nakashima O, Hattori S, et al. Clinicopathologic analysis of combined hepatocellular-cholangiocarcinoma according to the latest WHO classification. Am J Surg Pathol 2013;37:496-505.
18. Baumann U, Crosby HA, Ramani P, Kelly DA, Strain AJ. Expression of the stem cell factor receptor c-kit in normal and diseased pediatric liver: identification of a human hepatic progenitor cell? Hepatology 1999;30:112-7.
19. Nakamura H, Arai Y, Totoki Y, et al. Genomic spectra of biliary tract cancer. Nat Genet 2015;47:1003-10.
20. Israel MA, Danziger N, McGregor KA, et al. Comparative genomic analysis of intrahepatic cholangiocarcinoma: biopsy type, ancestry, and testing patterns. Oncologist 2021;25:1-10.
22. Farshidfar F, Zheng S, Gingras MC, et al. Cancer Genome Atlas Network. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cell Rep 2017;19:2878-80.
23. Saborowski A, Lehmann U, Vogel A. FGFR inhibitors in cholangiocarcinoma: what's now and what's next? Ther Adv Med Oncol 2020;12:1758835920953293.
24. Silverman IM, Hollebecque A, Friboulet L, et al. Clinicogenomic analysis of FGFR2-rearranged cholangiocarcinoma identifies correlates of response and mechanisms of resistance to pemigatinib. Cancer Discov 2021;11:326-39.
25. Wu YM, Su F, Kalyana-Sundaram S, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov 2013;3:636-47.
26. Cleary JM, Raghavan S, Wu Q, et al. FGFR2 extracellular domain in-frame deletions are therapeutically targetable genomic alterations that function as oncogenic drivers in cholangiocarcinoma. Cancer Discov 2021;11:2488-505.
27. Lee K, Song YS, Shin Y, et al. Intrahepatic cholangiocarcinomas with IDH1/2 mutation-associated hypermethylation at selective genes and their clinicopathological features. Sci Rep 2020;10:15820.
28. Goyal L, Govindan A, Sheth RA, et al. Prognosis and clinicopathologic features of patients with advanced stage isocitrate dehydrogenase (IDH) mutant and IDH wild-type intrahepatic cholangiocarcinoma. Oncologist 2015;20:1019-27.
29. Borger DR, Tanabe KK, Fan KC, et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 2012;17:72-9.
30. Brat DJ, Verhaak RG, Aldape KD, et al. Cancer Genome Atlas Research Network. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med 2015;372:2481-98.
31. Demols A, Rocq L, Charry M, et al. NTRK gene fusions in biliary tract cancers. J Clin Oncol 2020;38:574.
32. Kheder ES, Hong DS. Emerging targeted therapy for tumors with NTRK fusion proteins. Clin Cancer Res 2018;24:5807-14.
33. Marchiò C, Scaltriti M, Ladanyi M, et al. ESMO recommendations on the standard methods to detect NTRK fusions in daily practice and clinical research. Ann Oncol 2019;30:1417-27.
34. Ju JY, Dibbern ME, Mahadevan MS, Fan J, Kunk PR, Stelow EB. Mismatch repair protein deficiency/microsatellite instability is rare in cholangiocarcinomas and associated with distinctive morphologies. Am J Clin Pathol 2020;153:598-604.
35. Goeppert B, Roessler S, Renner M, et al. Mismatch repair deficiency is a rare but putative therapeutically relevant finding in non-liver fluke associated cholangiocarcinoma. Br J Cancer 2019;120:109-14.
36. Marin JJG, Prete MG, Lamarca A, et al. working group 6 of the COST-action 18122 (Euro-Cholangio-NET) as part of the European Network for the study of Cholangiocarcinoma (ENSCCA). Current and novel therapeutic opportunities for systemic therapy in biliary cancer. Br J Cancer 2020;123:1047-59.
37. Loeuillard E, Yang J, Buckarma E, et al. Targeting tumor-associated macrophages and granulocytic myeloid-derived suppressor cells augments PD-1 blockade in cholangiocarcinoma. J Clin Invest 2020;130:5380-96.
38. Spizzo G, Puccini A, Xiu J, et al. Frequency of BRCA mutation in biliary tract cancer and its correlation with tumor mutational burden (TMB) and microsatellite instability (MSI). J Clin Oncol 2019;37:4085.
39. Spizzo G, Puccini A, Xiu J, et al. Molecular profile of BRCA-mutated biliary tract cancers. ESMO Open 2020;5:e000682.
40. Goeppert B, Frauenschuh L, Renner M, et al. BRAF V600E-specific immunohistochemistry reveals low mutation rates in biliary tract cancer and restriction to intrahepatic cholangiocarcinoma. Mod Pathol 2014;27:1028-34.
41. Javle M, Lowery M, Shroff RT, et al. Phase II study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J Clin Oncol 2018;36:276-82.
42. Albrecht T, Rausch M, Rössler S, et al. HER2 gene (ERBB2) amplification is a rare event in non-liver-fluke associated cholangiocarcinogenesis. BMC Cancer 2019;19:1191.
43. Rüschoff J, Hanna W, Bilous M, et al. HER2 testing in gastric cancer: a practical approach. Mod Pathol 2012;25:637-50.
44. Montal R, Sia D, Montironi C, et al. Molecular classification and therapeutic targets in extrahepatic cholangiocarcinoma. J Hepatol 2020;73:315-27.
45. Cadamuro M, Lasagni A, Lamarca A, et al. Targeted therapies for extrahepatic cholangiocarcinoma: preclinical and clinical development and prospects for the clinic. Expert Opin Investig Drugs 2021;30:377-88.
47. Chang TT, Cheng JH, Tsai HW, Young KC, Hsieh SY, Ho CH. Plasma proteome plus site-specific N-glycoprofiling for hepatobiliary carcinomas. J Pathol Clin Res 2019;5:199-212.
48. Francaviglia I, Magliacane G, Lazzari C, et al. Identification and monitoring of somatic mutations in circulating cell-free tumor DNA in lung cancer patients. Lung Cancer 2019;134:225-32.
49. Fabris L, Cadamuro M, Cagnin S, Strazzabosco M, Gores GJ. Liver matrix in benign and malignant biliary tract disease. Semin Liver Dis 2020;40:282-97.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Pedica F, Grassini G. Pathology and molecular pathology of cholangiocarcinoma. Hepatoma Res 2021;7:71. http://dx.doi.org/10.20517/2394-5079.2021.89
AMA Style
Pedica F, Grassini G. Pathology and molecular pathology of cholangiocarcinoma. Hepatoma Research. 2021; 7: 71. http://dx.doi.org/10.20517/2394-5079.2021.89
Chicago/Turabian Style
Pedica, Federica, Greta Grassini. 2021. "Pathology and molecular pathology of cholangiocarcinoma" Hepatoma Research. 7: 71. http://dx.doi.org/10.20517/2394-5079.2021.89
ACS Style
Pedica, F.; Grassini G. Pathology and molecular pathology of cholangiocarcinoma. Hepatoma. Res. 2021, 7, 71. http://dx.doi.org/10.20517/2394-5079.2021.89
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 18 clicks
Cite This Article 18 clicks

 Like This Article 4
likes
Like This Article 4
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.