
C-kit expression in cancer cells or hematopoietic cells of the tumoral microenvironment: which is the basis for efficacy of TK inhibitors and immunotherapy in HCC?
 0
0Abstract
Twenty years ago, hematologists introduced imatinib, a tyrosine kinase inhibitor (TKI), as a long-term oral systemic therapy for patients suffering from chronic myeloid leukemia (CML) and gastrointestinal stroma tumor (GIST) (400 mg/day) who have normal liver function. The mechanism of action of imatinib was considered to be the specific interruption of the activation of the BCR-ABL kinase in CML patients and the autoactivated c-kit-tyrosine kinase in GIST patients. After the first successful long-term treatment of a c-kit-positive HCC patient with imatinib, 10 further patients with unresectable HCC were treated in Göttingen. After 18 months of therapy
Keywords
INTRODUCTION
The change in anticancer therapy: from intravenous to oral “targeted” therapy
History of c-kit in oncology and the origin of “targeted” therapy
C-kit is a transmembrane protein whose gene was first cloned by Yarden et al.[1]. By means of the reagents created thereafter, c-kit-gene expression was determined in some normal human tissues[2] as well as some solid tumors[3]. It was then found that c-kit acts as a receptor of the growth factor stem cell factor (SCF)[4].
After activation of the receptor by the ligand, the intracellular domain acts as tyrosine kinase (TK) and phosphorylates several proteins involved in proliferation and apoptosis processes[5]. Studying a mast cell leukemia cell line, mutations in the extracellular domain of the c-kit gene were identified, which were held responsible for autoactivation of the c-kit-phosphorylating activity without the need of ligand binding[6]. A previously produced compound, imatinib, shown to be the first inhibitor of ABL-TK and synthesized to inhibit proliferation of Philadelphia positive myeloid cells[7], was used.
In vitro studies on myeloid, c-kit-positive, and PDGFR-positive cells showed that imatinib was able to suppress the growth of those cells[6]. In 1998, Hirota et al.[8] studied c-kit expression in 49 GIST by immunohistochemistry, sequenced the cDNA of 6 tumor c-kit clones, and sequenced the juxta membrane domain. They found mutations within 11 amino acid sequence (Lys 550 to Val-560). They also demonstrated autophosphorylating activity of the mutated c-kit and supposed that the mutation would also be cancerogenic. Nakahara et al.[9] found an additional mutation not restricted to the 11-amino acid stretch at codon 579 of the extracellular/membrane domain of the c-kit gene. In this case as well, the mutant c-kit exhibited constitutive phosphorylating kinase activity. These findings in GIST confirmed previous findings in mast cell lines and human mast cell neoplasms[5]. Lasota et al.[10] found c-kit mutations of codon 11 of the c-kit gene in 12 of 24 malignant GIST that were c-kit positive by immunohistochemistry. Only 1 of 19 benign (less than 1 mitosis per 10 high power fields) GIST showed mutant bands. Lasota also found a point mutation at codon 576 and in one case a three-base pair in-frame deletion involving codons 560 and 561. The authors suggested that c-kit mutations could represent a genotypic marker for malignancy as mutations correlated with the histological and clinical patterns of malignancy.
The first trial results on treatment of CML[11] and CML with leukemic crisis[12] were published by hematologists and medical oncologists in 2001 in the same issue of the New England Journal of Medicine where the successful treatment of a metastasized gastrointestinal stroma tumor by Finnish oncologists was also reported[13].
Multiple liver metastases and increased uptake of F18-fluorodeoxyglucose in the right renal pelvis and ureter were seen on a PET scan performed four days before treatment with imatinib STI571 (400 mg/day) for compassionate use as a single experimental treatment was started. One month after therapy was begun, no abnormal uptake of the radioactive tracer was seen in the liver and kidney. Histological studies of biopsies from liver metastasis performed at one and two months after therapy showed a marked decrease of tumor cells, myxoid degeneration, and no signs of inflammation or necrosis.
This last success of the tyrosine kinase was attributed to a mutated c-kit gene[8], which became a diagnostic marker of GIST tumors.
The advantage of an oral therapy was immediately underlined by Goldman and Melo[14], and they also mentioned the possibility to extend the application of this novel therapy to other solid tumors in their editorial.
At that time, the cellular composition of the tumor was considered to be homogeneously and exclusively made of c-kit-positive spindle cells with more or less intense DNA synthesis in their nuclei.
Although the size of some nuclei was smaller than that of the tumor cells, the difference was not so evident because the cells were all c-kit positive.
Studies about cells of the tumor microenvironment “defined as the total functional and structural constellation of neoplastic and non-neoplastic cells in addition to the dynamic microenvironment in which they live”[15] were ongoing in different solid tumors. It was found that non-tumoral cells (mast cells, granulocytes, monocyte/macrophages, and T-lymphocyte mesenchymal cells) were inflammatory cells not able to release signals to turn off the inflammation leading to wound healing.
Instead, “initiated” tumor cells continue to proliferate and contribute to tumor growth.
Several cytokines are involved in recruitment of those cells: SCF, VEGF, EGF, FGF, and PDGF[15]. Although the positivity of the tumor after administration of radioactively labeled FDG at the PET-scan suggested the presence of mononuclear phagocytes (known to be avid “consumers” of glucose as the test was used to search for inflammatory foci[16]), their presence in the GIST microenvirement was not discussed.
When the case report on the successful treatment of the metastasized GIST in Helsinki was published[13], a patient with a duodenal GIST in Göttingen suffered a local relapse after surgery and no efficacious chemotherapy was available. The patient agreed to ask the company to provide imatinib for compassionate use. Therapy was started with 400 mg/day orally, which was well tolerated. The tumor disappeared, and the patient survived for longer than 10 years with oral administration of 400 mg/day of imatinib[17]. The treatment of several other patients with relapsed or metastasized GIST was started with 400 mg/day imatinib[18].
Imatinib was effective even in high-risk c-kit-positive GIST patients with multiple chromosomal aberrations with a mean of 9.9 losses and of 3.9 gains[18]. C-kit mutations were observed in all 11 patients who were tested but not all responded to imatinib therapy.
Several years after starting therapy with imatinib, it was justified to speak about advanced GIST as a “chronic disease” with a median survival of 86 months since first diagnosis[19] in the palliative setting and even longer in the adjuvant therapy after resection of high risk GIST[20].
The first observation that the many dendritic cells [Figure 1] distributed among and in close contact with the spindle cells in the GIST tissue do not behave as inflammatory cells was reported by Cameron et al.[21]. In fact, they do not accumulate as “inflammatory cells” normally do and do not express the classical cytokines of the inflammatory cells, e.g., interleukin-6[21]. For the first time, the immunostainings could deliver a different way of seeing the interaction between immune cells and tumor cells [Figure 2]. The consequence could be a reinterpretation of the real target of anticancer drugs[22-24].
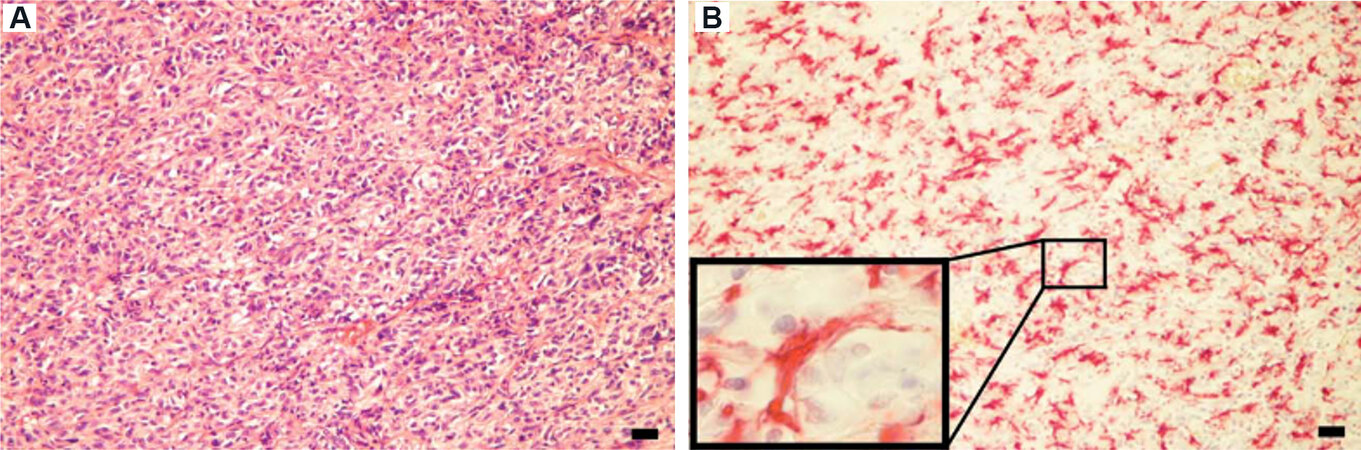
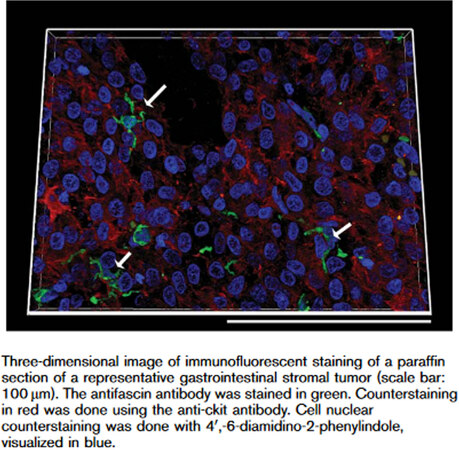
Figure 2. This is a further slice of the tissue studied in Figure 1. The slice is stained with an antibody against fascin (green) and counterstained with an antibody against c-kit. One dendritic cell (upper arrow left) is in contact with four tumor cells through his prolongations. From Cameron et al.[21] (with permission).
In fact, the immune cells of the microenvironment more than the tumor cells per se could be the main target of the anticancer therapy.
C-kit, HCC and imatinib
At the time when the efficacy of imatinib could be observed in a patient with relapse of the duodenal GIST after surgery, we were treating a 65-year-old patient with liver cirrhosis who developed a small tumoral nodule which, under observation, grew in size from 1.3 cm × 1.3 cm to 1.9 cm × 1.6 cm within 6 months. A biopsy of the tumor was first performed, and immunostaining showed positivity of c-kit in the tumoral and non-tumoral tissue.
As it was hypothesized that oval cells of the liver could become tumoral stem cells in the development of hepatocellular carcinoma[25] and it is known that those cells express c-kit[26], it was reasonable to try to treat the 66-year-old patient with liver cirrhosis and a small liver cancer who refused interventional therapies with imatinib. As imatinib is cleared by the liver and considering the advanced cirrhosis, imatinib therapy at 200 mg/day was started in March 2002. The drug was well tolerated and a follow-up control study was performed by abdominal ultrasound every eight weeks. After six months, an MRI with the administration of gadolinium was performed with no signs of tumor progression. HPLC measurement of the serum concentration of imatinib showed a normal value (550 ng/mL) with no signs of accumulation. The serum concentration of the main metabolite, N-DesM-imatinib, was 70 ng/mL, which corresponded to 50% of the expected serum concentration, indicating reduced hepatic metabolic activity.
After one year, MRI imaging and percutaneous biopsy were performed. There was a central hypodensity and a peripheral hyperdensity but no size reduction. Histology showed necrotic tissue material surrounded by cirrhotic septa and normal tissue[27].
FDG-PET was negative. Six months later, the tumor was no longer detectable and therapy was discontinued.
After this encouraging experience, an investigator initiated an observational study in 10 patients with c-kit-positive hepatocellular carcinoma and CHILD-A cirrhosis was started with 400 mg imatinib/day for the first eight weeks. Serum concentrations of alpha-fetoprotein and imatinib at one, two, and four weeks after starting therapy were measured. The tumor size was controlled by abdominal ultrasound. One patient stopped therapy because of intolerance (diarrhea). Nine patients continued after the first eight weeks but developed side effects thereafter. Then, treatment was discontinued for eight weeks and when the therapy was restarted, it was at a reduced dose (200 mg/day). Stable disease was found in two patients, complete response with normalization of AFP-serum level in one, and progressive disease in seven of nine patients[28]. After 18 months of therapy with 200 mg imatinib/day, 4 of 12 patients (33%) were alive, confirming that
A further phase II study, performed using a dosage of 400-600 mg imatinib/day, confirmed the occurrence of relevant side effects at such high dosage in patients with advanced tumor and reduced liver function[30].
The c-kit status in those patients was reported to be negative. Surprisingly, the pharmacokinetics of imatinib at a high dosage was not significantly affected by the reduced liver function. The results of that preliminary study show that imatinib is not effective in cirrhotic patients with reduced liver function and advanced c-kit-negative HCC when the “benchmark” dosage was used[30]. In California, a 63-year-old women suffering from advanced c-kit-positive cholangiocarcinoma was also treated with a daily dose of 400 mg of imatinib mesylate[31]. Severe side effects were first treated with corticosteroids, but drug administration had to be discontinued. In that report, 31% of the cholangiocarcinoma specimens were c-kit positive, 69% were PDGF-alpha postivie, and 46% were PDGF-beta positive. Furthermore, c-kit-receptor positivity was described in human cholangiocarcinoma, and imatinib efficacy was found in an animal model[32].
Baumhoer et al.[33] found that two small cancers which developed in two livers that had to be explanted because of fulminant hepatitis were c-kit positive.
Tanaka et al.[34] found c-kit positivity in 8 of 15 combined hepatocellular and intrahepatic cholangiocellular carcinomas. C-kit positivity of HCC was confirmed by Lee et al.[35] in 40 of 50 HCCs associated with HBV infection. Furthermore, c-kit expression was found in 25.6% of human HCC by both immunostaining and PCR analysis of total RNA[36]. In that study, c-kit expression was positively correlated with better survival.
Mansuroglu et al.[37] examined c-kit expression using an antibody from Dako in 72 human HCCs and the surrounding non-tumoral tissue.
C-kit expression was studied by immunohistochemistry and PCR analysis of total RNA from tumoral and peritumoral tissues. RNA from three HCC cell lines was also studied. Not only 70% of the tumors showed a different degree of c-kit positivity but also 90% of the non-cirrhotic and cirrhotic peritumoral tissues were c-kit-positive. Overall, 83% of the tumors, 75% of the non-cirrhotic peritumoral tissue, and 100% of the cirrhotic peritumoral tissue samples expressed c-kit-specific RNA. C-kit-specific RNA was also found in two of the tree cell lines (HUH-7 and SK-Hep 1). No mutation studies were performed. Mansuroglu et al.[38] also found that cholangiocarcinoma was c-kit positive in a rat model of cirrhosis and cholangiocarcinoma induction through repeated TAA administration.
It may be assumed that the positive effect of imatinib in some HCC is probably not dependent on c-kit mutations as was the case in benign GIST and half of the malignant GIST originating from the c-kit-positive cells of Cajal, as studied by Lasota et al.[10].
The era of oral administration of TKIs in advanced HCC in CHILD-A cirrhosis
After publication of a promising phase II study[39] which also included CHILD-B patients (1/3), the publication of the SHARP study[40] established the effectivity of a tyrosine kinase inhibitor as the first orally administrable systemic therapy for advanced hepatocellular carcinoma in patients with CHILD-A cirrhosis. In that study, only 5% of the treated patients had CHILD-B cirrhosis. The majority of the treated patients were male (87%) and the mean age of the patients was 64.9 years. The median serum level of albumin was 3.9 g/dL (normal value 3.5-5.0 g/dL) and bilirubin serum concentration was 0.7 mg/dL.
While the degree of c-kit expression in Hepatitis B-related HCC was found to negatively correlate with survival after surgery[41], the search for positive prognostic scores for efficacy of sorafenib in patients with advanced hepatocellular carcinoma and cirrhosis continued to develop 10 years after the introduction of sorafenib into HCC therapy[42-44].
Sunitinib, the second-line tyrosine kinase inhibitor in GIST patients, was tested in patients with advanced HCC in comparison to sorafenib at a dosage of 37.5 mg, which is the dosage used in patients with GIST who did not tolerate 50 mg/day as standard dosage. It is therefore not surprising that the side effects were considerable and overall survival was comparable to that of sorafenib only in the subgroup of Asian
Regorafenib was found to improve survival in HCC patients (88% male and mean age of 64 years) who were resistant to sorafenib but were still CHILD-A[46]. This study was followed by the publication of the next positive result of the treatment of advanced HCC with levantinib, a multikinase inhibitor, compared with sorafenib[47]. Overall survival of patients treated with levantinib (13.6 months) was comparable with that of the patients treated with sorafenib (12.3 months)[47]. Carbozantib was the next multikinase inhibitor which showed efficay in the treatment of advanced HCC after previous treatment with sorafenib. Overall, 80% of the patients were male and their median age was 64 years. Overall survival was 10.2 months with carbozantib, compared with 8.0 months in the placebo group[48]. Tyrosine kinase inhibitors at the dosages used, in mostly male patients with advanced HCC, cirrhosis CHILD-A, and median age of 64 years, had an efficacy comparable with that of sorafenib.
HCC systemic intravenous therapy with monoclonal antibodies
Monoclonal anti-VEGFR2 antibody was significantly effective in males with HCC and CHILF-A cirrhosis
No major advances were expected and the new line of therapy, that of monoclonal antibodies, was initiated. An antibody against VEGFR2 was administered intravenously to patients with advanced hepatocellular carcinoma in CHILD-A cirrhosis and a serum level of alpha-fetoprotein of ≥ 400 ng/mL who were already treated with sorafenib. Overall, 78% of the patients were male in the ramucirumab group and 83% in the placebo group, the median age was 64 years, and all had CHILD-A cirrhosis. The median overall survival was significantly improved (8.5 months vs. 7.3 months) in the ramucirumab group compared with the placebo group[49]. Although all subgroup analyses were in favor of the treatment with the antibody, this was not the case for the female subgroup as there were only 16 females in the control group. This has to be kept in mind when females with advanced HCC and cirrhosis need treatment after sorafenib resistance or intolerance[49].
Immunotherapy in solid tumors: from the beginning in 2010 to now in advanced HCC in cirrhosis
After the intratumoral immune cell microenvironment directed attention of scientists toward HCC[50-52], the therapeutic use of the so-called checkpoint inhibitors, also defined as immunotherapy, in patients with HCC in cirrhosis has matured[53].
Ipilimumab was the first antibody (checkpoint inhibitor) of this generation against the protein (PD-1) responsible for blocking the interaction of antigen-presenting cells with T-lymphocytes. This molecule was considered to be responsible for the lack of effective cellular immune reaction against tumor cells.
Ipilimumab was the first of this group to be approved for the treatment of metastatic melanoma[54].
In August 2010, the therapy became available for a patient with liver metastasis of a melanoma resistant to dacarbazine administration. The use of ipilimumab was made available by the company before official approval was released. The patient developed complete response after the third intravenous administration of the antibody. There were no side effects[55]. The patient is, as of June 2021, still alive and FDG-PET is completely negative. Abundant infiltration of T-lymphocytes, macrophages, and dendritic cells could be detected within the tumor stroma of the primary tumor and the metastasis. A strong 18-FDG uptake by the metastasis was found immediately before therapy with ipilimumab was started.
Since the release of ipilimumab, several other antibodies against different checkpoint inhibitors (PD-L1 and CTLA-4) have been approved for treatment of different solid tumors, and recently one of them, a PD-L1 antagonist, atezolizumab, has been successfully used for treatment of hepatocellular carcinoma[56,57]. It has been found to be cost-effective not only in the treatment of advanced HCC in patients with CHILD-A cirrhosis and good clinical conditions compared to sorafenib[58] but also in the treatment of other solid tumors[59,60], and the combination of atezolizumab and bevacizumab is now the first-line therapy for at least one third of patients with HCC without contraindications such as advanced cirrhosis and esophageal varices[61].
CONCLUSIONS
In the last 20 years, we have witnessed the introduction of two anticancer therapy options: (1) the oral tyrosine kinase inhibitors, which are supposed to block proliferation of myeloid cells in CML and tumor cells in solid tumors such as operated and non-operable GIST, as long-term palliative or adjuvant therapy[17-20], and unresectable[26-28,40] HCC; and (2) the intravenously applied checkpoint inhibitors, which are supposed to activate cellular immune reaction against solid tumor cells such as melanoma and now also advanced HCC in CHILD-A cirrhosis.
The above-mentioned tumors have qualitatively and quantitatively different chromosomal aberrations with gains and losses, starting with the oldest and maybe simplest one of the Philadelphia chromosome (CML) to the more complicated ones of GIST[18], melanoma[62], and advanced HCC in cirrhosis[63].
Despite that, the new antitumoral drugs may act at a similar level to reduce local production of their survival factors eventually by c-kit-positive[64] and PD-1- and PD-L1-positive hematopoietic cells of the tumor microenvironment[21,53] [Figure 3].
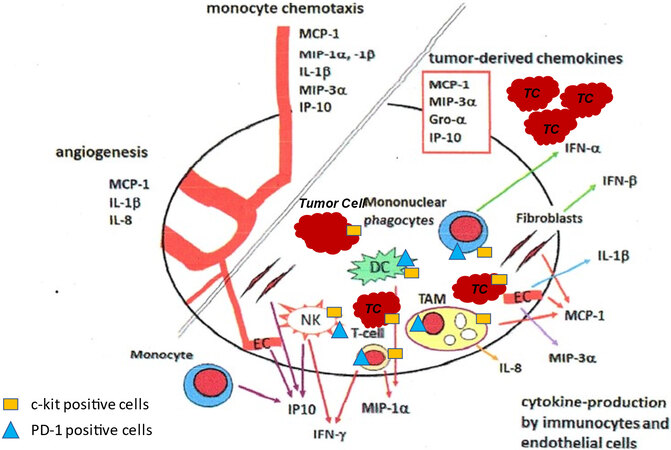
Figure 3. The cartoon, drawn on the basis of own data obtained from immunohistological and RNA studies on GIST and melanoma tissue and on published studies, indicates a hypothetical mechanism of how TKIs and anti-checkpoint antibodies may exert anticancer activity in different solid tumors including HCC. Immune cells, which are recruited by the chemokines released from the tumor cells, establish a symbiotic relationship with the tumor cells. Immune cells may be in close contact with tumor cells and continuously release factors necessary for tumor growth and survival. TKI and checkpoint inhibitors may induce tumor cell death by blocking the supply of those factors by means of different mechanisms. The success of the different drugs is dependent on the assembly and the metabolic status of the different cells of the tumor microenvironment which may be similar in different solid tumors such as GIST, HCC, or melanoma. The cartoon represents a modification of that published by Cameron et al.[22] (reprinted with permission). TKIs: Kinase inhibitors; NK: natural killer cell; DC: dendtric cell; TAM: tumor associated macrophages; TC: tumor cell; EC: endothelial cell.
However, before we jump into the next paradigm change, we may want to reconsider old oral drugs using a lower dosage for a longer time and accept stable disease for a longer time with a lower costs and better acceptance, especially in patients older than 65 years. The best would be a pill a day, as we have observed in the cases of relapsed GIST with sustained response under continuous therapy with 400 mg imatinib/day and healing of the small liver cancer in the cirrhotic patient after a year of 200 mg imatinib/day. It could also be possible to combine several oral drugs at a lower dose, as it has been established for “chronic” viral diseases or “autoimmune diseases” to maintain partial response or stable diseases even in older patients with stabile uncomplicated cirrhosis[65].
DECLARATIONS
AcknowledgmentsThe author is grateful to Dr. Eleonora Ramadori for her technical in performing modifications introduced in Figure 3.
Authors’ contributionsThe author contributed solely to the article.
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestThe author declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Yarden Y, Kuang WJ, Yang-Feng T, et al. Human proto-oncogene c-kit: a new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J 1987;6:3341-51.
2. Lammie A, Drobnjak M, Gerald W, Saad A, Cote R, Cordon-Cardo C. Expression of c-kit and kit ligand proteins in normal human tissues. J Histochem Cytochem 1994;42:1417-25.
3. Natali PG, Nicotra MR, Sures I, Santoro E, Bigotti A, Ullrich A. Expression of c-kit receptor in normal and transformed human nonlymphoid tissues. Cancer Res 1992;52:6139-43.
4. Besmer P, Manova K, Duttlinger R, et al. The kit -ligand (steel factor) and its receptor c- kit/W : pleiotropic roles in gametogenesis and melanogenesis. Development 1993;119:125-37.
5. Furitsu T, Tsujimura T, Tono T, et al. Identification of mutations in the coding sequence of the proto-oncogene c-kit in a human mast cell leukemia cell line causing ligand-independent activation of c-kit product. J Clin Invest 1993;92:1736-44.
6. Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med 1996;2:561-6.
7. Heinrich MC, Griffith DJ, Druker BJ, Wait CL, Ott KA, Zigler AJ. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood 2000;96:925-32.
8. Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998;279:577-80.
9. Nakahara M, Isozaki K, Hirota S, et al. A novel gain-of-function mutation of c-kit gene in gastrointestinal stromal tumors. Gastroenterology 1998;115:1090-5.
10. Lasota J, Jasinski M, Sarlomo-rikala M, Miettinen M. Mutations in exon 11 of c-kit occur preferentially in malignant versus benign gastrointestinal stromal tumors and do not occur in leiomyomas or leiomyosarcomas. Am J Pathol 1999;154:53-60.
11. Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 2001;344:1031-7.
12. Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med 2001;344:1038-42.
13. Joensuu H, Roberts PJ, Sarlomo-Rikala M, et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med 2001;344:1052-6.
14. Goldman JM, Melo JV. Targeting the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 2001;344:1084-6.
15. Kempen LC, Ruiter DJ, van Muijen GN, Coussens LM. The tumor microenvironment: a critical determinant of neoplastic evolution. Eur J Cell Biol 2003;82:539-48.
16. Love C, Tomas MB, Tronco GG, Palestro CJ. FDG PET of infection and inflammation. Radiographics 2005;25:1357-68.
17. Cameron S, Schaefer IM, Schwoerer H, Ramadori G. Ten years of treatment with 400 mg imatinib per day in a case of advanced gastrointestinal stromal tumor. Case Rep Oncol 2011;4:505-11.
18. Schindler CG, Armbrust T, Gunawan B, Langer C, Füzesi L, Ramadori G. Gastrointestinal stromal tumor (GIST) -- single center experience of prolonged treatment with imatinib. Z Gastroenterol 2005;43:267-73.
19. Armbrust T, Sobotta M, Gunawan B, et al. Does imatinib turn recurrent and/or metastasized gastrointestinal stromal tumors into a chronic disease? Eur J Gastroenterol Hepatol 2009;21:819-23.
20. Joensuu H, Wardelmann E, Sihto H, et al. Effect of KIT and PDGFRA mutations on survival in patients with gastrointestinal stromal tumors treated with adjuvant imatinib: an exploratory analysis of a randomized clinical trial. JAMA Oncol 2017;3:602-9.
21. Cameron S, Haller F, Dudas J, et al. Immune cells in primary gastrointestinal stromal tumors. Eur J Gastroenterol Hepatol 2008;20:327-34.
22. Cameron S, Gieselmann M, Blaschke M, Ramadori G, Füzesi L. Immune cells in primary and metastatic gastrointestinal stromal tumors(GIST). Int J Exp Pathol 2014;7:3563-79.
23. Vitiello GA, Bowler TG, Liu M, et al. Differential immune profiles distinguish the mutational subtypes of gastrointestinal stromal tumor. J Clin Invest 2019;129:1863-77.
24. Pantaleo MA, Tarantino G, Agostinelli C, et al. Immune microenvironment profiling of gastrointestinal stromal tumors (GIST) shows gene expression patterns associated to immune checkpoint inhibitors response. Oncoimmunology 2019;8:e1617588.
25. Braun L, Mikumo R, Fausto N. Production of hepatocellular carcinoma by oval cells: cell cycle expression of c-myc and p53 at different stages of oval cell transformation. Cancer Res 1989;49:1554-61.
26. Fujio K, Hu Z, Evarts RP, Marsden ER, Niu CH, Thorgeirsson SS. Coexpression of stem cell factor and c-kit in embryonic and adult liver. Exp Cell Res 1996;224:243-50.
27. Ramadori G, Füzesi L, Grabbe E, Pieler T, Armbrust T. Successful treatment of hepatocellular carcinoma with the tyrosine kinase inhibitor imatinib in a patient with liver cirrhosis. Anticancer Drugs 2004;15:405-9.
28. Ramadori G, Schleyer E, Armbrust T. Safety of imatinib in patients with liver cirrhosis and hepatocellular carcinoma. JCO 2004;22:4244.
29. Armbrust T, Baumhoer D, Werner J, Schleyer E, Ramadori G. Treatment of hepatocellular carcinoma (HCC) with the tyrosine kinase inhibitor Imatinib. JCO 2005;23:4210.
30. Eckel F, von Delius S, Mayr M, et al. Pharmacokinetic and clinical phase II trial of imatinib in patients with impaired liver function and advanced hepatocellular carcinoma. Oncology 2005;69:363-71.
31. Holcombe RF, Gu M, Imagawa D, Milovanovic T. Expression of Kit and platelet-derived growth factor receptors alpha and beta in cholangiocarcinoma, and case report of therapy with imatinib mesylate (STI571). Anticancer Drugs 2003;14:651-7.
32. Kamenz T, Caca K, Blüthner T, Tannapfel A, Mössner J, Wiedmann M. Expression of c-kit receptor in human cholangiocarcinoma and in vivo treatment with imatinib mesilate in chimeric mice. World J Gastroenterol 2006;12:1583-90.
33. Baumhoer D, Lorf T, Gunawan B, Armbrust T, Füzesi L, Ramadori G. Hepatic tumorigenesis in acute hepatic failure. Eur J Gastroenterol Hepatol 2005;17:1125-30.
34. Tanaka S, Yamamoto T, Tanaka H, et al. Potentiality of combined hepatocellular and intrahepatic cholangiocellular carcinoma originating from a hepatic precursor cell: Immunohistochemical evidence. Hepatol Res 2005;32:52-7.
35. Lee ES, Han EM, Kim YS, et al. Occurrence of c-kit+ tumor cells in hepatitis B virus-associated hepatocellular carcinoma. Am J Clin Pathol 2005;124:31-6.
36. Chung CY, Yeh KT, Hsu NC, et al. Expression of c-kit protooncogene in human hepatocellular carcinoma. Cancer Lett 2005;217:231-6.
37. Mansuroglu T, Baumhoer D, Dudas J, et al. Expression of stem cell factor receptor c-kit in human nontumoral and tumoral hepatic cells. Eur J Gastroenterol Hepatol 2009;21:1206-11.
38. Mansuroglu T, Ramadori P, Dudás J, et al. Expression of stem cell factor and its receptor c-Kit during the development of intrahepatic cholangiocarcinoma. Lab Invest 2009;89:562-74.
39. Abou-Alfa GK, Schwartz L, Ricci S, et al. Phase II study of sorafenib in patients with advanced hepatocellular carcinoma. J Clin Oncol 2006;24:4293-300.
40. Llovet JM, Ricci S, Mazzaferro V, et al. SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359:378-90.
41. Yan W, Zhu Z, Pan F, Huang A, Dai GH. Overexpression of c-kit(CD117), relevant with microvessel density, is an independent survival prognostic factor for patients with HBV-related hepatocellular carcinoma. Onco Targets Ther 2018;11:1285-92.
42. Pinyol R, Montal R, Bassaganyas L, et al. Molecular predictors of prevention of recurrence in HCC with sorafenib as adjuvant treatment and prognostic factors in the phase 3 STORM trial. Gut 2019;68:1065-75.
43. Marisi G, Cucchetti A, Ulivi P, et al. Ten years of sorafenib in hepatocellular carcinoma: Are there any predictive and/or prognostic markers? World J Gastroenterol 2018;24:4152-63.
44. Sansone V, Tovoli F, Casadei-Gardini A, et al. Comparison of prognostic scores in patients with hepatocellular carcinoma treated with sorafenib. Clin Transl Gastroenterol 2021;12:e00286.
45. Cheng AL, Kang YK, Lin DY, et al. Sunitinib versus sorafenib in advanced hepatocellular cancer: results of a randomized phase III trial. J Clin Oncol 2013;31:4067-75.
46. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017;389:56-66.
47. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet 2018;391:1163-73.
48. Abou-Alfa GK, Meyer T, Cheng AL, et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N Engl J Med 2018;379:54-63.
49. Zhu AX, Kang Y, Yen C, et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2019;20:282-96.
50. Hou J, Zhang H, Sun B, Karin M. The immunobiology of hepatocellular carcinoma in humans and mice: basic concepts and therapeutic implications. J Hepatol 2020;72:167-82.
51. Pan X, Lin P, Feng F, et al. Comparison of immune profiles between hepatocellular carcinoma subtypes. Biophys Rep 2020;6:19-32.
52. Yu S, Wang Y, Hou J, et al. Tumor-infiltrating immune cells in hepatocellular carcinoma: Tregs is correlated with poor overall survival. PLoS One 2020;15:e0231003.
53. Tsaknakis B, Schaefer IM, Schwörer H, et al. Long-lasting complete response of metastatic melanoma to ipilimumab with analysis of the resident immune cells. Med Oncol 2014;31:813.
54. Han Y, Liu D, Li L. PD-1/PD-L1 pathway:current researches in cancer. Am J Cancer Res ;2020:10:727-42.
55. Yau T, Kang YK, Kim TY, et al. Efficacy and safety of nivolumab plus ipilimumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib: the checkmate 040 randomized clinical trial. JAMA Oncol 2020;6:e204564.
56. Finn RS, Qin S, Ikeda M, et al. IMbrave150 Investigators. Atezolizumab plus Bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med 2020;382:1894-905.
57. Sonbol MB, Riaz IB, Naqvi SAA, et al. Systemic therapy and sequencing options in advanced hepatocellular carcinoma: a systematic review and network meta-analysis. JAMA Oncol 2020;6:e204930.
58. Zhang X, Wang J, Shi J, Jia X, Dang S, Wang W. Cost-effectiveness of atezolizumab plus bevacizumab vs sorafenib for patients with unresectable or metastatic hepatocellular carcinoma. JAMA Netw Open 2021;4:e214846.
59. Liu G, Kang S, Wang X, Shang F. Cost-effectiveness analysis of atezolizumab versus chemotherapy as first-line treatment for metastatic non-small-cell lung cancer with different PD-L1 expression status. Front Oncol 2021;11:669195.
60. Chisaki Y, Kuwada Y, Matsumura C, Yano Y. Cost-effectiveness analysis of atezolizumab plus nab-paclitaxel for advanced PD-L1 positive triple-negative breast cancer in Japan. Clin Drug Investig 2021;41:381-9.
61. Pelizzaro F, Ramadori G, Farinati F. Hepatoma Res 2021. p. 36. Systemic therapies for hepatocellular carcinoma: an evolving landscape
62. Bastian BC, LeBoit PE, Hamm H, Bröcker EB, Pinkel D. Chromosomal gains and losses in primary cutaneous melanomas detected by comparative genomic hybridisation. Cancer Res 1998;58:2170-5.
63. Homayounfar K, Gunawan B, Cameron S, et al. Pattern of chromosomal aberrations in primary liver cancers identified by comparative genomic hybridization. Hum Pathol 2009;40:834-42.
64. Sperling C, Schwartz S, Büchner T, Thiel E, Ludwig WD. Expression of the stem cell factor receptor C-KIT (CD117) in acute leukemias. Hematologica 1997;82:617-21.
65. . Robert Koch Institut. Leber. Krebs in Deutschland. Berlin, 2019. p. 44.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Ramadori G. C-kit expression in cancer cells or hematopoietic cells of the tumoral microenvironment: which is the basis for efficacy of TK inhibitors and immunotherapy in HCC?. Hepatoma Res 2021;7:69. http://dx.doi.org/10.20517/2394-5079.2021.80
AMA Style
Ramadori G. C-kit expression in cancer cells or hematopoietic cells of the tumoral microenvironment: which is the basis for efficacy of TK inhibitors and immunotherapy in HCC?. Hepatoma Research. 2021; 7: 69. http://dx.doi.org/10.20517/2394-5079.2021.80
Chicago/Turabian Style
Ramadori, Giuliano. 2021. "C-kit expression in cancer cells or hematopoietic cells of the tumoral microenvironment: which is the basis for efficacy of TK inhibitors and immunotherapy in HCC?" Hepatoma Research. 7: 69. http://dx.doi.org/10.20517/2394-5079.2021.80
ACS Style
Ramadori, G. C-kit expression in cancer cells or hematopoietic cells of the tumoral microenvironment: which is the basis for efficacy of TK inhibitors and immunotherapy in HCC?. Hepatoma. Res. 2021, 7, 69. http://dx.doi.org/10.20517/2394-5079.2021.80
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 7 clicks
Cite This Article 7 clicks

 Like This Article 30
likes
Like This Article 30
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.