
Epigenetics in hepatoblastoma
 0
0Abstract
Hepatoblastoma (HB) is the most frequent pediatric primary liver tumor. When the tumoral lesions can be resected, prognosis is generally favorable. However, there is a significant number of cases in which resection is not possible at diagnosis, and patients usually receive neo-adjuvant cisplatin-based chemotherapy prior to surgery. Unfortunately, some HBs develop resistance to initial chemotherapy or after recurrence, progressing to metastatic disease. Moreover, long-term side effects of chemotherapy remain a serious concern. Understanding the molecular bases of HB development and progression is thus essential for the identification of more efficacious therapies. HBs have a very low mutational burden, and the most frequent mutations occur in the CTNN1B gene (> 80% of cases) and to a lesser extent in NFE2L2 (~10% of cases). These observations suggest that other pathogenic processes besides genetic mutations may play a role in HB tumorigenesis. Epigenetic mechanisms encompass a variety of molecular processes with a tremendous potential to regulate gene expression. They include the covalent modifications of DNA and histones, the activity of enzymatic chromatin remodelers, and the expression of non-coding RNAs. Dysregulation of epigenetic processes has clearly become a hallmark of cancer. Regarding HB, recent studies have explored its epigenetic landscape, the expression of specific epigenetic effectors, and the tumorigenic consequences of epigenetic alterations. The reversible nature of most epigenetic modifications and the possibility to target non-coding RNAs may pave the way for new therapeutic avenues in HB. Here, we summarize and discuss the most relevant findings in this less explored aspect of HB.
Keywords
INTRODUCTION
Hepatoblastoma (HB) is the most common hepatic tumor among pediatric malignancies. The annual incidence increased from 1.89 per million in 2000 to 2.16 per million in 2015, and it usually develops before four years of age[1]. Even though HB appearance can be related to constitutional genetic abnormalities and malformations, such as Beckwith-Wiedemann syndrome and familial adenomatous polyposis, it is mainly sporadic[2,3]. Risk factors such as low birth weight (less than 1 kg) and premature birth have also been associated with HB incidence[2,3]. Fortunately, the survival rate of HB patients has increased by approximately 50% since 1990 due to the collaborative efforts of several multidisciplinary groups, including the International Childhood Tumor Strategy Group (SIOPEL), the Children Oncology Group (COG), the German Society for Pediatric Oncology and Hematology (GPOH), and the Japanese Study Group for Pediatric Liver Tumors (JPLT)[3,4]. SIOPEL provided the pretreatment extent of the tumor (PRETEXT) staging method, which is based on the grade of tumor occupancy in the liver[5]. These groups coordinate the Children’s Hepatic tumors International Collaboration (CHIC) that proposed a new global stratification system[6]. This system includes PRETEXT staging and its annotation factors (i.e., vessel invasion, extrahepatic growth, and multifocality), age of the patients, alpha-fetoprotein (AFP) serum levels, and the existence of metastasis[3,4,6]. However, the risk of HB patients is also determined by its histopathological characteristics, which were recently reviewed by Ranganathan et al.[7].
The histology of HBs recapitulates the stages of liver development, including epithelial and epithelial-mesenchymal mixed features[7]. The epithelial form includes pure fetal, mixed fetal-embryonal and embryonal HB subtypes. The pure fetal subtype is commonly associated with a better prognosis than more immature HBs[3,4,6]. The management of HB is based on the risk stratification[6]. According to the current Pediatric Hepatic International Tumor Trial (PHITT) strategy, there are four risk HB groups: very low risk, low risk, intermediate risk, and high risk. Very low- and low-risk HBs, characterized by well differentiated fetal histology, undergo cisplatin-based chemotherapy and resection, whereas intermediate- and high-risk HBs receive cisplatin in combination with other drugs such as doxorubicin, carboplatin, and vincristine
MOLECULAR MECHANISMS OF CARCINOGENESIS IN HB
The mutation rate of HB is very low (2.9-4.7 mutations/tumor), thereby indicating that HB is a genetically simple tumor[11-13]. The most recurrent mutation is found in the CTNNB1 gene, the key effector of the Wnt/β-catenin signaling pathway, mainly harboring exon 3 mutations or deletions[11,12]. Besides CTNNB1, additional mutations in other genes have been reported in different studies [Table 1][4,12,14-20].
Representative mutated genes in HB
Even though the Wnt/β-catenin pathway is the most dysregulated pathway in the pathogenesis of HB, as indicated by the mutation rate of some of its constituent genes (CTNNB1, AXIN1, and APC), there are further signaling pathways that have been found to be altered in this tumor, such as Hippo, Hedgehog, NFE2L2/KEAP1, HGF/c-Met, PI3K/AKT/mTOR, SP/NK-1R, and IGF pathways. The role of these pathways in the development and progression of HB was recently reviewed by Zhang et al.[21]. The low rate of mutations in HB together with the alteration of numerous signaling pathways provides evidence of the collaboration of additional mechanisms. In this line, epigenetic alterations have been proposed as important drivers of HB. Indeed, a recent study identified two epigenomic clusters (Epi-CA and Epi-CB) based on the degree of DNA CpG hypomethylation and hypermethylation that contribute to the molecular risk stratification of HB patients[18]. This molecular risk stratification approach based on the DNA methylation pattern of HB tumors could complement the clinical and biological risk stratification method that has been recently proposed[4], and therefore improve the management of HB patients. In addition, a very recent study proposed HB differentiation state groups that could further subdivide the Epi-CA and Epi-CB clusters. While Epi-CA mostly matched the “hepatocytic” HBs, Epi-CB was subclassified into three subgroups associated with “liver progenitor” and “mesenchymal” phenotypes and older age of patients. The differentiation state of HB was characterized by a unique DNA methylation landscape, thereby highlighting the importance of this epigenetic modification in the prognosis and management of HB[17]. Therefore, we next review the epigenetic landscape of HB pointing out the importance of these modifications in the diagnosis and management of HB [Figure 1].
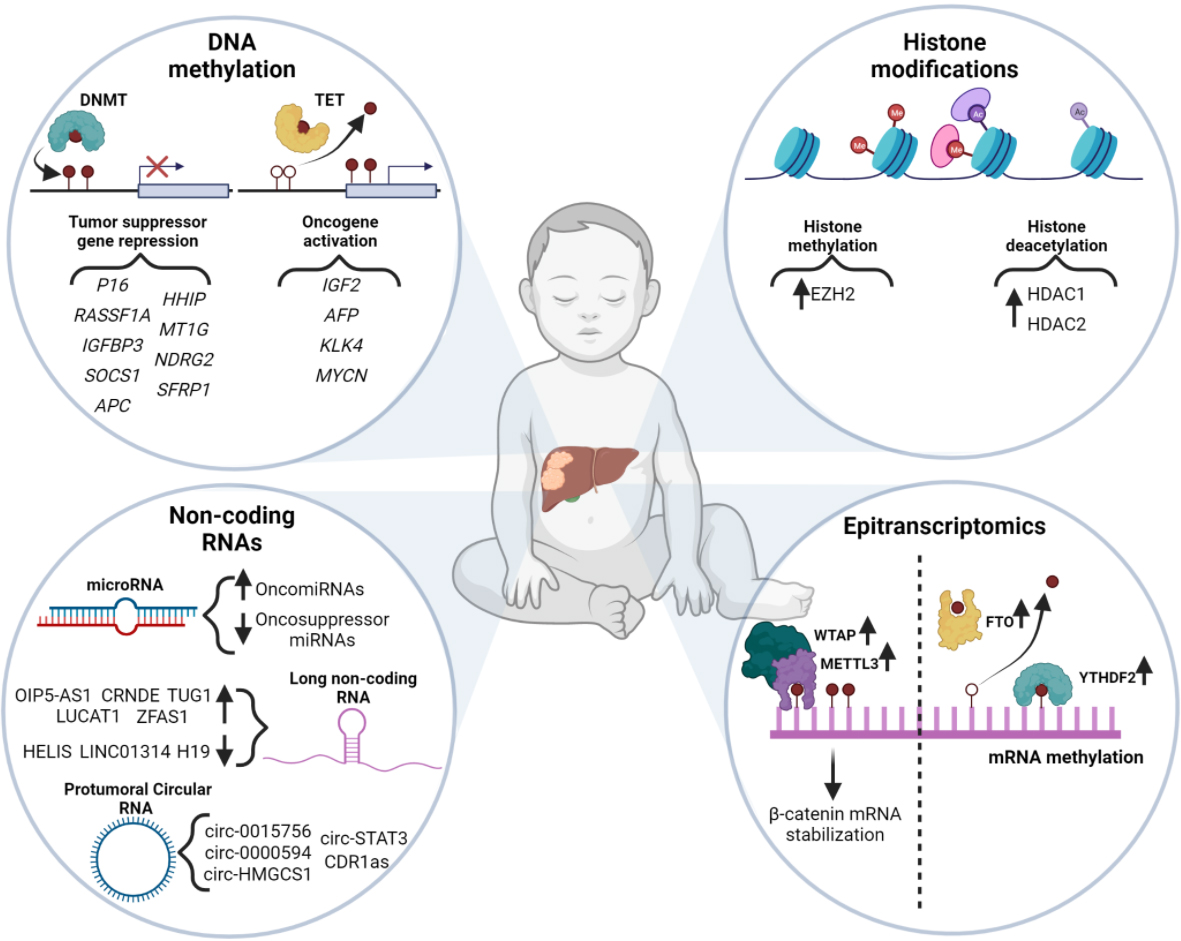
Figure 1. Summary of the epigenetic mechanisms dysregulated in HB.
DNA METHYLATION IN HEPATOBLASTOMA
DNA methylation is a process mainly regulated by two enzymes, namely DNA methyltransferases (DNMTs) and ten-eleven translocation (TET) enzymes, which are responsible for the transfer and elimination of methyl groups CpG dinucleotides of DNA, respectively[22,23]. This epigenetic modification has usually been associated with gene downregulation. However, the outcome of DNA methylation depends on the methylated region within a gene. When the methylation occurs in the CpG islands of the gene promoter, it results in gene expression decay. On the contrary, hypermethylation of low-methylated CpG islands and methylation in gene bodies are associated with the upregulation of gene expression[23,24] [Figure 1].
HB shows a global pattern of DNA hypomethylation together with hypermethylation of specific CpG islands[18,25]. Even though HBs display increased expression of oncogenes, there are few studies demonstrating that hypomethylation is the cause. However, several reports highlight that in HB DNA hypermethylation is usually found in the CpG islands of tumor suppressor gene (TSG) promoters[22]. The first evidence of methylation-associated variations of gene expression in HB was described in 1998[26]. The authors demonstrated that the expression of IGF2 and H19, which are known dysregulated genes in HB[27,28], was altered due to changes in the methylation of their promoters. H19 low expression was associated with increased methylation of the promoter, while IGF2 high expression was linked to a decreased methylation of its promoter[26]. Therefore, in HB as well, this epigenetic modification works both as an inactivator of TSGs and an activator of oncogenes [Table 2][22].
Genes regulated by DNA methylation in HB
| DNA methylation | Gene | Gene expression | Ref. |
| Hypermethylation | H19 | ↓ | [26,28] |
| P16 | ↓ | [34] | |
| RASSF1A | ↓ | [48-50] | |
| RASSF5 | ↓ | [52] | |
| IGFBP3 | ↓ | [64,68] | |
| SOCS1 | ↓ | [69,77] | |
| APC | Unknown | [77] | |
| HHIP | ↓ | [83,84] | |
| MT1G | ↓ | [77] | |
| NDRG2 | ↓ | [88] | |
| SFRP1 | ↓ | [95] | |
| GPR180 | Unknown | [97] | |
| MST1R | Unknown | [97] | |
| OCIAD2 | Unknown | [97] | |
| PARP6 | Unknown | [97] | |
| NNMT | ↓ | [98] | |
| CSF3R | Unknown | [101] | |
| Hypomethylation | IGF2 | ↑ | [26,28] |
| AFP | ↑ | [105] | |
| KLK4 | ↑ | [109] | |
| MYCN | ↑ | [112] |
P16/INK4a is a tumor suppressor gene that inhibits proliferation through G1 cell-cycle arrest. It binds to cyclin-dependent kinase 4 (CDK4) and 6 (CDK6) and inhibits their recruitment of cyclin D1[29]. This interaction abrogates CDK4/6-dependent phosphorylation of retinoblastoma (Rb) family members, which triggers cell-cycle arrest[30]. The hypermethylation of P16/INK4a promoter has been described as a crucial event to repress its expression in several cancers such as hepatocellular carcinoma (HCC)[31], non-small cell lung carcinoma[32], and gastric cancer[33]. However, the role of P16/INK4a hypermethylation in HB remains controversial. A study reported increased methylation of P16/INK4a gene followed by reduced p16 protein expression in 12 of 24 HB tissues, suggesting that p16 is an important tumor suppressor gene in HB formation[34]. On the contrary, another study analyzing the methylation status of various tumor suppressor genes in HB did not find evidence of hypermethylation of the P16/INK4a promoter in 27 cases[35]. Further studies are needed to assess the role of p16 in hepatoblastoma tumorigenesis.
RASSF1A (Ras-association domain family 1, isoform A) is one of the best-characterized TSGs displaying methylation-mediated regulation in cancer. It is involved in apoptosis, cytoskeletal organization, cell cycle, and DNA repair[36], mainly forming complexes with other proteins[37]. It may interact with MST-1[38] and MOAP-1[39,40] to trigger apoptosis, and it can associate with p120E4F to inhibit G1-S phase transition[41]. Methylation of RASSF1A gene promoter drives its repression in cancer[36,37]. Indeed, hypermethylation of RASSF1A has been reported in liver tumors such as HCC[42-44], cholangiocarcinoma (CCA)[45-47], and HB[35,48-50]. The rate of methylation of RASSF1A in hepatoblastoma is 33.8%-44.3%[48-50] and is associated with metastasis and poor outcome of HB patients[48-50]. Moreover, it predicts low responsiveness to chemotherapy and could serve as a prognostic tool to identify patients who are liable to suffer recurrence[49]. Finally, a study demonstrated that RASSF1A methylation levels increase in parallel with the histological subtype (normal liver < fetal type < embryonal type)[50]. It is possible to measure RASSF1A methylation levels in plasma cell-free DNA from HCC patients[51], a liquid biopsy which could also be applied in HB studies. Therefore, RASSF1A methylation may be a prognostic factor as well as a therapeutic target for HB. RASSF5, another gene from the RASSF family, has also been implicated in HB, being downregulated in tumor tissues in association with its aberrant methylation[52].
It is well known that the IGF (insulin-like growth factor) pathway is involved in HB development[21]. The activation of IGF signaling stimulates growth and metastasis in numerous human tumors such as myeloma[53], HCC[54], gastric cancer[55], and lung cancer[56], among others. The binding of IGF1/IGF2 to their receptors activates the PI3K-AKT pathway, which promotes cell proliferation and cell survival and inhibits apoptosis[57]. Indeed, several components of the IGF pathway are upregulated in HB tumors, such as IGF1, IGF1R, and IGF2[58]. In some cases, HB is characterized by biallelic expression of IGF2 due to a loss of imprinting associated with hypomethylation of the gene, which leads to IGF2 overexpression[28,59]. IGF-binding proteins (IGFBPs) inhibit this pathway through the interaction with IGFs, which hinders their interaction with the IGF receptors[60]. In line with this, the levels of IGFBP2 and IGFBP3 are downregulated in HB tissues[61]. A study reported that IGFBP3 enhances p53-dependent apoptosis in several colorectal cell lines in response to DNA damage[62]. Consistently, a previous work showed that IGFBP3 is transcriptionally regulated by p53[63]. According to this, p53-dependent transcription of IGFBP3 may be silenced by methylation of the p53 binding sites in the IGFBP3 promoter in HepG2 cells[64], which is a human HB cell line[65]. The methylation of IGFBP3 promoter was postulated as a key mechanism mediating its downregulation in several human tumors, including HCC, gastric cancer, and colorectal cancer[66,67]. Indeed, the downregulation of IGFBP3 expression is associated in part with hypermethylation of its promoter in HB tissues and cell lines, which is rescued after a treatment with the demethylating agent 5-Aza-2’-deoxycytidine (also known as decitabine)[68]. This methylation-dependent downregulation of IGFBP3 seems to be a late event in the development of HB and is associated with high-risk HB characterized by metastatic and vascular-invasive features. Following this, strategies that restore IGFBP3 expression have long-term effects on cell growth, cell migration, and apoptosis in HB cell lines[68], and therefore may constitute potential therapeutic approaches for HB.
The suppressor of cytokine signaling 1 (SOCS1) has also been implicated in HB[69]. SOCS1 is a negative regulator of the JAK-STAT signaling pathway[69], whose activation drives cancer growth and metastasis through the induction of STAT3 transcription factor activity[70]. However, the role of SOCS1 in cancer development is controversial. SOCS1 may act as a tumor suppressor gene in several tumors such as HCC, preventing the activation of STAT3, and melanoma, in which it inhibits STAT3-induced upregulation of MMP2, VEGF, and bFGF, thereby blocking brain metastasis[71,72]. On the contrary, SOCS1 is overexpressed in human breast, epidermal, and brain cancers[73,74]. The best characterized oncogenic activity of SOCS1 is its crucial role regulating the resistance to the anti-cancer effects of IFNs by blocking their capacity to induce the immune response[75,76]. Although SOCS1 is regulated by histone modifications in HCC[71], its expression is mainly regulated by promoter hypermethylation in various cancers, including 39%-60% of HCCs, 50% of pancreatic cancers, and 75% of melanomas, among others[73]. In HB, the SOCS1 gene promoter is hypermethylated, which is associated with a decreased expression[49,69,77]. Even though SOCS1 has a dual role in cancer, in HB, it seems to be acting as a tumor suppressor gene[69,77].
As aforementioned, Wnt/β-catenin is the most important pathway dysregulated in HB due to the high rate of mutations of CTNNB1 and APC genes[12,16,78]. APC constitutes part of the repressive complex in charge of the degradation of β-catenin in normal conditions[79]. However, in some HBs, downregulation of APC leads to the activation of β-catenin due to lack of degradation of the protein. This occurs as a consequence of APC gene mutation or deletion or its hypermethylation[22]. Indeed, a study analyzing the methylation status of 25 genes in HB reported that APC is hypermethylated in 30% of HB tissues[77]. Therefore, it is possible that APC mutation or methylation-dependent downregulation might be associated with the activation of the Wnt/β-catenin pathway in β-catenin wild-type HBs[16]. Therefore, experimental studies are needed to assess the importance of APC gene methylation in HB development.
The hedgehog (Hh) signaling pathway is a critical mediator of embryonic liver development and liver regeneration[80]. In normal conditions, PTCH receptor inhibits the G protein-coupled receptor Smo in the absence of Hh ligands. After the binding of Hh protein to PTCH, Smo releases GLI proteins from a complex, which translocate to the nucleus to act as transcription factors. GLI’s target genes are mainly involved in cell proliferation, stem cell-mediated regeneration, cell survival, and EMT. Indeed, the Hh pathway is often aberrantly activated in several cancers such as basal cell carcinoma, gastrointestinal cancer, pancreatic cancer, malignant lymphoma, and HCC[81]. The expression of various components of the pathway is controlled in part by epigenetic mechanisms, principally DNA methylation. This includes hypermethylation of the promoters of hedgehog interacting protein (HHIP) and PTCH genes, as well as hypomethylation of the SHH (Sonic hedgehog ligand) promoter[81]. The hypomethylation of the SHH promoter has been demonstrated to be associated with its increased expression in gastric cancer[82]. Moreover, HHIP, which is a known negative regulator of Hh signaling, is downregulated in HCC and HB cell lines, as well as HCC tissues, and is associated with hypermethylation of its promoter. Accordingly, treatment of HepG2 and HuH6 HB cell lines with a demethylating agent increased the expression of HHIP in parallel with a decrease in GLI1 and PTCH mRNA levels[83]. This is in accordance with a study showing that the HHIP promoter is hypermethylated in 26% of HB cases. Consequently, HHIP is downregulated in HB, and its expression can be rescued upon treatment with the demethylating drug decitabine[84]. Considering the importance of the Hh signaling pathway and the epigenetic regulation of its components, it could also constitute an attractive therapeutic target in HB.
The epigenetic repression of other TSGs has also been reported in HB. Hypermethylation of metallothionein-1G gene (MT1G) promoter is linked with poor prognosis in HB patients, suggesting that it could be therapeutically targeted[77]. The N-myc downstream regulated gene 2 (NDRG2) is a known TSG that acts as a negative regulator of the Wnt/β-catenin pathway, downregulating the expression of cyclin D1 and c-myc induced by the β-catenin pathway[85-87]. In line with this, HB tumors are characterized by the downregulation of NDRG2, which is associated with the hypermethylation of its promoter. Indeed, low NDRG2 expression is linked to tumor metastasis, while high NDRG2 expression correlates with an increased survival rate[88]. The secreted frizzled-related protein 1 (SFRP1) is another known antagonist of the Wnt pathway, which directly binds to WNT proteins or the frizzled receptor, and is commonly downregulated in several cancers such as colorectal cancer, bladder cancer, non-small cell lung cancer, gastric cancer, and HCC[89]. The major mechanism driving the downregulation of SFRP1 in these tumors is the hypermethylation of its promoter[90-94], which also occurs in HB tissues and cell lines[95]. Consistently, overexpression of SFRP1 hampers the Wnt/β-catenin pathway in human HB cell lines, resulting in inhibition of tumor cell growth, colony formation, and migration, whereas SFRP1 knockdown stimulates tumor cell growth[95,96]. Moreover, hypermethylation of the promoters of G protein-coupled receptor 180
Most of the studies on HB have focused their attention on overexpressed proteins such as glypican 3 (GPC3)[16,102], delta-like 1 homolog (DLK-1)[16], NFE2L2[12], and LIN28B[103]. As mentioned above, the increased expression of certain proteins may be associated with a reduced methylation of their respective genes. Indeed, tumorigenesis is also characterized by hypomethylation of oncogenes[22]. Alpha-fetoprotein has been widely used as a biomarker for diagnosis and prognosis of HCC and HB[2,104], and it is one of the bases of the tumor staging system[3]. A study reported that increased AFP mRNA expression is associated with hypomethylation of the gene promoter, suggesting that DNA methylation modulates AFP expression in HB[105]. Moreover, kallikrein-related peptidase 4 (KLK4) is found to be upregulated in several types of cancer, including prostate cancer[106], ovarian cancer[107], and colorectal adenocarcinoma[108]. KLK4 gene expression is also upregulated in HB in association with aberrant promoter hypomethylation[109]. The MYCN gene has an important function in cancer development and its upregulation occurs in several tumors, including lung and breast cancer, medulloblastoma, and neuroblastoma[110]. Abnormal activation of MYCN expression induces cell cycle progression and inhibits apoptosis[111]. In line with this, a recent study demonstrated that MYCN is upregulated in HB tissues due to hypomethylation, correlating with a more undifferentiated tumor histology. Consistently, MYCN knockdown and MYCN inhibitors block the growth of HB cell lines[112]. However, further studies are required to fully understand if DNA methylation is the main modulator of MYCN gene expression alterations in HB.
As introduced above, DNA methylation is regulated by enzymes that transfer or eliminate methyl groups in the CpG dinucleotides within the DNA sequence. The expression pattern of these enzymes can change in certain situations such as cancer. In fact, the expression of DNMT1, which is in charge of the maintenance of the DNA methylation pattern during mitosis, has been reported to be upregulated in hepatic fibrosis and HCC[113,114]. It has also been demonstrated that DNMTs are upregulated in others types of cancer such as gastric cancer[115], prostate cancer[116], CCA[117], and pancreatic cancer[118]. Indeed, DNMT1, DNMT3A, and DNMT3B are known to also be upregulated in HB tumors[18,119]. On the other hand, evidence suggests that TETs, enzymes involved in DNA demethylation, may act as tumor suppressors in some solid tumors such as melanoma, prostate, breast, and lung tumors[120]. However, it has been demonstrated that hypoxia, which promotes tumor malignancy, induces the upregulation of TET enzymes in a HIF-1α-dependent manner in HepG2 HB cells, resulting in an increase in 5-hydroxymethylcytosine (5hmC) levels in DNA[121]. Indeed, recent studies reported that TETs are upregulated in HB tissues in comparison with non-tumoral tissues[18,119]. TET1 and TET3 are overexpressed in Epi-CB tumors, which are associated with poor prognosis[18]. In line with this, TET1 and TET2 upregulation leads to an enrichment in 5hmC content and global DNA hypomethylation. Intriguingly, low 5hmC levels are associated with poor overall survival in HB patients[119]. This is in accordance with the role described for TETs in others tumors such as HCC, whose 5hmC levels positively correlate with overall survival of the patients[122]. Therefore, the role of TETs in HB requires further investigation to understand if they act as tumor suppressors or tumor promoters.
Finally, UHRF1 is a master regulator of epigenetic modifications, acting as a coordinator of histone modifications and DNA methylation. UHRF1 helps in the maintenance of DNA methylation during cell proliferation through the recruitment of DNMT1 to the hemi-methylated DNA[123]. Indeed, UHRF1 has been described as a crucial mediator in the DNA methylation-mediated silencing of TSGs in several cancers such as breast cancer[124], colorectal cancer[125], HCC[126], bladder cancer[127], and gastric cancer[128]. Furthermore, it was demonstrated that UHRF1 is upregulated and promotes the silencing of TSGs in HB[119,129]. Specifically, a recent study reported that the UHRF1-DNMT1 complex silences the expression of HHIP, IGFBP3, and SFRP1, which have been previously described as important TSGs in HB[129].
THE ROLE OF HISTONE MODIFICATIONS IN HB
Histone modifications are a highly dynamic process regulated by chromatin-modifying enzymes in a cell context-dependent manner. These modifications modulate the chromatin structure, leading to changes in gene transcription, which may be transcription activation or repression. The outcome may result from alterations in the interaction between and within nucleosomes. However, there are proteins with specialized domains that are able to bind to these modifications and recruit other chromatin modifiers, which act as effectors of the modification. Histones are a target of several post-translational modifications including methylation, acetylation, phosphorylation, SUMOylation, ADP ribosylation, and O-GlcNAcylation, among others, that may be involved in cancer[130].
Even though histone modifications are well-characterized players in tumor development and progression, little is known about their role in HB. The first evidence on the role of these epigenetic modifications in HB was from studies focused on histone acetylation[131,132] [Figure 1]. Histone acetylation is regulated by histone acetyltransferases (HAT) and histone deacetylases (HDACs), which drive the deposition and elimination of acetyl groups, respectively. The histones’ acetylation weakens their interaction with DNA, thereby enhancing gene transcription. On the contrary, histone deacetylation leads to gene transcription repression due to the reinforcement of this interaction. Dysregulation of the histone acetylation process has been broadly described in numerous cancers[133]. Indeed, resveratrol, a natural histone deacetylase inhibitor[134], blocks cell proliferation and induces p53-dependent apoptosis in HepG2 HB cells[132,135]. HDACs are commonly dysregulated in several cancers and represent an important therapeutic target[133]. Studies have reported overexpression of HDAC1 and HDAC2 in HB tumors and cell lines. In line with this, pharmacological inhibition of HDACs hampers cell proliferation and induces apoptosis of HepG2, HuH6, and HepT1 HB cells. Furthermore, HDAC inhibition enhances cisplatin sensitivity, thereby suggesting a marked synergistic effect with the routine chemotherapy for HB. Therefore, HDAC inhibition may represent an important therapeutic strategy for high-risk HB tumors with cisplatin-resistant features[136]. As previously mentioned, erasers (HDACs) and writers (HATs) are the effectors of the epigenetic modification, whereas other proteins act as readers. Acetylated histones are recognized by proteins that contain bromodomains and extraterminal (BET) protein-protein interaction motifs. This family includes BRD2, BRD3, and BRD4, which are the best-characterized BET proteins in cancer. Consequently, BET inhibitors are being tested in clinical trials for numerous cancers such as multiple myeloma, breast cancer, non-small cell lung cancer, and pancreatic ductal adenocarcinoma[137]. In fact, BET inhibition may also show therapeutic potential for the treatment of HB, as shown by the inhibitory effects of BRD4 suppression in the migration and proliferation of HepG2 cells[138]. Indeed, JQ1, which is an inhibitor of BRD4, induces cell-cycle arrest of HB cell lines through the downregulation of MYCN gene and protein expression and hampers the growth of HB patient-derived organoids[112,139]. Therefore, targeting the acetylation/deacetylation process may be a useful strategy for HB treatment, although the side effects of HDAC and BET inhibition must be considered due to their impact on global gene expression. In this regard, cancer-tissue targeted therapies should be implemented to avoid toxicity[23,137].
Histone methylation also plays an important role in the progression of HB. This epigenetic modification is regulated by histone methyltransferases (HMTs) and histone demethylases (HDMTs), and the outcome depends on the specific position of the lysine residue and the extent of the methylation. Indeed, trimethylation of lysine 27 of histone 3 (H3K27me3) involves gene silencing, whereas H3K4me3 results in transcriptional activation[23]. Histone methylation often shows an aberrant pattern in the context of cancer and frequently results in the activation of proto-oncogenes and the inhibition of TSGs[140]. Enhancer of zeste homolog 2 (EZH2) is part of the polycomb repressive complex 2 (PRC2), which is involved in the methylation of H3K27 to induce transcriptional silencing and plays a crucial role in a variety of cancers[141]. EZH2 is overexpressed in HB tumors and is associated with metastasis and poor survival. According to this, shRNA-mediated silencing of EZH2 induces cell-cycle arrest and reduces cell proliferation of HB cell lines[142,143]. Finally, HB tumors generally display downregulated TSGs expression, which is driven by DNA methylation together with histone methylation alterations. In line with this, knockdown of UHRF1 enhances the expression of important TSGs in hepatoblastoma (HHIP, IGFBP3, and SFRP1) followed by the loss of repressive histone marks in these loci[129]. Histone methylation is a crucial event in the regulation of gene expression in cancer and could represent a potential therapeutic target in HB.
THE ROLE OF NON-CODING RNAS IN HB
Only 2% of genomic DNA is eventually translated into proteins, and most of the transcriptome is dedicated to the expression of non-coding RNAs[144]. Non-coding RNAs comprise microRNAs (miRNA), small-nucleolar RNAs (snoRNAs), small interferent RNAs (siRNAs), Piwi-interacting RNAs (piRNAs), long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs). Even though lncRNAs are the most abundant non-coding RNAs in mammalians, miRNAs remain the best characterized non-coding RNAs[23,144] [Figure 1].
miRNAs IN HB
miRNAs are critical modulators of the cellular response to environmental stress through the regulation of the gene expression. miRNAs form complexes with target mRNAs to block their translation through the interaction with their 3’-untranslated region (3’-UTR), thereby producing a decrease in the protein levels. The regulation of gene expression by miRNAs in response to environmental factors is a key event in cancer[145]. Indeed, miRNAs can act as oncogenes (also called “oncomiRNAs”) or tumor suppressors (“oncosuppressors miRNAs”), and their dysregulation is associated with tumor formation, progression, and metastasis[146].
Numerous studies demonstrate that miRNAs are involved in HB as oncogenes and tumor suppressors and show a marked differential expression. A recent study reported the altered expression of miRNAs in HB, identifying 13 miRNAs differentially expressed between tumor and non-tumor tissues[147]. In line with this, another study highlighted that differential expression of miRNAs may exist among HB subtypes. The study shows that miR-18a levels are higher in embryonal HB, which entails the worst prognosis, than in the fetal subtype. Moreover, while miR-122 expression does not show any differences between non-tumor tissue and fetal HB, it is markedly downregulated in the embryonal subtype in comparison to surrounding liver tissue[148]. The authors showed for the first time that the expression of various miRNAs is correlated with the overall survival of patients with epithelial type HB[148]. It was also demonstrated that embryonal HBs display increased levels of miR-19b in comparison with fetal HBs[149]. miRNAs that are upregulated or downregulated are shown in Table 3.
miRNA expression in HB tumors
| miRNA expression | miRNA | Ref. |
| Upregulated | Let-7a, miR-125a-5p, miR-15b, miR-181a, miR-199a-5p, miR-214, miR-10a, miR-10b | [147] |
| miR-221 | [148] | |
| miR-21 | [148,165] | |
| miR-492 | [152,153] | |
| miR-483 | [161] | |
| Downregulated | miR-885-5p, miR-148a, miR-30b, miR-455-3p | [147] |
| miR-122 | [147,148] | |
| miR-17-5p, miR-195, miR-210, miR-214, miR-222, miR-224 | [148] | |
| miR-17 | [149] | |
| miR-125b | [159] | |
| miR-4510, miR-203-3p, miR-548aa, miR-376-3p | [162] | |
| miR-203 | [167] | |
| miR-217 | [168] | |
| miR-503-5p | [169] | |
| miR-29a/b/c-3p | [170] |
miRNAs may function as inhibitors of several genes that affect the growth and progression of HB. PLAG-1 is an important transcription factor upregulated during HB development, which in turn regulates the expression of several HB biomarkers such as IGF2 and keratin 19 (KRT19)[150,151]. According to this, overexpression of KRT19 is associated with that of miR-492, which originates from the KRT19 coding sequence[152]. Consistently, miR-492 overexpression promotes cell proliferation, migration, and invasion through the inhibition of CD44 expression, thereby enhancing the metastatic features of the tumor in patients with decreased survival[153]. DKK3 is a negative regulator of the Wnt/β-catenin pathway whose role in carcinogenesis is controversial. Although it is downregulated in some tumors such as breast cancer and papillary thyroid carcinoma[154,155], it is upregulated in other cancers such as glioma, colorectal carcinoma, HCC, and dedifferentiated HB[156-158]. In HB, DKK3 is upregulated due to the GATA4-mediated transcriptional repression of miR-125b, which targets DKK3 mRNA. In fact, overexpression of miR-125b induces cell-cycle arrest and inhibits the migration of HB HuH6 cells, suggesting that miR-125b may act as a tumor suppressor miRNA in HB[159]. miR-125b downregulation occurs in parallel with the decreased expression of miR-100 and Let-7a-2 and the upregulation of miR-371-3, which constitute a four-miRNA signature in undifferentiated aggressive HBs[160]. As previously mentioned, IGF2 and GPC3 are known oncogenes upregulated in HB. In line with this, IGF2 overexpression correlates with the expression of miR-483, which derives from the intronic sequence of IGF2 and may act as a biomarker of poor outcome in HB patients[161]. However, miR-4510, which is downregulated in HB tumors, may target GPC3 through direct binding to its mRNA, thereby decreasing GPC3 expression. Consistently, miR-4510 overexpression stimulates cell apoptosis and hampers tumor growth through the inhibition of the Wnt/β-catenin pathway, suggesting a role for this miRNA as a tumor suppressor in HB[162]. Apoptosis-stimulating p53 protein-2 (ASPP2) supports the apoptotic role of p53 through the formation of a complex and subsequent induction of proapoptotic genes[163]. In fact, high levels of ASPP2 are associated with a better prognosis in HCC patients[164]. It has been recently demonstrated that ASPP2 is downregulated in HB due to its targeted inhibition by miR-21, whose levels are found to be increased in this tumor. Moreover, siRNA-mediated inhibition of ASPP2 promotes migration and invasion of HB HuH6 cells. Indeed, treatment with an miR-21 inhibitor induces ASPP2 levels in parallel with a decreased tumor growth in HuH6 cells mouse xenografts[165]. Finally, several miRNAs in the 14q32.2 locus included in the DLK1-DIO3 cluster are upregulated in HB, including lung metastatic HB tumors[166] [Table 3][147-149,152,153,159,161,162,165,167-170]. Furthermore, the overexpression of these ncRNAs correlates with the outcome of the patients, suggesting the utility of miRNAs in diagnosis and prognosis to improve their management[18].
Exosomal microRNAs can be detected in serum of cancer patients and may represent important biomarkers for prognosis and clinical management of the disease. Dysregulation of the levels of serum exosomal miRNAs has been documented in several pediatric malignancies such as neuroblastoma, osteosarcoma, rhabdomyosarcoma, and leukemias[171]. Following this, a recent study found that the serum levels of miR-34 family members are significantly reduced in HB patients in comparison with healthy controls, suggesting that exosomal miRNAs could also be used as prognosis factors for patients with HB[172].
LONG NON-CODING RNAs IN HB
lncRNAs are defined as RNAs that are > 200 nucleotides long. They are involved in the regulation of numerous biological events such as epigenetic modifications, transcriptional regulation, post-transcriptional regulation, translational regulation, and post-translational regulation. Their mechanisms of action are based on their capacity to bind to DNA, mRNA, proteins, and even miRNAs. lncRNAs have been characterized as important dysregulated factors in the development and progression of cancer[173]. Indeed, certain lncRNAs may function as oncogenes such as MALAT1 in osteosarcoma, SNHG6 in gastric cancer, or HNF1A-AS1 in colon cancer, while others may act as tumor suppressors such as CANT1 in melanoma, GAS5 in HCC, and PCAT29 in prostate cancer[174].
Genome-wide analysis of HB tissues showed a dysregulation in the expression of lncRNAs that was associated with the differential expression of several genes, suggesting an important role for lncRNAs in HB tumorigenesis[175]. Moreover, in a subcutaneous xenograft model of HB in nude mice undergoing incomplete radio frequency ablation (RFA), a model of residual disease, changes in the expression of specific lncRNAs were observed in comparison to untreated mice, suggesting that lncRNAs may also serve as potential markers of residual HB[176]. H19 is an imprinted gene that is maternally expressed whose role in cancer remains controversial. It has been shown to be upregulated and to act as an oncogene in many solid tumors[177]. However, as previously mentioned, H19 is known to be downregulated in HB due to hypermethylation of its promoter. Nevertheless, mechanistic studies are lacking to better understand the role of H19 in this tumor[26]. Taurine-upregulated gene 1 (TUG1) is a lncRNA with altered expression in cancer. It can act as a tumor suppressor in several tumors such as glioma[178], non-small cell lung cancer[179], and breast cancer[180]. However, it may also function as an oncogene in other cancers such as osteosarcoma[181], colon cancer[182], and HCC[183]. In line with this, it has been demonstrated that TUG1 is upregulated in HB tissues and cell lines and its inhibition blocks tumor growth and angiogenesis in vivo. Mechanistically, TUG1 blocks miR-34a-5p-mediated silencing of vascular endothelial growth factor A (VEGFA), thereby increasing its expression and allowing vascularization of the tumor[184]. Furthermore, colorectal neoplasia differentially expressed (CRNDE) is characterized as an oncogenic lncRNA that is upregulated in several cancers such as glioma, HCC, lung cancer, breast cancer, and gastric cancer. It is involved in a variety of biological processes that are crucial for tumor formation and progression, including cell proliferation, inhibition of apoptosis, invasion and migration, inflammatory response, chemoresistance, and radioresistance[185]. According to this, CRNDE has been found to be upregulated in HB tumors and cell lines. Consistently, CRNDE knockdown inhibits tumor growth and angiogenesis in vivo and reduces cell proliferation and viability in vitro in HuH6 cells through the activation of the mTOR pathway, which has a key role in cell growth and proliferation[186]. Moreover, CRNDE also regulates the expression of VEGFA through the inhibition of miR-203, which is downregulated in HB tissues, thereby promoting tumor angiogenesis and growth[167]. As previously mentioned, the hippo-YAP pathway is frequently activated and is a key signaling pathway in the tumorigenesis of HB. Following this, LINC01314 is a lncRNA that has been reported to be downregulated in HB. LINC01314 blocks the activation of the YAP pathway via promoting the expression of MST1 and subsequent phosphorylation of LATS1 and YAP, which hampers the translocation of YAP into the nucleus. In accordance, overexpression of LIN01314 decreases cell proliferation and migration of HepG2 cells, thereby proposing LINC01314 as a novel TSG in HB[187]. OPA-interacting protein 5 antisense 1 (OIP5-AS1) is a lncRNA broadly studied in several cancers. A study reported that exosomal transfer of OIP5-AS1 between cells enhances trastuzumab resistance in breast cancer via sponging miR-381-3p, blocking miR-381-3p-mediated inhibition of HMGB3, which is associated to cell growth and progression in breast cancer[188]. Moreover, it has been demonstrated that OIP5-AS1 may also act as sponge against miR-186-5p in pancreatic cancer and HB[189,190]. In pancreatic cancer, the absence of miR-186-5p due to the inhibition of OIP5-AS1 causes the upregulation of nerve growth factor receptor (NGFR), which is associated with the progression and metastasis of pancreatic cancer[190]. However, in HB, OIP5-AS1 is upregulated in tumor tissues and correlates with poor prognosis of the patients. In this context, OIP5-AS1-mediated inhibition of miR-186-5p induces the expression of ZEB1, which is linked to cell proliferation, metastasis, and EMT in various HB cell lines[189]. The sponging phenomenon is a common interaction between lncRNAs and miRNA, and recent experimental studies further demonstrate that this event is common in HB tumors. Indeed, lung cancer-associated transcript 1 (LUCAT1) and zinc finger antisense 1 (ZFAS1) are lncRNAs that are able to sponge and inhibit miR301b and miR-193a-3p, respectively[191,192]. LUCAT1 levels are increased in HB tumors and are associated to a poor overall survival. It blocks miR-301b-mediated inhibition of STAT3, which is able to enhance the expression of LUCAT1, thereby perpetuating the feedback loop. Consequently, LUCAT1 activation promotes cell proliferation and migration in HB cells, highlighting the oncogenic role of this lncRNA[191]. ZFAS1 expression is markedly upregulated in HB tissues and cells, which is linked to an aggressive phenotype of the tumor and poor outcome of the patients[192]. The mechanism underlying the function of ZFAS1 is based on the activation of the HGF/c-Met pathway, which is a key pathway in HB development[21,192]. Finally, a very recent study reported that HELIS and LIN01093 are lncRNAs that are downregulated in HB tumors, suggesting a possible role as biomarkers of this tumor[193].
CIRCULAR RNAs IN HB
Circular RNAs (circRNAs) were considered as “splicing noise” due to the closed conformation constituted by the union of the 5’ caps and 3’ tails. However, further studies demonstrated that they are abundant and highly conserved, thereby discarding the possibility of being mere accidental byproducts. circRNAs originate from variable splicing of pre-mRNA, which is followed by an RNA cyclization[194]. These ncRNAs are involved in several biological functions as a consequence of their capacity to function as miRNAs sponges[195], translate into proteins[196], bind to proteins[197], and regulate mRNA splicing[198]. Indeed, circRNAs are dysregulated in numerous human diseases including cancer, and there are recent studies describing the upregulation or downregulation of various circRNAs in several tumors[194]. For instance, circRNAs hsa-circ-002059 and circFAM169A are downregulated in gastric cancer and bladder cancer, respectively[199,200]. On the contrary, circPTK2 and circTCF25 are upregulated in bladder cancer[199]. In line with this, a recently published review describes the role of circRNAs in liver cancer with special focus in HCC[201]. Regarding HB, the first study evaluating the role of circRNAs concluded that there are 421 upregulated circRNAs and 448 downregulated circRNAs in this tumor [Figure 1]. Furthermore, the study found circ-0015756 to be upregulated in HB tissues and metastatic cell lines. In fact, circ-0015756 promotes cell proliferation and invasion via sponging miR-1250-3p, a mechanism that is blocked by siRNA-mediated inhibition of circ-0015756. Consistently, overexpression of miR-1250-3p can mimic the effects of circ-0015756 silencing[202]. Moreover, hsa-circ-0000594 is also upregulated in epithelial HB tumors and is associated with cell viability and proliferation. Accordingly, hsa-circ-0000594 inhibition reduces cell migration and induces apoptosis of HuH6 and HepG2 HB cells. Mechanistically, hsa-circ-0000594 is able to sponge miR-217, whose target is SIRT1 mRNA[168]. Therefore, hsa-circ-0000594 induces the expression of SIRT1 protein, which is implicated as an oncoprotein in several cancer such as ovarian cancer and HCC[203,204], providing evidence of the tumor promoter role of hsa-circ-0000594. circRNAs have also been reported to be implicated in the activation of important pathways in HB such as IGF and JAK/STAT signaling pathways[169,170]. In line with this, circHMGCS1 (hsa-circ-0072391) is upregulated in HB tissues, correlating with poor patient prognosis. This circRNA is able to sponge and inhibit tumor suppressor miR-503-5p, whose target is IGF2 mRNA, thereby increasing IGF2 levels and activating the IGF-PI3k-AKT. The activation of this pathway enhances the expression of the enzyme glutamine synthetase (GLS), which is also upregulated in HB tissues, thereby inducing the production of glutamine[169]. Glutamine metabolism favors tumor growth and adaptation, representing a crucial event in cancer growth[205]. According to this, circHMGCS1 knockdown abrogates cell proliferation and induces apoptosis, accompanied by downregulation of the AKT pathway and a decline in GLS mRNA levels in HuH6 and HepG2 HB cells. Therefore, circHMGCS1 may serve as a diagnostic and prognostic factor, and targeting the circHMGCS1-miR-503-5p axis may represent a novel therapeutic target for the treatment of HB[169]. circ-0043800 (circ-STAT3) is a circRNA that is originated from the sequence of STAT3 and is consistently upregulated in HB tissues and cells. circ-STAT3 induces the expression of STAT3 and GLI2 via sponging miR-29 family members, which are responsible for the inhibition of STAT3 expression. Indeed, STAT3 and GLI2 are also involved in the upregulation of circ-STAT3, thereby establishing a positive feedback loop. In accordance, STAT3 or GLI2 overexpression rescue the suppressive effects of silencing circ-STAT3 on cell proliferation, migration, invasion, and stemness. Furthermore, circ-STAT3 also enhances tumor growth in vivo, whereas circ-STAT3 depletion hampers it, being accompanied by GLI2 and STAT3 downregulation[170]. Finally, circ-RNA CDR1as has been recently reported to promote HB cells proliferation through the sponging of miR-7-5p, which targets and downregulates the transcription factor KLF4. Consequently, KLF4 is activated and enhances the stem cell phenotype of HB cells, thereby promoting HB cell proliferation. Indeed, CDR1as knockdown induces miR-7-5p levels in parallel with the downregulation of KLF4[206]. In view of all these relevant interactions, it is important to fully understand the role of circRNAs in the development of HB, as they may also be interesting prognostic and diagnostic factors as well as therapeutic targets.
THE ROLE OF EPITRANSCRIPTOMICS IN HB
In addition to DNA and histones, RNAs can also be targets of several post-transcriptional modifications, which impact their stability, fate, and function. These modifications are highly dynamic and reversible, suggesting they may be involved in important regulatory functions. The most prevalent RNA modification is the methylation of adenosine residues in the N6 position, resulting in N6-methyladenosine (m6A), which constitutes 0.1%-0.4% of the total adenosine nucleotides in mammals. This process is tightly regulated by the activity of writer and eraser enzymes. The writer activity is carried out by a complex mainly constituted by methyltransferase-like 3 (METTL3), METTL14, and Wilms’ tumor 1-associating protein (WTAP). On the contrary, the eraser activity is accomplished by fat mass and obesity-associated protein (FTO) and alkB homolog 5 (ALKBH5)[207]. The m6A modification is preferably deposited in the long internal exons and around stop codon, thereby indicating a potential regulatory function in mRNA[208]. However, the recognition of the m6A modification by m6A reader proteins will determine the effected biological function. The most characterized m6A readers are the members of the YT521-B homology (YTH) N6-methyladenosine RNA binding proteins (YTHDFs) and YTH domain containing (YTHDCs) protein families. While YTHDF1 promotes the translation of the target mRNA, YTHDF2, which is the first identified m6A reader, transforms the m6A modification into a degradation signal to induce mRNA decay. In addition, YTHDF3 can contribute to either the translation or the degradation of mRNA. Furthermore, m6A readers can also interact with splicing factors such as in the case of YTHDC1. The dysregulation of the m6A balance is frequently associated to several cancers, such as lung cancer, breast cancer, and liver cancer, and other tumors[209,210].
The first evidence of the role of RNA methylation in HB was reported in 2018. The study described that METTL3 knockdown reduces cell proliferation, colony formation, and migration of HepG2 cells[211]. In line with this, a recent study demonstrated that METTL3 is upregulated in HB cell lines and HB tissues, correlating with recurrence and poor outcome, and is associated with aberrant m6A mRNA methylation. Moreover, the authors also found that METTL3 upregulation is accompanied by increased expression of WTAP, FTO, and YTHDF2 [Figure 1]. Accordingly, METTL3, WTAP, FTO, and YTHDF2 siRNA-mediated silencing suppresses cell proliferation and colony formation in parallel with an induction of apoptosis in HepG2 cells. Mechanistically, METTL3 expression increases in parallel with CTNNB1 expression due to the increased m6A methylation of the CTNNB1 mRNA, which enhances its stability. Indeed, METTL3 knockdown reduces the m6A modification and the expression levels of the CTNNB1 mRNA. Furthermore, METTL3 expression knockdown reduces tumor growth in HuH6 mouse subcutaneous xenografts[212]. It has been recently reported that METTL3 expression is in part regulated by miR-186, which is downregulated in HB tumors. miR-186 overexpression reverses the expression of METTL3 and inhibits aggressiveness of HB cells in vitro and in vivo[213]. Finally, it has been shown that WTAP variants may be associated with the development of HB[214]. Further studies are required to shed light on the role of the epitranscriptomics in HB formation and progression.
RNA EDITING
RNA editing is a post-transcriptional mechanism based on the modification of the mRNA sequence in a specific, dynamic, and flexible way. These changes may result in missense codon changes, alteration of alternative splicing, and modification of regulatory RNAs. The outcomes of these alterations can be similar to genomic mutations[215]. This process is mainly regulated by the adenosine deaminase acting on RNA (ADAR) family of enzymes, which mediate the conversion of adenosine to inosine (A-to-I editing)[215]. In the context of cancer, a dysregulation in the RNA editing process has been reported in several tumors, showing elevated A-to-I editing levels in parallel with an increased expression of ADAR1 enzyme[216]. Indeed, a recent study showed an imbalance in ADAR1 and ADAR2 enzyme expression in HB tumors, accompanied by global RNA hypoediting. However, the authors also reported specific hyperediting of the BLCAP transcript, which is a highly conserved gene with tumor suppressor activities[18,217]. Despite the fact that this field requires deeper understanding, the study of this new epigenetic mechanism could contribute to the understanding of HB development and the design of novel “epidrugs” for the treatment of this and other tumors.
CONCLUSION
The application of high-throughput analyses has produced significant advances in the understanding of the molecular features of HB. These studies, together with more targeted approaches, have identified a number of epigenetic mechanisms that can significantly contribute to the emergence and progression of this pediatric tumor. Harnessing this knowledge may deliver new therapeutic opportunities for HB. Indeed, small molecules that inhibit epigenetic effectors or interfere with epigenetic readers, the so-called “epidrugs”, are actively being developed[218]. On the other hand, interfering with oncogenic non-coding RNAs or delivering tumor-suppressing non-coding RNAs to tumoral cells is also feasible[219]. Nevertheless, epigenetic mechanisms are complex and intertwined, and thus the true epigenetic drivers in HB need to be carefully identified to maximize the efficacy of their inhibition while minimizing toxicity. Epigenetic mechanisms are also likely to participate in cancer chemoresistance[220-222]. Therefore, their manipulation could increase the efficacy of chemotherapeutic drugs administered in combination and reduce the burden of their long-term side effects[223]. Interference with epigenetic mechanisms outside tumor cells may also entail toxicity. In this regard, cancer cell-targeted drug delivery strategies are actively pursued for epigenetic drugs that are administered systemically[224,225]. Altogether, the evidence put forward in this review suggests that the dysregulation of epigenetic mechanisms play an important role in the pathogenesis of HB, and despite its limitations this field holds promise for the development of new therapies for this tumor. Nevertheless, to reach this goal, more fundamental research is necessary.
DECLARATIONS
AcknowledgementFigure has been created with the BioRender program (BioRender.com).
Authors’ contributionsDrafted the manuscript: Clavería-Cabello A
Revised the draft: Arechederra M, Berasain C
Produced the final version: Fernández-Barrena MG, Ávila MA
Availability of data and materialsNot applicable.
Financial support and sponsorshipWork in the authors’ laboratory is supported by: CIBERehd; grant PI19/00163 from Instituto de Salud Carlos III (ISCIII) co-financed by “Fondo Europeo de Desarrollo Regional” (FEDER) “Una manera de hacer Europa”; grants PID2019-104878RB-100/AEI/10.13039/501100011033 and PID2019-104265RB-100/AEI/10.13039/501100011033, from FEDER/Ministerio de Ciencia, Innovación y Universidades-Agencia Estatal de Investigación; AECC LAB 2020 project: Novel epigenetic approaches for the treatment of hepatic and digestive cancers, from Spanish Association Against Cancer (AECC); Fundación Eugenio Rodríguez Pascual; Fundación Mario Losantos, Fundación M. Torres; POSTD18014AREC post-doctoral fellowship from AECC to MA, and Ramón y Cajal Program contract RYC-2018-024475-1 to MGFB; FIMA-AC pre-doctoral fellowship to AC-C. The generous support of Mr. Eduardo Avila is acknowledged.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Feng J, Polychronidis G, Heger U, Frongia G, Mehrabi A, Hoffmann K. Incidence trends and survival prediction of hepatoblastoma in children: a population-based study. Cancer Commun (Lond) 2019;39:62.
3. Calvisi DF, Solinas A. Hepatoblastoma: current knowledge and promises from preclinical studies. Transl Gastroenterol Hepatol 2020;5:42.
4. Cairo S, Armengol C, Maibach R, et al. A combined clinical and biological risk classification improves prediction of outcome in hepatoblastoma patients. Eur J Cancer 2020;141:30-9.
5. Aronson DC, Schnater JM, Staalman CR, et al. Predictive value of the pretreatment extent of disease system in hepatoblastoma: results from the International Society of Pediatric Oncology Liver Tumor Study Group SIOPEL-1 study. J Clin Oncol 2005;23:1245-52.
6. Meyers RL, Maibach R, Hiyama E, et al. Risk-stratified staging in paediatric hepatoblastoma: a unified analysis from the Children's Hepatic tumors International Collaboration. Lancet Oncol 2017;18:122-31.
7. Ranganathan S, Lopez-Terrada D, Alaggio R. Hepatoblastoma and pediatric hepatocellular carcinoma: an update. Pediatr Dev Pathol 2020;23:79-95.
8. Perilongo G, Maibach R, Shafford E, et al. Cisplatin versus cisplatin plus doxorubicin for standard-risk hepatoblastoma. N Engl J Med 2009;361:1662-70.
9. Freyer DR, Brock PR, Chang KW, et al. Prevention of cisplatin-induced ototoxicity in children and adolescents with cancer: a clinical practice guideline. Lancet Child Adolesc Health 2020;4:141-50.
10. Marin JJG, Cives-Losada C, Asensio M, Lozano E, Briz O, Macias RIR. Mechanisms of anticancer drug resistance in hepatoblastoma. Cancers (Basel) 2019;11:407.
11. Jia D, Dong R, Jing Y, et al. Exome sequencing of hepatoblastoma reveals novel mutations and cancer genes in the Wnt pathway and ubiquitin ligase complex. Hepatology 2014;60:1686-96.
12. Eichenmüller M, Trippel F, Kreuder M, et al. The genomic landscape of hepatoblastoma and their progenies with HCC-like features. J Hepatol 2014;61:1312-20.
13. Gröbner SN, Worst BC, Weischenfeldt J, et al. ICGC PedBrain-Seq Project. The landscape of genomic alterations across childhood cancers. Nature 2018;555:321-7.
14. Cairo S, Armengol C, De Reyniès A, et al. Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell 2008;14:471-84.
15. Taniguchi K, Roberts LR, Aderca IN, et al. Mutational spectrum of beta-catenin, AXIN1, and AXIN2 in hepatocellular carcinomas and hepatoblastomas. Oncogene 2002;21:4863-71.
16. Sumazin P, Chen Y, Treviño LR, et al. Genomic analysis of hepatoblastoma identifies distinct molecular and prognostic subgroups. Hepatology 2017;65:104-21.
17. Hirsch TZ, Pilet J, Morcrette G, et al. Integrated genomic analysis identifies driver genes and cisplatin-resistant progenitor phenotype in pediatric liver cancer. Cancer Discov ;2021:candisc.
18. Carrillo-Reixach J, Torrens L, Simon-Coma M, et al. Epigenetic footprint enables molecular risk stratification of hepatoblastoma with clinical implications. J Hepatol 2020;73:328-41.
19. Aretz S, Koch A, Uhlhaas S, et al. Should children at risk for familial adenomatous polyposis be screened for hepatoblastoma and children with apparently sporadic hepatoblastoma be screened for APC germline mutations? Pediatr Blood Cancer 2006;47:811-8.
20. Yang A, Sisson R, Gupta A, Tiao G, Geller JI. Germline APC mutations in hepatoblastoma. Pediatr Blood Cancer 2018;65:e26892.
21. Zhang Y, Solinas A, Cairo S, Evert M, Chen X, Calvisi DF. Molecular mechanisms of hepatoblastoma. Semin Liver Dis 2021;41:28-41.
22. Shen G, Shen H, Zhang J, Yan Q, Liu H. DNA methylation in hepatoblastoma-a literature review. Ital J Pediatr 2020;46:113.
23. Claveria-Cabello A, Colyn L, Arechederra M, et al. Epigenetics in liver fibrosis: could HDACs be a therapeutic target? Cells 2020;9:2321.
24. Barcena-Varela M, Colyn L, Fernandez-Barrena MG. Epigenetic mechanisms in hepatic stellate cell activation during liver fibrosis and carcinogenesis. Int J Mol Sci 2019;20:2507.
25. Maschietto M, Rodrigues TC, Kashiwabara AY, et al. DNA methylation landscape of hepatoblastomas reveals arrest at early stages of liver differentiation and cancer-related alterations. Oncotarget 2017;8:97871-89.
26. Li X, Kogner P, Sandstedt B, Haas OA, Ekström TJ. Promoter-specific methylation and expression alterations of igf2 and h19 are involved in human hepatoblastoma. Int J Cancer 1998;75:176-80.
27. Hartmann W, Waha A, Koch A, et al. p57KIP2 is not mutated in hepatoblastoma but shows increased transcriptional activity in a comparative analysis of the three imprinted genes p57KIP2, IGF2, and H19. Am J Pathol 2000;157:1393-403.
28. Honda S, Arai Y, Haruta M, et al. Loss of imprinting of IGF2 correlates with hypermethylation of the H19 differentially methylated region in hepatoblastoma. Br J Cancer 2008;99:1891-9.
30. Goel S, DeCristo MJ, McAllister SS, Zhao JJ. CDK4/6 inhibition in cancer: beyond cell cycle arrest. Trends Cell Biol 2018;28:911-25.
31. Zang JJ, Xie F, Xu JF, et al. P16 gene hypermethylation and hepatocellular carcinoma: a systematic review and meta-analysis. World J Gastroenterol 2011;17:3043-8.
32. Sterlacci W, Tzankov A, Veits L, et al. A comprehensive analysis of p16 expression, gene status, and promoter hypermethylation in surgically resected non-small cell lung carcinomas. J Thorac Oncol 2011;6:1649-57.
34. Shim YH, Park HJ, Choi MS, et al. Hypermethylation of the p16 gene and lack of p16 expression in hepatoblastoma. Mod Pathol 2003;16:430-6.
35. Harada K, Toyooka S, Maitra A, et al. Aberrant promoter methylation and silencing of the RASSF1A gene in pediatric tumors and cell lines. Oncogene 2002;21:4345-9.
36. Malpeli G, Innamorati G, Decimo I, et al. Methylation dynamics of RASSF1A and its impact on cancer. Cancers (Basel) 2019;11:959.
37. Hesson LB, Cooper WN, Latif F. The role of RASSF1A methylation in cancer. Dis Markers 2007;23:73-87.
38. Khokhlatchev A, Rabizadeh S, Xavier R, et al. Identification of a novel Ras-regulated proapoptotic pathway. Current Biology 2002;12:253-65.
39. Law J, Salla M, Zare A, et al. Modulator of apoptosis 1 (MOAP-1) is a tumor suppressor protein linked to the RASSF1A protein. J Biol Chem 2015;290:24100-18.
40. Baksh S, Tommasi S, Fenton S, et al. The tumor suppressor RASSF1A and MAP-1 link death receptor signaling to Bax conformational change and cell death. Mol Cell 2005;18:637-50.
41. Fenton SL, Dallol A, Agathanggelou A, et al. Identification of the E1A-regulated transcription factor p120 E4F as an interacting partner of the RASSF1A candidate tumor suppressor gene. Cancer Res 2004;64:102-7.
42. Zhang YJ, Ahsan H, Chen Y, et al. High frequency of promoter hypermethylation of RASSF1A and p16 and its relationship to aflatoxin B1-DNA adduct levels in human hepatocellular carcinoma. Mol Carcinog 2002;35:85-92.
43. Wong IH, Chan J, Wong J, Tam PK. Ubiquitous aberrant RASSF1A promoter methylation in childhood neoplasia. Clin Cancer Res 2004;10:994-1002.
44. Villanueva A, Portela A, Sayols S, et al. HEPTROMIC Consortium. DNA methylation-based prognosis and epidrivers in hepatocellular carcinoma. Hepatology 2015;61:1945-56.
45. Wong N, Li L, Tsang K, Lai PB, To K, Johnson PJ. Frequent loss of chromosome 3p and hypermethylation of RASSF1A in cholangiocarcinoma. J Hepatol 2002;37:633-9.
46. Yang B, House MG, Guo M, Herman JG, Clark DP. Promoter methylation profiles of tumor suppressor genes in intrahepatic and extrahepatic cholangiocarcinoma. Mod Pathol 2005;18:412-20.
47. Chen YJ, Tang QB, Zou SQ. Inactivation of RASSF1A, the tumor suppressor gene at 3p21.3 in extrahepatic cholangiocarcinoma. World J Gastroenterol 2005;11:1333-8.
48. Sugawara W, Haruta M, Sasaki F, et al. Promoter hypermethylation of the RASSF1A gene predicts the poor outcome of patients with hepatoblastoma. Pediatr Blood Cancer 2007;49:240-9.
49. Honda S, Haruta M, Sugawara W, et al. The methylation status of RASSF1A promoter predicts responsiveness to chemotherapy and eventual cure in hepatoblastoma patients. Int J Cancer 2008;123:1117-25.
50. Honda S, Miyagi H, Suzuki H, et al. RASSF1A methylation indicates a poor prognosis in hepatoblastoma patients. Pediatr Surg Int 2013;29:1147-52.
51. Yeo W, Wong N, Wong WL, Lai PB, Zhong S, Johnson PJ. High frequency of promoter hypermethylation of RASSF1A in tumor and plasma of patients with hepatocellular carcinoma. Liver Int 2005;25:266-72.
52. Liu G, Liu B, Zheng S, Dong K, Dong R. Aberrant RASSF5 gene transcribed region hypermethylation in pediatric hepatoblastomas. Int J Clin Exp Pathol 2018;11:3612-7.
53. Sprynski AC, Hose D, Kassambara A, et al. Insulin is a potent myeloma cell growth factor through insulin/IGF-1 hybrid receptor activation. Leukemia 2010;24:1940-50.
54. Enguita-Germán M, Fortes P. Targeting the insulin-like growth factor pathway in hepatocellular carcinoma. World J Hepatol 2014;6:716-37.
55. Abdel-Rahman O. Insulin-like growth factor pathway aberrations and gastric cancer; evaluation of prognostic significance and assessment of therapeutic potentials. Med Oncol 2015;32:431.
56. Dziadziuszko R, Camidge DR, Hirsch FR. The insulin-like growth factor pathway in lung cancer. J Thorac Oncol 2008;3:815-8.
57. Nitulescu GM, Van De Venter M, Nitulescu G, et al. The Akt pathway in oncology therapy and beyond (Review). Int J Oncol 2018;53:2319-31.
58. Gray SG, Eriksson T, Ekström C, et al. Altered expression of members of the IGF-axis in hepatoblastomas. Br J Cancer 2000;82:1561-7.
59. Rumbajan JM, Maeda T, Souzaki R, et al. Comprehensive analyses of imprinted differentially methylated regions reveal epigenetic and genetic characteristics in hepatoblastoma. BMC Cancer 2013;13:608.
61. Akmal SN, Yun K, Maclay J, Higami Y, Ikeda T. Insulin-like growth factor 2 and insulin-like growth factor binding protein 2 expression in hepatoblastoma. Human Pathology 1995;26:846-51.
62. Williams AC, Collard TJ, Perks CM, et al. Increased p53-dependent apoptosis by the insulin-like growth factor binding protein IGFBP-3 in human colonic adenoma-derived cells. Cancer Res 2000;60:22-7.
63. Buckbinder L, Talbott R, Velasco-Miguel S, et al. Induction of the growth inhibitor IGF-binding protein 3 by p53. Nature 1995;377:646-9.
64. Hanafusa T, Shinji T, Shiraha H, et al. Functional promoter upstream p53 regulatory sequence of IGFBP3 that is silenced by tumor specific methylation. BMC Cancer 2005;5:9.
65. López-Terrada D, Cheung SW, Finegold MJ, Knowles BB. Hep G2 is a hepatoblastoma-derived cell line. Hum Pathol 2009;40:1512-5.
66. Hanafusa T, Yumoto Y, Nouso K, et al. Reduced expression of insulin-like growth factor binding protein-3 and its promoter hypermethylation in human hepatocellular carcinoma. Cancer Letters 2002;176:149-58.
67. Tomii K, Tsukuda K, Toyooka S, et al. Aberrant promoter methylation of insulin-like growth factor binding protein-3 gene in human cancers. Int J Cancer 2007;120:566-73.
68. Regel I, Eichenmüller M, Joppien S, et al. IGFBP3 impedes aggressive growth of pediatric liver cancer and is epigenetically silenced in vascular invasive and metastatic tumors. Mol Cancer 2012;11:9.
69. Nagai H, Naka T, Terada Y, et al. Hypermethylation associated with inactivation of the SOCS-1 gene, a JAK/STAT inhibitor, in human hepatoblastomas. J Hum Genet 2003;48:65-9.
71. Bagnyukova TV, Tryndyak VP, Muskhelishvili L, Ross SA, Beland FA, Pogribny IP. Epigenetic downregulation of the suppressor of cytokine signaling 1 (Socs1) gene is associated with the STAT3 activation and development of hepatocellular carcinoma induced by methyl-deficiency in rats. Cell Cycle 2008;7:3202-10.
72. Huang FJ, Steeg PS, Price JE, et al. Molecular basis for the critical role of suppressor of cytokine signaling-1 in melanoma brain metastasis. Cancer Res 2008;68:9634-42.
73. Zhang J, Li H, Yu JP, Wang SE, Ren XB. Role of SOCS1 in tumor progression and therapeutic application. Int J Cancer 2012;130:1971-80.
74. Raccurt M, Tam SP, Lau P, et al. Suppressor of cytokine signalling gene expression is elevated in breast carcinoma. Br J Cancer 2003;89:524-32.
75. Guenterberg KD, Lesinski GB, Mundy-Bosse BL, et al. Enhanced anti-tumor activity of interferon-alpha in SOCS1-deficient mice is mediated by CD4+ and CD8+ T cells. Cancer Immunol Immunother 2011;60:1281-8.
76. Zimmerer JM, Lesinski GB, Kondadasula SV, et al. IFN-alpha-induced signal transduction, gene expression, and antitumor activity of immune effector cells are negatively regulated by suppressor of cytokine signaling proteins. J Immunol 2007;178:4832-45.
77. Sakamoto LH, DE Camargo B, Cajaiba M, Soares FA, Vettore AL. MT1G hypermethylation: a potential prognostic marker for hepatoblastoma. Pediatr Res 2010;67:387-93.
78. Tomlinson GE, Kappler R. Genetics and epigenetics of hepatoblastoma. Pediatr Blood Cancer 2012;59:785-92.
79. Russell JO, Monga SP. Wnt/β-catenin signaling in liver development, homeostasis, and pathobiology. Annu Rev Pathol 2018;13:351-78.
80. Gao L, Zhang Z, Zhang P, Yu M, Yang T. Role of canonical Hedgehog signaling pathway in liver. Int J Biol Sci 2018;14:1636-44.
81. Zheng X, Zeng W, Gai X, et al. Role of the Hedgehog pathway in hepatocellular carcinoma (review). Oncol Rep 2013;30:2020-6.
82. Wang LH, Choi YL, Hua XY, et al. Increased expression of sonic hedgehog and altered methylation of its promoter region in gastric cancer and its related lesions. Mod Pathol 2006;19:675-83.
83. Tada M, Kanai F, Tanaka Y, et al. Down-regulation of hedgehog-interacting protein through genetic and epigenetic alterations in human hepatocellular carcinoma. Clin Cancer Res 2008;14:3768-76.
84. Eichenmüller M, Gruner I, Hagl B, et al. Blocking the hedgehog pathway inhibits hepatoblastoma growth. Hepatology 2009;49:482-90.
85. Xu X, Li J, Sun X, et al. Tumor suppressor NDRG2 inhibits glycolysis and glutaminolysis in colorectal cancer cells by repressing c-Myc expression. Oncotarget 2015;6:26161-76.
86. Kim YJ, Yoon SY, Kim JT, et al. NDRG2 expression decreases with tumor stages and regulates TCF/beta-catenin signaling in human colon carcinoma. Carcinogenesis 2009;30:598-605.
87. Lusis EA, Watson MA, Chicoine MR, et al. Integrative genomic analysis identifies NDRG2 as a candidate tumor suppressor gene frequently inactivated in clinically aggressive meningioma. Cancer Res 2005;65:7121-6.
88. Gödeke J, Luxenburger E, Trippel F, et al. Low expression of N-myc downstream-regulated gene 2 (NDRG2) correlates with poor prognosis in hepatoblastoma. Hepatol Int 2016;10:370-6.
89. Baharudin R, Tieng FYF, Lee LH, Ab Mutalib NS. Epigenetics of SFRP1: the dual roles in human cancers. Cancers (Basel) 2020;12:445.
90. Zhao CH, Bu XM, Zhang N. Hypermethylation and aberrant expression of Wnt antagonist secreted frizzled-related protein 1 in gastric cancer. World J Gastroenterol 2007;13:2214-7.
91. Wang X, Wang H, Bu R, Fei X, Zhao C, Song Y. Methylation and aberrant expression of the Wnt antagonist secreted Frizzled-related protein 1 in bladder cancer. Oncol Lett 2012;4:334-8.
92. Taguchi YH, Iwadate M, Umeyama H. SFRP1 is a possible candidate for epigenetic therapy in non-small cell lung cancer. BMC Med Genomics 2016;9 Suppl 1:28.
93. Kim J, Kim S. In silico identification of SFRP1 as a hypermethylated gene in colorectal cancers. Genomics Inform 2014;12:171-80.
94. Shih YL, Shyu RY, Hsieh CB, et al. Promoter methylation of the secreted frizzled-related protein 1 gene SFRP1 is frequent in hepatocellular carcinoma. Cancer 2006;107:579-90.
95. Regel I, Eichenmüller M, Mahajan UM, et al. Downregulation of SFRP1 is a protumorigenic event in hepatoblastoma and correlates with beta-catenin mutations. J Cancer Res Clin Oncol 2020;146:1153-67.
96. Shih YL, Hsieh CB, Lai HC, et al. SFRP1 suppressed hepatoma cells growth through Wnt canonical signaling pathway. Int J Cancer 2007;121:1028-35.
97. Honda S, Minato M, Suzuki H, et al. Clinical prognostic value of DNA methylation in hepatoblastoma: four novel tumor suppressor candidates. Cancer Sci 2016;107:812-9.
98. Rivas MP, Aguiar TFM, Maschietto M, et al. Hepatoblastomas exhibit marked NNMT downregulation driven by promoter DNA hypermethylation. Tumour Biol 2020;42:1010428320977124.
99. Li S, Qiao L, Yang Z, He C. Prognostic value of nicotinamide N-methyltransferase expression in patients with solid tumors: a systematic review and meta-analysis. Front Physiol 2018;9:1407.
100. Ulanovskaya OA, Zuhl AM, Cravatt BF. NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat Chem Biol 2013;9:300-6.
101. Fujiyoshi S, Honda S, Minato M, et al. Hypermethylation of CSF3R is a novel cisplatin resistance marker and predictor of response to postoperative chemotherapy in hepatoblastoma. Hepatol Res 2020;50:598-606.
102. Zynger DL, Gupta A, Luan C, Chou PM, Yang GY, Yang XJ. Expression of glypican 3 in hepatoblastoma: an immunohistochemical study of 65 cases. Hum Pathol 2008;39:224-30.
103. Nguyen LH, Robinton DA, Seligson MT, et al. Lin28b is sufficient to drive liver cancer and necessary for its maintenance in murine models. Cancer Cell 2014;26:248-61.
104. Meng W, Bai B, Bai Z, et al. The immunosuppression role of alpha-fetoprotein in human hepatocellular carcinoma. Discov Med 2016;21:489-94.
105. Cui X, Liu B, Zheng S, Dong K, Dong R. Genome-wide analysis of DNA methylation in hepatoblastoma tissues. Oncol Lett 2016;12:1529-34.
106. Fuhrman-Luck RA, Stansfield SH, Stephens CR, Loessner D, Clements JA. Prostate cancer-associated kallikrein-related peptidase 4 activates matrix metalloproteinase-1 and thrombospondin-1. J Proteome Res 2016;15:2466-78.
107. Wang P, Magdolen V, Seidl C, et al. Kallikrein-related peptidases 4, 5, 6 and 7 regulate tumour-associated factors in serous ovarian cancer. Br J Cancer 2018;119:1-9.
108. Kontos CK, Chantzis D, Papadopoulos IN, Scorilas A. Kallikrein-related peptidase 4 (KLK4) mRNA predicts short-term relapse in colorectal adenocarcinoma patients. Cancer Lett 2013;330:106-12.
109. Liu B, Cui X, Zheng S, Dong K, Dong R. Aberrant KLK4 gene promoter hypomethylation in pediatric hepatoblastomas. Oncol Lett 2017;13:1360-4.
110. Vita M, Henriksson M. The Myc oncoprotein as a therapeutic target for human cancer. Semin Cancer Biol 2006;16:318-30.
111. Kress TR, Sabò A, Amati B. MYC: connecting selective transcriptional control to global RNA production. Nat Rev Cancer 2015;15:593-607.
112. Eberherr C, Beck A, Vokuhl C, et al. Targeting excessive MYCN expression using MLN8237 and JQ1 impairs the growth of hepatoblastoma cells. Int J Oncol 2019;54:1853-63.
113. Barcena-Varela M, Paish H, Alvarez L, et al. Epigenetic mechanisms and metabolic reprogramming in fibrogenesis: dual targeting of G9a and DNMT1 for the inhibition of liver fibrosis. Gut 2021;70:388-400.
114. Bárcena-Varela M, Caruso S, Llerena S, et al. Dual targeting of histone methyltransferase G9a and DNA-methyltransferase 1 for the treatment of experimental hepatocellular carcinoma. Hepatology 2019;69:587-603.
115. Ding WJ, Fang JY, Chen XY, Peng YS. The expression and clinical significance of DNA methyltransferase proteins in human gastric cancer. Dig Dis Sci 2008;53:2083-9.
116. Tzelepi V, Logotheti S, Efstathiou E, et al. Epigenetics and prostate cancer: defining the timing of DNA methyltransferase deregulation during prostate cancer progression. Pathology 2020;52:218-27.
117. Colyn L, Bárcena-Varela M, Álvarez-Sola G, et al. Dual targeting of G9a and DNA Methyltransferase-1 for the treatment of experimental cholangiocarcinoma. Hepatology 2021;73:2380-96.
118. Li A, Omura N, Hong SM, Goggins M. Pancreatic cancer DNMT1 expression and sensitivity to DNMT1 inhibitors. Cancer Biol Ther 2010;9:321-9.
119. Rivas MP, Aguiar TFM, Fernandes GR, et al. TET upregulation leads to 5-hydroxymethylation enrichment in hepatoblastoma. Front Genet 2019;10:553.
120. Huang Y, Rao A. Connections between TET proteins and aberrant DNA modification in cancer. Trends Genet 2014;30:464-74.
121. Lin G, Sun W, Yang Z, Guo J, Liu H, Liang J. Hypoxia induces the expression of TET enzymes in HepG2 cells. Oncol Lett 2017;14:6457-62.
122. Liu C, Liu L, Chen X, et al. Decrease of 5-hydroxymethylcytosine is associated with progression of hepatocellular carcinoma through downregulation of TET1. PLoS One 2013;8:e62828.
123. Sidhu H, Capalash N. UHRF1: The key regulator of epigenetics and molecular target for cancer therapeutics. Tumour Biol 2017;39:1010428317692205.
124. Jin W, Chen L, Chen Y, et al. UHRF1 is associated with epigenetic silencing of BRCA1 in sporadic breast cancer. Breast Cancer Res Treat 2010;123:359-73.
125. Sabatino L, Fucci A, Pancione M, et al. UHRF1 coordinates peroxisome proliferator activated receptor gamma (PPARG) epigenetic silencing and mediates colorectal cancer progression. Oncogene 2012;31:5061-72.
126. Zhuo H, Tang J, Lin Z, et al. The aberrant expression of MEG3 regulated by UHRF1 predicts the prognosis of hepatocellular carcinoma. Mol Carcinog 2016;55:209-19.
127. Ying L, Lin J, Qiu F, et al. Epigenetic repression of regulator of G-protein signaling 2 by ubiquitin-like with PHD and ring-finger domain 1 promotes bladder cancer progression. FEBS J 2015;282:174-82.
128. Zhou L, Shang Y, Jin Z, et al. UHRF1 promotes proliferation of gastric cancer via mediating tumor suppressor gene hypermethylation. Cancer Biol Ther 2015;16:1241-51.
129. Beck A, Trippel F, Wagner A, et al. Overexpression of UHRF1 promotes silencing of tumor suppressor genes and predicts outcome in hepatoblastoma. Clin Epigenetics 2018;10:27.
131. Gray SG, Hartmann W, Eriksson T, et al. Expression of genes involved with cell cycle control, cell growth and chromatin modification are altered in hepatoblastomas. Int J Mol Med 2000;6:161-9.
132. Kuo P, Chiang L, Lin C. Resveratrol- induced apoptosis is mediated by p53-dependent pathway in Hep G2 cells. Life Sciences 2002;72:23-34.
133. West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest 2014;124:30-9.
134. Singh AK, Bishayee A, Pandey AK. Targeting histone deacetylases with natural and synthetic agents: an emerging anticancer strategy. Nutrients 2018;10:731.
135. Venturelli S, Berger A, Böcker A, et al. Resveratrol as a pan-HDAC inhibitor alters the acetylation status of histone [corrected] proteins in human-derived hepatoblastoma cells. PLoS One 2013;8:e73097.
136. Beck A, Eberherr C, Hagemann M, et al. Connectivity map identifies HDAC inhibition as a treatment option of high-risk hepatoblastoma. Cancer Biol Ther 2016;17:1168-76.
137. French C. . Small-molecule targeting of BET proteins in cancer. Elsevier; 2016. p. 21-58.
138. Wang YH, Sui XM, Sui YN, et al. BRD4 induces cell migration and invasion in HCC cells through MMP-2 and MMP-9 activation mediated by the Sonic hedgehog signaling pathway. Oncol Lett 2015;10:2227-32.
139. Saltsman JA, Hammond WJ, Narayan NJC, et al. A human organoid model of aggressive hepatoblastoma for disease modeling and drug testing. Cancers (Basel) 2020;12:2668.
140. Chen Y, Ren B, Yang J, et al. The role of histone methylation in the development of digestive cancers: a potential direction for cancer management. Signal Transduct Target Ther 2020;5:143.
142. Wang Y, Xiao Y, Chen K, et al. Enhancer of zeste homolog 2 depletion arrests the proliferation of hepatoblastoma cells. Mol Med Rep 2016;13:2724-8.
143. Schlachter K, Gyugos M, Halász J, et al. High tricellulin expression is associated with better survival in human hepatoblastoma. Histopathology 2014;65:631-41.
144. Wong LS, Wong CM. Decoding the roles of long noncoding RNAs in hepatocellular carcinoma. Int J Mol Sci 2021;22:3137.
145. Syeda Z, Langden SSS, Munkhzul C, Lee M, Song SJ. Regulatory mechanism of microRNA expression in cancer. Int J Mol Sci 2020;21:1723.
146. Iorio MV, Croce CM. MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. a comprehensive review. EMBO Mol Med 2012;4:143-59.
147. Magrelli A, Azzalin G, Salvatore M, et al. Altered microRNA expression patterns in hepatoblastoma patients. Transl Oncol 2009;2:157-63.
148. Gyugos M, Lendvai G, Kenessey I, et al. MicroRNA expression might predict prognosis of epithelial hepatoblastoma. Virchows Arch 2014;464:419-27.
149. Ecevit ÇÖ, Aktaş S, Tosun Yildirim H, et al. MicroRNA-17, microRNA-19b, microRNA-146a, microRNA-302d expressions in hepatoblastoma and clinical importance. J Pediatr Hematol Oncol 2019;41:7-12.
150. Zatkova A, Rouillard JM, Hartmann W, et al. Amplification and overexpression of the IGF2 regulator PLAG1 in hepatoblastoma. Genes Chromosomes Cancer 2004;39:126-37.
151. Voz ML, Mathys J, Hensen K, et al. Microarray screening for target genes of the proto-oncogene PLAG1. Oncogene 2004;23:179-91.
152. Frowein J, Pagel P, Kappler R, von Schweinitz D, Roscher A, Schmid I. MicroRNA-492 is processed from the keratin 19 gene and up-regulated in metastatic hepatoblastoma. Hepatology 2011;53:833-42.
153. von Frowein J, Hauck SM, Kappler R, et al. MiR-492 regulates metastatic properties of hepatoblastoma via CD44. Liver Int 2018;38:1280-91.
154. Yin DT, Wu W, Li M, et al. DKK3 is a potential tumor suppressor gene in papillary thyroid carcinoma. Endocr Relat Cancer 2013;20:507-14.
155. Veeck J, Wild PJ, Fuchs T, et al. Prognostic relevance of Wnt-inhibitory factor-1 (WIF1) and Dickkopf-3 (DKK3) promoter methylation in human breast cancer. BMC Cancer 2009;9:217.
156. Li Q, Shen K, Zhao Y, Ma C, Liu J, Ma J. MiR-92b inhibitor promoted glioma cell apoptosis via targeting DKK3 and blocking the Wnt/beta-catenin signaling pathway. J Transl Med 2013;11:302.
157. Farkas SA, Vymetalkova V, Vodickova L, Vodicka P, Nilsson TK. DNA methylation changes in genes frequently mutated in sporadic colorectal cancer and in the DNA repair and Wnt/β-catenin signaling pathway genes. Epigenomics 2014;6:179-91.
158. Pei Y, Kano J, Iijima T, Morishita Y, Inadome Y, Noguchi M. Overexpression of Dickkopf 3 in hepatoblastomas and hepatocellular carcinomas. Virchows Arch 2009;454:639-46.
159. Pei Y, Yao Q, Yuan S, et al. GATA4 promotes hepatoblastoma cell proliferation by altering expression of miR125b and DKK3. Oncotarget 2016;7:77890-901.
160. Cairo S, Wang Y, de Reyniès A, et al. Stem cell-like micro-RNA signature driven by Myc in aggressive liver cancer. Proc Natl Acad Sci U S A 2010;107:20471-6.
161. Weiss JBW, Wagner AE, Eberherr C, et al. High expression of IGF2-derived intronic miR-483 predicts outcome in hepatoblastoma. Cancer Biomark 2020;28:321-8.
162. Cartier F, Indersie E, Lesjean S, et al. New tumor suppressor microRNAs target glypican-3 in human liver cancer. Oncotarget 2017;8:41211-26.
163. Samuels-lev Y, O'connor DJ, Bergamaschi D, et al. ASPP proteins specifically stimulate the apoptotic function of p53. Molecular Cell 2001;8:781-94.
164. Liang B, Chen R, Song S, et al. ASPP2 inhibits tumor growth by repressing the mevalonate pathway in hepatocellular carcinoma. Cell Death Dis 2019;10:830.
165. Liu L, Wang L, Li X, et al. Effect of miR-21 on apoptosis in hepatoblastoma cell through activating ASPP2/p38 signaling pathway in vitro and in vivo. Artif Cells Nanomed Biotechnol 2019;47:3729-36.
166. Honda S, Chatterjee A, Leichter AL, et al. A microRNA cluster in the DLK1-DIO3 imprinted region on chromosome 14q32.2 is dysregulated in metastatic hepatoblastomas. Front Oncol 2020;10:513601.
167. Chen LJ, Yuan MX, Ji CY, et al. Long non-coding RNA CRNDE regulates angiogenesis in hepatoblastoma by targeting the MiR-203/VEGFA axis. Pathobiology 2020;87:161-70.
168. Song H, Bian ZX, Li HY, et al. Characterization of hsa_circ_0000594 as a new biomarker and therapeutic target for hepatoblastoma. Eur Rev Med Pharmacol Sci 2019;23:8274-86.
169. Zhen N, Gu S, Ma J, et al. CircHMGCS1 promotes hepatoblastoma cell proliferation by regulating the IGF signaling pathway and glutaminolysis. Theranostics 2019;9:900-19.
170. Liu Y, Song J, Liu Y, Zhou Z, Wang X. Transcription activation of circ-STAT3 induced by Gli2 promotes the progression of hepatoblastoma via acting as a sponge for miR-29a/b/c-3p to upregulate STAT3/Gli2. J Exp Clin Cancer Res 2020;39:101.
171. Galardi A, Colletti M, Di Paolo V, et al. Exosomal miRNAs in pediatric cancers. Int J Mol Sci 2019;20:4600.
172. Jiao C, Jiao X, Zhu A, Ge J, Xu X. Exosomal miR-34s panel as potential novel diagnostic and prognostic biomarker in patients with hepatoblastoma. J Pediatr Surg 2017;52:618-24.
173. Nandwani A, Rathore S, Datta M. LncRNAs in cancer: regulatory and therapeutic implications. Cancer Lett 2021;501:162-71.
175. Dong R, Jia D, Xue P, et al. Genome-wide analysis of long noncoding RNA (lncRNA) expression in hepatoblastoma tissues. PLoS One 2014;9:e85599.
176. Wang XD, Peng JB, Zhou CY, et al. Potential therapies for residual hepatoblastoma following incomplete ablation treatment in a nude mouse subcutaneous xenograft model based on lncRNA and mRNA expression profiles. Oncol Rep 2020;43:1915-27.
177. Alipoor B, Parvar SN, Sabati Z, Ghaedi H, Ghasemi H. An updated review of the H19 lncRNA in human cancer: molecular mechanism and diagnostic and therapeutic importance. Mol Biol Rep 2020;47:6357-74.
178. Li J, Zhang M, An G, Ma Q. LncRNA TUG1 acts as a tumor suppressor in human glioma by promoting cell apoptosis. Exp Biol Med (Maywood) 2016;241:644-9.
179. Zhang EB, Yin DD, Sun M, et al. P53-regulated long non-coding RNA TUG1 affects cell proliferation in human non-small cell lung cancer, partly through epigenetically regulating HOXB7 expression. Cell Death Dis 2014;5:e1243.
180. Fan S, Yang Z, Ke Z, et al. Downregulation of the long non-coding RNA TUG1 is associated with cell proliferation, migration, and invasion in breast cancer. Biomed Pharmacother 2017;95:1636-43.
181. Ma B, Li M, Zhang L, et al. Upregulation of long non-coding RNA TUG1 correlates with poor prognosis and disease status in osteosarcoma. Tumour Biol 2016;37:4445-55.
182. Zhai HY, Sui MH, Yu X, et al. Overexpression of long non-coding RNA TUG1 promotes colon cancer progression. Med Sci Monit 2016;22:3281-7.
183. Huang MD, Chen WM, Qi FZ, et al. Long non-coding RNA TUG1 is up-regulated in hepatocellular carcinoma and promotes cell growth and apoptosis by epigenetically silencing of KLF2. Mol Cancer 2015;14:165.
184. Dong R, Liu GB, Liu BH, et al. Targeting long non-coding RNA-TUG1 inhibits tumor growth and angiogenesis in hepatoblastoma. Cell Death Dis 2016;7:e2278.
185. Zhang J, Yin M, Peng G, Zhao Y. CRNDE: an important oncogenic long non-coding RNA in human cancers. Cell Prolif 2018;51:e12440.
186. Dong R, Liu XQ, Zhang BB, Liu BH, Zheng S, Dong KR. Long non-coding RNA-CRNDE: a novel regulator of tumor growth and angiogenesis in hepatoblastoma. Oncotarget 2017;8:42087-97.
187. Lv B, Zhang L, Miao R, et al. Comprehensive analysis and experimental verification of LINC01314 as a tumor suppressor in hepatoblastoma. Biomed Pharmacother 2018;98:783-92.
188. Yu Q, Li Y, Peng S, Li J, Qin X. Exosomal-mediated transfer of OIP5-AS1 enhanced cell chemoresistance to trastuzumab in breast cancer via up-regulating HMGB3 by sponging miR-381-3p. Open Med (Wars) 2021;16:512-25.
189. Zhang Z, Liu F, Yang F, Liu Y. Kockdown of OIP5-AS1 expression inhibits proliferation, metastasis and EMT progress in hepatoblastoma cells through up-regulating miR-186a-5p and down-regulating ZEB1. Biomed Pharmacother 2018;101:14-23.
190. Li A, Feng L, Niu X, Zeng Q, Li B, You Z. Downregulation of OIP5-AS1 affects proNGF-induced pancreatic cancer metastasis by inhibiting p75NTR levels. Aging (Albany NY) 2021;13:10688-702.
191. Wang X, Guo S, Zhao R, Liu Y, Yang G. STAT3-activated long non-coding RNA lung cancer associated transcript 1 drives cell proliferation, migration, and invasion in hepatoblastoma through regulation of the miR-301b/STAT3 axis. Hum Gene Ther 2019;30:702-13.
192. Cui X, Wang Z, Liu L, et al. The long non-coding RNA ZFAS1 sponges miR-193a-3p to modulate hepatoblastoma growth by targeting RALY via HGF/c-Met pathway. Front Cell Dev Biol 2019;7:271.
193. Burenina OY, Lazarevich NL, Kustova IF, et al. Panel of potential lncRNA biomarkers can distinguish various types of liver malignant and benign tumors. J Cancer Res Clin Oncol 2021;147:49-59.
194. Jiao S, Wu S, Huang S, Liu M, Gao B. Advances in the identification of circular RNAs and research into circRNAs in human diseases. Front Genet 2021;12:665233.
195. Hansen TB, Jensen TI, Clausen BH, et al. Natural RNA circles function as efficient microRNA sponges. Nature 2013;495:384-8.
196. Yang Y, Fan X, Mao M, et al. Extensive translation of circular RNAs driven by N6-methyladenosine. Cell Res 2017;27:626-41.
197. Li Z, Huang C, Bao C, et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol 2015;22:256-64.
198. Conn VM, Hugouvieux V, Nayak A, et al. A circRNA from SEPALLATA3 regulates splicing of its cognate mRNA through R-loop formation. Nat Plants 2017;3:17053.
199. Zhong Z, Lv M, Chen J. Screening differential circular RNA expression profiles reveals the regulatory role of circTCF25-miR-103a-3p/miR-107-CDK6 pathway in bladder carcinoma. Sci Rep 2016;6:30919.
200. Li P, Chen S, Chen H, et al. Using circular RNA as a novel type of biomarker in the screening of gastric cancer. Clin Chim Acta 2015;444:132-6.
201. Tang Y, Jiang M, Jiang HM, et al. The roles of circRNAs in liver cancer immunity. Front Oncol 2020;10:598464.
202. Liu BH, Zhang BB, Liu XQ, Zheng S, Dong KR, Dong R. Expression profiling identifies circular RNA signature in hepatoblastoma. Cell Physiol Biochem 2018;45:706-19.
203. Lee H, Kim KR, Noh SJ, et al. Expression of DBC1 and SIRT1 is associated with poor prognosis for breast carcinoma. Hum Pathol 2011;42:204-13.
204. Zhang ZY, Hong D, Nam SH, et al. SIRT1 regulates oncogenesis via a mutant p53-dependent pathway in hepatocellular carcinoma. J Hepatol 2015;62:121-30.
205. Matés JM, Campos-Sandoval JA, Santos-Jiménez JL, Márquez J. Dysregulation of glutaminase and glutamine synthetase in cancer. Cancer Lett 2019;467:29-39.
206. Chen L, Shi J, Wu Y, et al. CircRNA CDR1as promotes hepatoblastoma proliferation and stemness by acting as a miR-7-5p sponge to upregulate KLF4 expression. Aging (Albany NY) 2020;12:19233-53.
207. Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol 2017;18:31-42.
208. Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012;485:201-6.
209. Wang X, Lu Z, Gomez A, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014;505:117-20.
210. Xu Y, Zhang W, Shen F, et al. YTH domain proteins: a family of m6A readers in cancer progression. Front Oncol 2021;11:629560.
211. Chen M, Wei L, Law CT, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology 2018;67:2254-70.
212. Liu L, Wang J, Sun G, et al. m6A mRNA methylation regulates CTNNB1 to promote the proliferation of hepatoblastoma. Mol Cancer 2019;18:188.
213. Cui X, Wang Z, Li J, et al. Cross talk between RNA N6-methyladenosine methyltransferase-like 3 and miR-186 regulates hepatoblastoma progression through Wnt/β-catenin signalling pathway. Cell Prolif 2020;53:e12768.
214. Zhuo ZJ, Hua RX, Chen Z, et al. WTAP gene variants confer hepatoblastoma susceptibility: a seven-center case-control study. Mol Ther Oncolytics 2020;18:118-25.
215. Han L, Diao L, Yu S, et al. The genomic landscape and clinical relevance of A-to-I RNA editing in human cancers. Cancer Cell 2015;28:515-28.
216. Paz-Yaacov N, Bazak L, Buchumenski I, et al. Elevated RNA editing activity is a major contributor to transcriptomic diversity in tumors. Cell Rep 2015;13:267-76.
217. Galeano F, Leroy A, Rossetti C, et al. Human BLCAP transcript: new editing events in normal and cancerous tissues. Int J Cancer 2010;127:127-37.
218. Feng S, De Carvalho DD. Clinical advances in targeting epigenetics for cancer therapy. FEBS J 2021; doi: 10.1111/febs.15750.
219. El Sayed S, Cristante J, Guyon L, Denis J, Chabre O, Cherradi N. MicroRNA Therapeutics in cancer: current advances and challenges. Cancers (Basel) 2021;13:2680.
220. Oronsky B, Oronsky N, Knox S, Fanger G, Scicinski J. Episensitization: therapeutic tumor resensitization by epigenetic agents: a review and reassessment. Anticancer Agents Med Chem 2014;14:1121-7.
221. Oronsky BT, Oronsky AL, Lybeck M, et al. Episensitization: defying time's arrow. Front Oncol 2015;5:134.
222. Li GH, Qu Q, Qi TT, et al. Super-enhancers: a new frontier for epigenetic modifiers in cancer chemoresistance. J Exp Clin Cancer Res 2021;40:174.
223. Yang T, Yang Y, Wang Y. Predictive biomarkers and potential drug combinations of epi-drugs in cancer therapy. Clin Epigenetics 2021;13:113.
224. Wei QY, Xu YM, Lau ATY. Recent Progress of Nanocarrier-Based Therapy for Solid Malignancies. Cancers (Basel) 2020;12:2783.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Clavería-Cabello A, Arechederra M, Berasain C, Fernández-Barrena MG, Ávila MA. Epigenetics in hepatoblastoma. Hepatoma Res 2021;7:65. http://dx.doi.org/10.20517/2394-5079.2021.77
AMA Style
Clavería-Cabello A, Arechederra M, Berasain C, Fernández-Barrena MG, Ávila MA. Epigenetics in hepatoblastoma. Hepatoma Research. 2021; 7: 65. http://dx.doi.org/10.20517/2394-5079.2021.77
Chicago/Turabian Style
Clavería-Cabello, Alex, María Arechederra, Carmen Berasain, Maite G. Fernández-Barrena, Matías A. Ávila. 2021. "Epigenetics in hepatoblastoma" Hepatoma Research. 7: 65. http://dx.doi.org/10.20517/2394-5079.2021.77
ACS Style
Clavería-Cabello, A.; Arechederra M.; Berasain C.; Fernández-Barrena MG.; Ávila MA. Epigenetics in hepatoblastoma. Hepatoma. Res. 2021, 7, 65. http://dx.doi.org/10.20517/2394-5079.2021.77
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 19 clicks
Cite This Article 19 clicks

 Like This Article 32
likes
Like This Article 32
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.