
HCC in metabolic syndrome: current concepts and future directions

Abstract
Hepatocellular carcinoma (HCC) is one of the leading causes of cancer-related deaths globally. In recent years, the metabolic syndrome epidemic is changing the etiological landscape of HCC, with metabolic liver disease comprising an exponentially increasing proportion of HCC cases. In this review, we discuss HCC in the context of metabolic syndrome, including its epidemiology, its unique clinical and pathological characteristics, and its multifactorial pathogenesis. We also discuss HCC prevention and management as relates to these patients.
Keywords
INTRODUCTION
Globally, hepatocellular carcinoma (HCC) is the fifth most common cancer and the third most frequently reported cause of cancer-related death[1]. The leading etiologies of liver disease in patients with HCC have historically been chronic hepatitis B and C, and alcoholic liver disease (ALD); however, with the advent of curative treatments for HCV and large scale vaccination efforts for HBV, the proportional incidence of viral hepatitis is projected to decline[2]. Inversely, the metabolic syndrome is anticipated to become the most common cause of HCC in developed countries in the near future[3].
Metabolic syndrome (MS) refers to the co-occurrence of several cardiometabolic risk factors including insulin resistance, obesity, atherogenic dyslipidemia, and hypertension[4]. This multiplex of risk factors tend to cluster and have a shared responsiveness to lifestyle modifications, suggesting that they are not independent of one another but rather that they share underlying mediators, mechanisms, and pathways[5].
Non-alcoholic fatty liver disease (NAFLD) is defined by the presence of steatosis in > 5% of hepatocytes alongside the exclusion of all other causes of secondary hepatic fat accumulation, particularly alcohol consumption and viral hepatitis, and is strongly associated with features of MS[6]. Although NAFLD was documented more than 30 years ago[7], its pathogenesis remains incompletely understood. Initially, Day and James (1998)[8] proposed a two-hit hypothesis to define NAFLD pathogenesis. The “first hit” corresponds to the appearance of hepatic steatosis; the “second hit” comprises an additional oxidative injury which in turn leads to necroinflammation and fibrosis[8]. The prolonged inflammation and fibrosis they theorized, in turn predisposes the liver to HCC development. Today, this hypothesis has been expanded in support of a multifactorial explanation which suggests that NAFLD is the result of several conditions acting in parallel, perhaps synergistically in genetically predisposed individuals, with insulin resistance as the key mechanism leading to hepatic steatosis, and other factors such as genetic factors, lifestyle factors, altered immune response, and intestinal dysbiosis among others acting as contributing factors to oxidative stress[9].
However, the current definition of NAFLD falls short in incorporating our current knowledge on the pathogenesis of this hepatic disease, which is strongly tied to metabolic dysfunction. Furthermore, the current definition of NAFLD may exclude certain cirrhotic patients missing histological evidence of steatosis despite being comorbid with several metabolic risk factors. Most likely, these patients’ histological evidence of steatosis, inflammation or hepatocellular injury becomes replaced with abundant fibrotic tissue during cirrhotic progression. In light of these nomenclatural shortcomings, a group of experts recently proposed a new term, “metabolic-associated fatty liver disease (MAFLD)”. This term is designed to more accurately reflect the pathogenesis and natural history of the disease, in hopes that this more appropriate overarching term may aid in patient stratification and management[10,11]. This definition allows for a positive diagnosis, based on histological, imaging or serum biomarker evidence of hepatic steatosis plus one of the following three criteria: overweight/obese, the presence of type 2 diabetes mellitus, or evidence of metabolic dysregulation. Metabolic dysregulation here is defined by the presence of two or more features of the MS and other metabolic risk abnormalities. Shifting from a negative diagnosis framework (based on the exclusion of all other liver pathologies) provides a major clinical advantage. Given the increasing incidence of metabolic liver disease, it is often found in adjunct to other liver conditions such as alcoholic liver disease or viral hepatitis. A positive diagnostic framework allows us to identify metabolic liver disease as a comorbid condition. Additionally, it allows cirrhotic patients with low or undetectable levels of steatosis who meet the diagnostic criteria proposed for MAFLD to be considered as MAFLD-associated cirrhosis, thus avoiding the use of the term “cryptogenic cirrhosis”, which is an ambiguous host definition to many more accurately defined MAFLD patients. For the purpose of this review, we will draw from studies utilizing both criteria of hepatic metabolic disease to describe MS’s complex relationship to HCC. This review will focus on the epidemiology, clinical features, and contributing factors in the pathogenesis of HCC related to the metabolic syndrome, in particular in patients with NAFLD.
EPIDEMIOLOGY OF HCC IN THE CONTEXT OF THE METABOLIC SYNDROME
With NAFLD affecting up to 25% of the general population globally[12-15], it is not surprising that NAFLD-HCC is on the rise. A United States-based 2015 study[16] examining the annual incidence of NAFLD-related HCC based on a SEER-Medicare cohort, found that NAFLD-HCC increased at an annual rate of 9% during the six years between 2004 and 2009. This increasing trend is mirrored on a global scale. In South Korea, a retrospective cohort study[17] involving 329 HCC subjects found an increase from 3.8% of HCC cases being attributed to NAFLD during the 2001-2005 period to 12.2% during the 2006-2010 period (P = 0.008). A recent Swiss population-based study[18], examining all HCC cases resident in the canton of Geneva between 1990-2014, also found an increase in NAFLD and MAFLD-related HCCs, particularly in women. Between 1990-1994 and 2010-2014, we observed a significant increase in HCC incidence in women [standardized incidence ratio (SIR) = 1.83, 95%CI: 1.08-3.13, P = 0.026], which was not observed in men (SIR = 1.10, 95%CI: 0.85-1.43, P = 0.468). During the same timeframe, the proportion of NAFLD-HCC in respect to non-NAFLD causes of HCC increased more in women (0% to 29%, P = 0.037) than in men (2% to 12%, P = 0.010), while the proportion of MAFLD increased from 21% to 68% in both sexes and 7% to 67% in women
This rise in the incidence of NAFLD-related HCC has impacted trends in liver transplantation as well. In a US-based 2018 study[20] analyzing 26,121 HCC patients between 2002 and 2016 in the Scientific Registry of Transplant Recipients database, the proportion of patients with NASH-HCC increased 7.7-fold during the 14-year period from 2.1% to 16.2% (P < 0.0001). Furthermore since 2002, the prevalence of HCC in liver transplant candidates with NASH increased 11.8-fold, steeper than that of any other liver etiology (P < 0.0001 in a trend regression model) with the second highest being chronic hepatitis B (6.0-fold increase), followed by alcoholic liver disease (3.4-fold increase) and chronic hepatitis C (2.3-fold increase) (all P < 0.0001).
A large recent meta-analysis[13] estimated the annual incidence of HCC in NAFLD to be 0.44 per 1000 person‐years (95%CI: 0.29‐0.66), however this value rises significantly when we consider HCC in NASH which the study estimates have an incidence rate of 5.29 per 1000 person‐years (95%CI: 0.75‐37.56). Indeed, liver inflammation and especially liver fibrosis has been shown to be the most important predictor of mortality in NAFLD patients[21]. The risk of HCC is highest when we consider cirrhotic NAFLD patients[13,22-25] and in particular Hispanic ones[22]. In a retrospective cohort study comparing 296,707 NAFLD patients with an equal number of controls, researchers found that among NAFLD patients with cirrhosis, the annual incidence of HCC was 10.6 per 1000 person-years. In Hispanics with NAFLD-cirrhosis, the annual incidence of HCC was 23.76 per 1000 person-years (95%CI: 12.27-41.50). Moreover, while this proportion of HCC progression still remains lower than that reported for other chronic liver disorders, the overall burden NAFLD-HCC remains an important public health concern given the sheer pervasiveness and projected increase of NAFLD. Several studies have aimed to forecast the NAFLD disease burden. A 2018 study modeling NAFLD disease burden in eight countries (China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States)[26] between 2016-2030 predicted that even if obesity and diabetes type 2 diabetes level out in the next several years, total NAFLD growth is estimated to reach up to 30%. The model also suggests that despite modest NAFLD growth, NASH prevalence will increase by up to 56%, with NAFLD-HCC estimated to increase 122% in the United States, from 5510 to 12,240 cases. In Europe, France was projected to have the largest increase (117%), while the UK had the smallest increase (88%). Similarly, in a Swiss modeling study[27], researchers used the close link between obesity and type 2 diabetes with NAFLD to quantify fibrotic progression among the NAFLD and NASH populations and predict disease burden up to 2030. Results suggest an increase in NASH prevalence of 40% between 2018 and 2030, with the number of deaths as a result of NAFLD surging by 41%.
However, a number of studies have shown that unlike other etiologies of liver disease, 30%-50% of HCCs in the context of the MS and NAFLD occur in the absence of cirrhosis[18,22,28-34]. Paradis et al.[35] were among the first to show that HCCs in patients with features of MS often occurred in the absence of advanced fibrosis in a retrospective study of patients who had undergone hepatic resection for HCC. Some studies suggest this finding may be associated with gender[29,31,32,36]; for instance, a recent retrospective study in Japan[29] comprising 104 patients in a cohort spanning nearly 17 years, found that while 80.5% of women with NAFLD-HCC presented with advanced fibrosis (F3-F4), only 58.8% of males did (P = 0.03). In another Japanese study[31], the histological review of 209 NAFLD-HCC cases revealed an even larger gender disparity in the proportion of NAFLD-HCC patients with cirrhosis (72.7% female vs. 37.6% males). Whether the prognosis of NAFLD-HCC occurring in the absence of cirrhosis is significantly different to that occurring in cirrhosis remains unknown. In a Japanese prospective study evaluating the characteristics of HCC in NAFLD patients without cirrhosis[37] with a median follow-up period of 52.7 months, researchers found that, liver function was better preserved in the non-cirrhotic HCC group, with platelet count and prothrombin time being significantly higher than in the cirrhotic HCC group. Following curative treatment, the 5-year recurrence rate was significantly lower in the non-cirrhotic group (40.9%) than in the cirrhotic HCC group (85.7%) and the 5-year survival was improved. Alternatively, in a retrospective cohort of 225 NAFLD-HCC patients treated in Sweden, 37% were non-cirrhotic and these patients were significantly older, had larger tumors, less frequently underwent liver transplantation but more frequently resection, and overall mortality was not significantly different between the 2 groups[36]. Therefore, it remains unclear if the benefit of a non-cirrhotic liver allowing, for example, more aggressive curative liver resection is offset by later diagnosis (presumably, in part, due to less common HCC screening) and older age.
American Association for the Study of Liver Diseases and European Association for the Study of the Liver guidelines recommend screening for HCC by performing liver ultrasonography every six months in at-risk populations, defined by the presence of advanced fibrosis or cirrhosis[1,28]. Although the evidence base for such recommendations is mostly based on observational studies, a meta-analysis comprising 47 retrospective and prospective studies demonstrated that HCC surveillance is associated with improved early tumor detection, an increased likelihood to receive curative therapy and overall survival in cirrhotic patients, regardless of etiology[38]. However, while over 90% of the patients in this meta-analysis had underlying cirrhosis, only around half of NAFLD-HCC patients have cirrhosis at diagnosis[18,22,28-34], creating an inherent challenge to the existing surveillance framework based on fibrotic progression. We have shown in a modelling study that risk-score stratified HCC screening was cost-effective, at least in cirrhotic patients and outperformed currently recommended non-stratified biannual ultrasound screening in all patients with cirrhosis[39]. Other researchers have proposed using polygenic risk scores based on current genetic knowledge in order to help stratify the risk of hepatocellular carcinoma independently of fibrosis staging[40,41]. For instance, in one study assessing polygenic risk scores in NAFLD subjects combining variants in PNPLA3, TM6SF2, GCKR, and MBOAT7 genes and adjusted for HSD17B13, Bianco et al.[42] found that their score improved accuracy in detecting HCC and may be a useful tool to aid HCC risk stratification in NAFLD. Potential future alternative strategies for stratifying HCC risk in NAFLD subjects include scores utilizing readily available clinical information and serum tests, gene signatures, liver elastometry, blood based biomarkers such as AFP, lens culinaris agglutinin-reactive AFP, and des-γ-carboxy prothrombin[43-46].
MECHANISMS OF HEPATOCARCINOGENESIS IN THE CONTEXT OF MS
The risk factors and pathogenetic mechanisms of HCC development in patients with metabolic liver disease are multifactorial and remain incompletely understood; however, a number of mechanisms have been implicated [Figure 1][47].
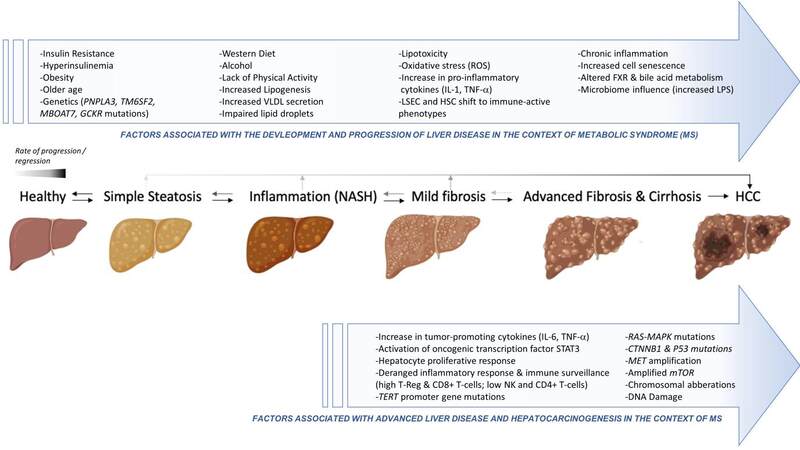
Figure 1. Multifactorial processes involved in metabolic liver disease evolution, from steatosis to HCC development.
Insulin resistance, diet and physical activity
Since the 1990s, insulin resistance has been implicated as the key mechanism driving NAFLD progression and has been frequently reported as a risk factor for HCC in NAFLD patients[48,49]. In a population-based case-control study examining SEER-Medicare records[50], researchers found that 43% of the 2061 HCC patients identified had diabetes, a value significant greater than non-cancer controls (19%). Even after adjusting for other HCC risk factors (HCV, HBV, alcoholic liver disease, and hemochromatosis), diabetes was associated with a threefold increase in HCC risk. In fact, researchers found a significant positive interaction between HCV and diabetes (P < 0.0001). In a recent study from the Mayo Clinic[51] aiming to quantify the association between diabetes and the risk of HCC, researchers found that 71% of the 354 patients admitted with NASH and cirrhosis between 2006 and 2015 had diabetes. Following a median follow-up 47 months, 30 patients developed HCC. Diabetes was independently associated with an increased risk of developing HCC (HR = 4.2, 95%CI: 1.2-14.2, P = 0.02), along with age (per decade, HR per decade 1.8, 95%CI: 1.2-2.6, P < 0.01) and low serum albumin (HR = 2.1, 95%CI: 1.5-2.9, P < 0.01). Other metabolic risk factors, including body mass index (BMI), hyperlipidemia, and hypertension, were not associated with HCC risk. These results were externally validated in a liver transplant registration cohort (UNOS) including all registrants with NASH between 2004 and 2017, where diabetes was still found to be an independent predictor of HCC (HR = 1.3, 95%Cl: 1.0-1.7, P = 0.3).
A strong link between MS and cancer deaths, including HCC has been long well-established. A 2003 landmark New England Journal of Medicine study[52] calculated that obesity confers a relative risk of liver cancer development of 1.90 (95%CI: 1.46-2.47) in individuals with BMIs of 30.0 to 34.9 and up to 4.52 (95%CI: 2.94-6.94) in individuals with BMIs of 35.0 to 39.9. Given the strong link between obesity and liver cancer, many studies have seeked to examine the impact of lifestyle interventions on HCC risk in both mouse models and human studies. In a murine study seeking mechanistic insights into obesity’s HCC promoting activity[53], researchers fed mice either normal chow (LFD) or a high-fat diet (HFD) for nine months. As expected, the mice on the HFD gained more weight, saw an increase in relative liver weight, and had increased liver triglycerides, serum transaminases, glucose intolerance, and hepatosteatosis. Importantly, when analyzed at 9 months of age, HFD mice exhibited many more HCCs per liver than their LFD-maintained mice counterparts. Dietary obesity also increased tumor size and incidence. Obesity-promoted HCC development here was dependent on enhanced production of the tumor promoting cytokines IL-6 and TNF, which cause hepatic inflammation and activation of the oncogenic transcription factor STAT3. In another murine study investigating whether nutrients like sugar or fat drive HCC tumorigenesis independently of obesity[54], researchers found that mice fed high-sugar diets had the greatest liver tumor incidence while dietary fat intake was not associated with tumorigenesis, suggesting an independent role of sugar metabolism in the pathophysiology of diet-induced hepatic tumorigenesis. Reversely, increased physical activity appears to have a beneficial effect on HCC risk. In a 2015 study[55], mice fed standard chow (10% fat diet) were randomly divided into sedentary or exercise groups (the exercise group ran on a motorized treadmill for 1 h per day throughout the duration of the study). After 32 weeks, the exercise-group demonstrated significantly fewer tumors per liver, as well as a smaller total tumoral volume per liver. Notably, exercise did not affect steatosis and had no effect on the non-alcoholic fatty liver disease activity score (NAS). Exercise effectively led to decreased tumor cell proliferation and stimulated the phosphorylation of AMPK and its substrate raptor, which decreased the kinase activity of mTOR. Human studies have confirmed these experimental observations. A recent study utilized one of the world’s largest international cohorts spanning 10 nations, the European Prospective Investigation into Cancer and Nutrition[56] to assess the impact of vigorous physical activity on different types of liver cancer in 467,336 men and women with a median follow up of 14.9 years. Here, researchers found that the multivariable-adjusted HR of HCC was 0.55 (95%CI: 0.38-0.80) comparing active and inactive individuals, with waist circumference and BMI accounting for ~40% and 30%, respectively of the overall association of total physical activity and HCC. For individuals reporting vigorous physical activity (> 2 h/week) compared to those reporting no vigorous activity, the HR for HCC was 0.50 (95%CI: 0.33-0.76), after accounting for potential confounding factors. Overall, a 45% lower risk of HCC was observed when comparing high and low levels of physical activity. Regularly engaging in vigorous physical activity was associated with a 50% lower risk of HCC.
Genetic factors
In the last two decades, much research has been conducted on the hereditary and specific genetic contributors to HCC development in the context of NAFLD. Various family studies have shown that the risk of developing NAFLD increases with the number of ancestors affected by the disease, with hepatic fibrosis being more common in the same family group[57,58]. Next generation sequencing, genome wide association studies, and advanced computational data analyses have offered the first few candidates genes involved in HCC pathogenesis in NAFLD[40]. A range of single nucleotide polymorphisms and other genetic variants in genes involved in the regulation of hepatic lipid metabolism, such as the patatin-like phospholipase domain-containing protein 3 (PNPLA3), transmembrane 6 superfamily member 2
Single nucleotide polymorphisms associated with NAFLD, advanced fibrosis, and HCC development
| Gene | SNP | Postulated pathogenesis | NAFLD | Advanced fibrosis (F3-F4) | HCC | Ref. |
| PNPLA3 | rs738409C>G | Protein involved in the control of triglycerides, phospholipids and retinoic acid axis in lipid droplets Rs738409C>G leads to accumulation of polyunsaturated fatty acids and retinoids in lipid droplets, leading to steatosis and activation of hepatic stellate cells | CG × CC: OR = 1.757, (95%CI: 1.037-2.977), P = 0.0044 GG × CC: OR = 3.296, (95%CI: 1.504-7.225), P = 0.0044[61] | OR = 2.33, (95%CI: 1.66-3.27, Chi squared 24.8, P < 0.0001) (Cochran-Armitage Chi squared for trend 22.68, P < 0.0001)[62] | GC vs. CC: unadjusted OR = 2.52, (95%CI: 1.55-4.10), P = 0.0002 GG vs. CC: OR = 12.19, (95%CI: 6.89-21.58), P < 0.0001)[62] | Mazo et al.[61] Liu et al.[62] |
| TM6SF2 | rs58542926C>T (E167K variant) | Protein involved in the secretion of very low-density lipoprotein from the hepatocyte Rs58542926 C>T causes a reduced expression of protein, leading to accumulation of VLDL in hepatocytes | OR = 2.13, (95%CI: 1.36-3.30) P = 0.0009; n = 3273[79] | OR = 1.88, (95%CI: 1.41-2.5) P = 1.63 × 10-5, n = 1074[63] | OR = 1.922, (95%CI: 1.31-2.81), P = 6.81 × 10-4 (Univariate analysis of homozygote, significance lost at multivariate analysis)[63] | Pirola et al.[79] Liu et al.[63] |
| MBOAT7 | rs641738C>T | Six-transmembrane protein involved in phospholipids and intracellular lipid remodeling in Lands cycle Rs641738C>T reduces MBOAT7 activity | OR = 1.17, (95%CI: 1.05-1.3), pz = 0.003 (recessive model CC+CT vs. TT in overall population)[86] | OR = 1.22, (95%CI: 1.03- 1.45), pz = 0.021 (recessive model CC+CT vs. TT in Caucasian Population)[86] | OR = 1.64, (95%CI: 1.18-2.27), pz = 0.003 [dominant model (CC vs. CT+TT) of inheritance in overall population][86] | Teo et al.[86] |
| GCKR | rs1260326 (protein variant P446L) | Regulates the flux of glucose and de novo lipogenesis in hepatocytes | OR = 1.38, (95%CI: 1.25- 1.53), P = 9.6 × 10-10[91] | ~ | OR = 1.84, (95%CI: 1.23- 2.75), P = 0.0031, significance = (P = 5.3×10-7)[91] | Romeo et al.[40] Kawaguchi et al.[91] |
| HSD17B13 | rs72613567T>TA | Regulates retinoic acid enzymatic activity and availability The two variants determine lower activity of the protein function | NAFLD progression risk was reduced by 17% (95%CI: 8-25) in heterozygotes and by 30% (95%CI: 13-43) in homozygotes[95] | Reduced risk of developing significant fibrosis OR = 0.77, (95%CI: 0.58-1.03) or any fibrosis OR = 0.42, (95%CI: 0.17-1.00)[95] | Reduced HCC risk demonstrated in total liver disease group (not NAFLD only) pooled OR = 0.64 (95%CI: 0.53-0.77), P heterogeneity = 0.236, I2 = 27.9% (P Egger = 0.741, P Begg = 0.806)[95] | Wang et al.[95] |
The most significant variant uncovered in recent years is the rs738409C>G variant in the PNPLA3 gene, which is considered a major determinant of hepatic fat content[59]. The protein encoded by the PNPLA3 gene is involved in the retinoic acid axis and it is responsible for the mobilization of triglycerides from hepatic lipid droplets, with a lipase-like activity[60]. This protein’s function is impaired in the rs738409C>G variant, leading to the accumulation of polyunsaturated fatty acids and retinoids in lipid droplets as well as steatosis[61]. This in turn activates hepatic stellate cells, inducing a more inflammatory and fibrogenic phenotype involving GM-CSF and CCL-5 signaling[61]. In a Brazilian multicenter cross-sectional study[61] evaluating 248 patients with biopsy-proven NAFLD and 134 healthy controls, researchers found that the presence of rs738409C>G in both heterozygous (CG) individuals and homozygous (GG) individuals was associated with an increased risk of developing NAFLD by a 1.8-fold and 3.3-fold increase respectively. Interestingly, this study suggests there is a dose effect in this variant. Further research has confirmed that this variant is also associated with clinically relevant hepatic fibrosis (advanced fibrosis and cirrhosis) in a genetic dose-dependent manner [OR = 2.33 (95%CI: 1.66-3.27), P < 0.0001][62]. Notably it has also been associated with a 3-fold increased risk of developing HCC[63-66]. Although the association between the rs738409C>G variant and HCC is particularly clear in patients with alcoholic liver disease, the effect in subjects with NAFLD appears lower and whether this association is independent of fibrosis remains unclear[67,68]. Recent studies have also evaluated the role of the PNPLA3 variant in diminishing the response to certain treatments, such as with statins or omega 3[59,69-72]. Accordingly, screening patients for the presence of this variant in order to better tailor therapy is becoming of particular interest[40].
The TM6SF2 gene rs58542926C>T variant, named the TM6SF2 E167K variant has been shown to be strongly linked to ALD and hepatocellular carcinoma[73]. Recently, its role in metabolic liver disease has also been uncovered, even in the absence of alcohol consumption (NAFLD)[74-77]. The loss of function of this Golgi membrane protein, expressed mainly in the liver and intestine is responsible for lipid retention in the hepatocyte via reduction in lipidation and number of lipoprotein particles[74-77]. The TM6SF2 E167K variant increases the risk of NAFLD by causing lipids to accumulate in intracellular droplets[78]. While this variant poses an increased risk for liver damage, it is protective against cardiovascular disease due to the reduction in circulating lipoproteins[78]. A meta-analysis reviewing 10 studies spanning pooled estimates of random effects in over 100,000 individuals confirmed the role of the E167K variant as an important modifier of blood lipid traits in different populations[79]. In the study, homozygous carriers of this minor T allele variant were found to have a moderately higher risk of NAFLD (OR = 2.13, 95%CI: 1.36-3.30, P = 0.0009, n = 3273)[79]. This variant was also associated advanced fibrosis [(F0-F1 vs. F2-F4), OR = 1.88, 95%CI: 1.41-2.5, P = 1.63 × 10-5, n = 1074][63] and HCC (OR = 1.922, 95%CI: 1.31-2.81, P = 6.81 × 10−4)[63]. Overall the association of the rs58542926C>T SNP with HCC remains incompletely understood, in particular in NAFLD patients, although this polymorphism is part of the previously discussed polygenic risk score[42] and some data suggests an independent association between the polymorphism and NAFLD-HCC[80,81].
Another SNP known for its involvement in hepatic disease is rs641738C>T in MBOAT7, initially studied for its role in ALD and cirrhosis development[82]. Recent research has also assessed the SNP’s role also in promoting NAFLD, fibrosis, cirrhosis and HCC[80,83]. MBOAT7 encodes lysophosphatidylinositol acyltransferase 1, a six-transmembrane protein anchored to lipid droplets, the endoplasmic reticulum and mitochondrial membranes[84,85]. Emerging evidence suggests the enzymatic activity of this protein is likely related to the remodeling of phospholipids with polyunsaturated fatty acids in the Lands Cycle[40,83]. A landmark 2021 meta-analysis analyzing 1,066,175 individuals of European descent across 42 studies[86], found that the rs641738C>T variant was significantly associated with NAFLD in Caucasian adults as modelled by a recessive model of inheritance (CC + CT vs. TT) (OR = 1.17, 95%CI: 1.05-1.3, P = 0.003)[86]. Here researchers demonstrated that the presence of any fibrosis (F0 vs. F1-4) is positively associated with rs641738C>T in all populations (OR = 1.27, 95%CI: 1.04-1.54, pz = 0.018) and that advanced fibrosis (F0-F2 vs. F3-F4) also exhibits a positive association with rs641738C>T in Caucasian populations (OR = 1.22, 95%CI: 1.03-1.45, pz = 0.021). In this study[86] the rs641738C>T was also associated with HCC, significantly increasing the odds of malignant transformation in NAFLD when utilizing dominant model (CC vs. CT+TT) of inheritance (OR = 1.64, 95%CI: 1.18-2.27, pz = 0.003).
Candidate gene studies have led to the identification of the GCKR gene as a modulator of circulating triglycerides, glucose and have confirmed the gene’s role in NAFLD[40,41,87]. The intracellular glucokinase activity of this gene regulates the flux of glucose into hepatocytes by controlling the levels of glucose phosphate and fructose-6-phosphate which exert a negative feedback on GCKR. The reduction in activity of the glucokinase in turn reduces malonyl-CoA synthesis and consequent de novo lipogenesis via the induction of glycolysis and the stimulation of carbohydrate-responsive element-binding protein[88,89]. The GCKR rs1260326 variant encodes a loss of function variant (P446L) protein which increases the activity of intracellular glucokinase, thereby increasing intracellular glucose phosphate, and leading to de novo lipogenesis via the induction of glycolysis and the stimulation of carbohydrate-responsive element-binding protein[40,90]. This process results in higher liver fat content and higher levels of circulating triglycerides, but lower insulin resistance and a reduced predisposition to diabetes in carriers[40]. In a 2018 Japanese risk estimation model for NAFLD[91], researchers found a significant association [rs1260326 in GCKR (OR = 1.38, 95%CI: 1.25-1.53, P = 9.6 × 10-10], between GCKR rs1260326 and NAFLD in 936 histologically confirmed patients (ntotal = 8608). However, in this same study the authors found that the association between GCKR rs1260326 and HCC was non-significant, and it remains unclear whether there is an independent association with HCC development in NAFLD.
In juxtaposition to all these hepato-detrimental variants, a recent study utilizing data from the DiscovEHR cohort[92] revealed that the common loss‐of‐function rs72613567T>TA variant in the 17β‐hydroxysteroid dehydrogenase 13 (HSD17B13) gene has a protective effect on the liver for NAFLD and ALD, as well as on associated liver fibrosis/cirrhosis[93], and HCC[94,95]. The 17β‐hydroxysteroid dehydrogenases are a family of 14 enzymes with retinol dehydrogenase activity that localize to lipid droplets in hepatocytes where they’re involved in the conversion of retinol to retinoic acids[96-98]. A recent large meta-analysis[95] investigating the role of the rs72613567 variant in NAFLD progression and any liver disease progression toward HCC found that the loss of function conferred by the variant was associated with a reduced risk of NASH development, but not steatosis [NAFLD progression risk was reduced by 17% (95%CI: 8-25) in heterozygotes and by 30% (95%CI: 13-43) in homozygotes], and it was associated with a reduced risk of developing significant fibrosis (OR = 0.77, 95%CI: 0.58-1.03) or any fibrosis (OR = 0.42, 95%CI: 0.17-1.00) even when adjusting for BMI, sex, age, and PNPLA3 variants. This study[95] also suggests a protective effect of the variant against HCC development. The NAFLD cohort in this study was too small to conclusively determine the HCC risk reduction in this subgroup, however the reduced HCC risk was demonstrated in the total liver disease group (pooled OR = 0.64, 95%CI: 0.53-0.77).
Immune response and dysbiosis
Obesity and MS can lead to chronic inflammation, which is independently associated with an increased cancer risk in general[99]; however, metabolic dysfunction also leads to a particular immune dysfunction underlying the pathogenesis of NAFLD and NASH, thereby directly promoting HCC development[99,100]. In addition to its metabolic and detoxifying functions, the liver is also a pivotal immunological organ, home to a coordinated network of innate immune cells, including Kupffer cells, dendritic cells, and lymphocytes[101]. Hepatocytes and liver sinusoidal endothelial cells are not formally innate immune cells, but in response to stress, they transition from immune-tolerant states (correlated with the production of IL-10, TGF-β, etc.) to immune-active phenotypes (characterized by the secretion of IL-1, TNF-α, etc.)[102]. This complex immune milieu employs pattern recognitions receptors ligation and activation of complement receptors or scavenger receptors to function as the liver’s first line of defense against gut-derived microbial compounds and circulating pathogens[101]. Recent research has emphasized the key role of innate immune mechanisms as pivotal drivers of inflammation in NASH[101]. In particular, natural killer (NK) cells seem to play a crucial role in metabolic liver disease and HCC development[103]. In response to the hepatic inflammation associated with NASH, NK cells increase in quantity and the number of ligands of various NK cells in the liver is increased[104,105]. NK cells work to prevent fibrotic development in the liver by killing hepatic stellate cells responsible for causing fibrosis[105]. In NASH, hepatic stellate cells are chronically activated due to dysregulated senescence and thus are able to resist NK cell-mediated cytolytic attacks[105]. In HCC, an increase in Treg cells and subsequent impaired production IFN-γ and cytotoxic function, reduces the number of NK which limits their tumoral surveillance function, a crucial component to in fact combat HCC[106]. In a German murine study, researchers found that myeloid-derived suppressor cells work to disarm the innate immune system, specifically they inhibit NK cell’s antitumoral function, further enabling the development of HCC[107].
Sterile inflammation, a form pathogen-free inflammation triggered by damage-associated molecular patterns (DAMPs) such as nuclear DNA or uric acid released by distressed cells, can activate hepatic inflammation and represents an important mechanism of injury in NASH[108,109]. DAMPs prompt the assembly of a cytosolic protein complex termed the inflammasome, which is required for the conversion of DAMP signals into the activation secretion pro-inflammatory cytokines such as IL-1β, IL-18, and other cytokines[110].
Several features of MS, such as obesity and high-fat, high-fructose western diets are recognized as major risk factors for HCC; however, the precise molecular mechanisms linking these events remains poorly understood. The first part of this link, between dysbiosis and the development of obesity, MS, NAFLD, and NASH has been established across many studies[111-121]. Limited evidence also suggests that the gut microbiota is also involved in HCC development, particularly by increasing LPS levels and contributing to a pro-inflammatory microenvironment in the liver[122]. In a Japanese study examining hepatocarcinogenesis in obese mice[123], researchers found that the administration of antibiotics and gut sterilization led to a decreased risk of developing HCC in the mice; however, it had no regression effect on already established tumors. This implicates that gut sterilization and antibiotic treatment, which works to counter dysbiosis and thus diminishes the release of pro-inflammatory and pro-carcinogenic factors, may be used as a possible treatment to prevent the development of HCC.
Accumulating evidence has also implicated bile acid signaling in the pathogenesis and progression of HCC[124]. Bile acids are endogenous signaling molecules which can activate the nuclear farnesoid X receptor (FXR), a receptor responsible for regulating host metabolism, bile acid homeostasis, immune responses, lipid and glucose homeostasis, insulin signaling, and known for its role in activating hepatoprotective signaling pathways[125-127]. A 2017 study comparing germ-free and conventionally raised wild type vs. Fxr-/- mice fed high fat diets for 10 weeks, found that gut microbiota promoted weight gain and hepatic steatosis in an FXR-dependent manner. Furthermore, the researchers transferred the cecal microbiota from the high fat diet-fed mice to germ-free wild type mice and found that the obese phenotype was in fact a transferable phenomenon, and was associated with increased β-cell mass, increased adipose inflammation, increased steatosis, and expression of genes involved in lipid uptake in the colonized mice. In human studies, the FXR receptor has been implicated as one of the main factors mediating the beneficial effects on NAFLD following bariatric surgery[111,112,126,128]. This hepatoprotective effect also appears to have a role in the context of HCC, with studies evidencing a decrease in FXR expression in human HCC samples[129]. Several studies aiming to better characterize the role of FXR in hepatocarcinogenesis utilizing the same Fxr-/- mice models have found that both these male and female knockout mice spontaneously developed HCC in tandem with bile acid homeostasis disruption and activation of the Wnt/β-catenin signaling pathway[124,129-131].
PATHOLOGICAL ASPECTS OF NAFLD-HCC
HCCs arising in the context of MS or NASH can present with a wide array of morphological patterns, including well or moderately differentiated carcinomas with a trabecular pattern [Figure 2A], pseudoglandular pattern [Figure 2B], mild fatty changes [Figure 2C], or mixed patterns [Figure 2D]. These patterns frequently exhibit aspects of the steatohepatitic subtype of HCC (SH-HCC, Figure 2E). This SH-HCC variant was not included in WHO classification 2010[132], but rather only a cytological variant nominated “fatty change” was described. However, in the most recent WHO 2019 classification, SH-HCC is now recognized at a formal HCC subtypes[133](WHO 2019). This subtype of HCC is characterized by steatosis, ballooning of the hepatocytes, pericellular fibrosis, Mallory-Bodies, and inflammation, recapitulating the characteristics of NASH, as described first by Salomao et al.[134]. In this study, the subjects were HCV positive, however, the authors observed that this subtype was mainly present in patients comorbid with NAFLD/NASH, suggesting an association between the underlying metabolic condition and the SH-HCC subtype. A series of subsequent studies further found that this HCC subtype was common not only in patients with non-alcoholic steatohepatitis or alcoholic steatohepatitis, but also in patients with stigmata of MS without established NASH[135]. These data were also confirmed by Shibahara et al.[136] in a separate study that found a statistical correlation with diabetes mellitus and levels of cholesterol and triglycerides, as compared to other subtypes of HCC. Notably, a very recent study spanning 2 different cohorts demonstrated that the SH-HCC variant is associated with steatohepatitis in general, independently of the etiology[137]. This subtype has been shown to be associated with the activation of the IL-6/STAT3 signaling pathway[138], whilst it rarely harbors the activated Wnt/β-catenin pathway[139].
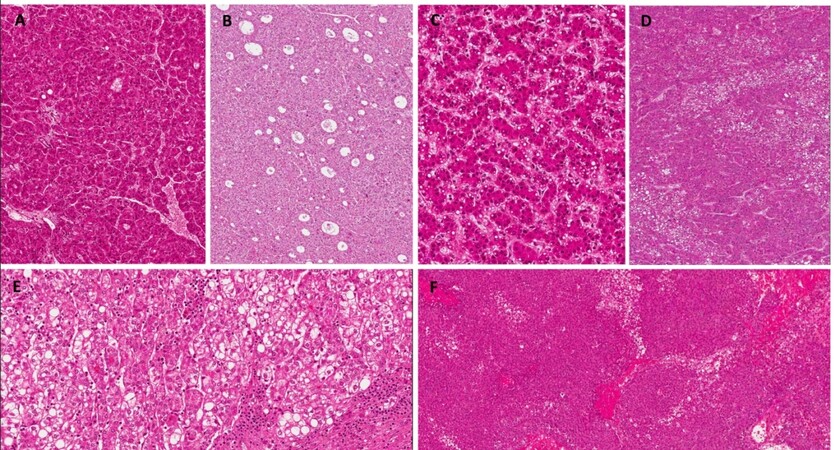
Figure 2. Histological patterns of HCC. (A) Trabecular pattern of HCC (Haematoxylin Eosin staining, 10×); (B) Pseudoglandular pattern of HCC (Haematoxylin Eosin staining, 10×); (C) Fatty change aspects of HCC (Haematoxylin Eosin staining, 20×); (D) Mixed pattern trabecular and fatty change (Haematoxylin Eosin staining, 10×); (E) Steatohepatitic HCC (Haematoxylin Eosin staining, 20×); and (F) Macrotrabecular HCC (Haematoxylin Eosin staining, 40×).
Although the SH-HCC subtype does not seem to influence the prognosis of patients[136], one must consider that these patients often come to our attention with larger tumoral masses because of the lack of a definite screening program able to detect small HCC. Larger HCC tend to be more heterogeneous, exhibiting different morphologies, typically higher grade and with more frequent vascular invasion. The most relevant subtype of HCC prognostically speaking is the macrotrabecular subtype, characterized by macrotrabeculae of neoplastic hepatocytes (> 6 trabeculae)[139] [Figure 2F], with an increased frequency of vascular invasions, thus conditioning a poor prognosis. This subtype has been associated with the mutation of TP53 gene, the amplification of FGF19 gene, the activation of angiogenesis because of overexpression of angiopoietin 2, and with vascular endothelial growth factor A[140]. As previously discussed, another important histological aspect that must be highlighted is the peculiar absence of a direct correlation between fibrotic stage and the activity of steatohepatitis in patients with metabolic liver disease. While livers with more active steatohepatitis
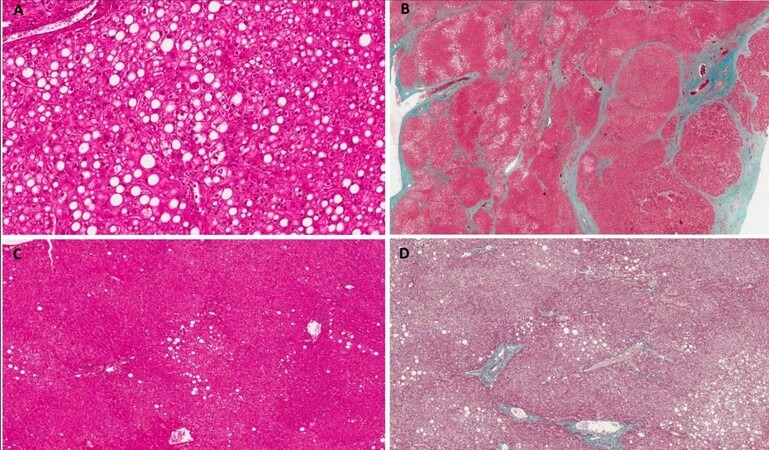
Figure 3. Histological findings of non-tumoral tissue in patients with NAFLD-HCC. (A) Example of NASH with diffuse steatosis, mainly macrovesicular, with ballooning, apoptotic hepatocytes and foci of inflammation (Haematoxylin Eosin staining, 10×); (B) Advanced fibrosis demonstrated with Masson’s thricrome staining (4×); (C) Mild steatosis in the context of NAFLD (Haematoxylin Eosin staining, 10×); and (D) Fibrosis limited to portal tract in a case of NASH (Masson’s thricrome, 10×).
PREVENTION AND MANAGEMENT OF HCC IN THE CONTEXT OF MS
Prevention of HCC
Given the pathogenesis of HCC in context of MS, current preventative measures focus on preventing NAFLD progression. To this effect, weight loss has been implicated as a key factor for improvement in both liver histology and liver biochemical tests on serology, as well as quality of life[144,145]. For example, in one of the largest prospective studies to date conducted in Cuba, 293 subjects with histologically documented NASH underwent a 52-week period of lifestyle changes including modified hypocaloric diet and increased exercise[146]; 25% of these patients achieved resolution of steatohepatitis, and 47% had reductions in nonalcoholic fatty liver disease activity score, while 19% had regression of fibrosis. The degree of weight loss here was independently associated with improvements in all NASH-related histological parameters while the extent of weight loss was proportional to the NASH resolution. In patients that lost ≥ 5% of their body weight, 58% had NASH resolution and 82% had a 2-point reduction in the NAFLD activity score (NAS). In comparison, of the patients who lost ≥ 10% of their body weight, 100% demonstrated reductions in NAS and 90% has resolution of NASH with 45% displaying regression of fibrosis. Discouragingly, only a minority of patients in this study (30%) achieved meaningful weight loss through lifestyle intervention alone and even fewer sustained this weight loss long term, matching real-world clinical practice. In particular, the Mediterranean diet has been associated to have a particularly protective effect in the development HCC[147], and lines of evidence suggest this may be mediated by a reduced hepatic fat content[148]. As discussed earlier, regular physical activity, may reduce the risk of developing HCC by up to 50% compared to sedentary individuals[149]. Bariatric surgery is perhaps the most effective NASH intervention and method of HCC prevention, with significant histological and clinical improvement evidenced across studies[150,151]. In a study involving 109 morbidly obese subjects with biopsy-proven NASH, clinical, biological, and histological data were collected before surgery and 1 year after surgery. In the 1-year follow up 85% of subjects had a complete resolution of NASH, especially subjects that had a milder form of NASH[152]. A prospective study recently published from the same research group in France conducted a long term follow up of patients with NASH who underwent bariatric surgery and found that the reduction in fibrosis following bariatric surgery is progressive, continuing for at least 5 years[153]. A 2020 US-based study[154] analyzing 4112 matches of bariatric patients with obese controls found that bariatric surgery was preventative for HCC, with the operated cohort reporting fewer new cases of HCC (0.05% vs. 0.34%, P = 0.03) than their non-operated counterparts over a median follow up of 7.1 years.
As regards pharmacological prevention measures, an Italian 2015 cross-sectional study[69] demonstrated a protective effect of statin use in individuals at high risk of HCC development. Notably, the protective effect of statins on steatohepatitis from steatosis was not significant in subjects carrying the I148M PNPLA3 risk variant, however in wild type individuals, statin use was found to be protective against steatosis, steatohepatitis, and fibrosis stage F2-F4 in a dose-dependent manner in in at-risk individuals (adjusted P < 0.05 for all). This is in accordance with previous research[79] which also found statin use is associated with a significant reduction in the risk of HCC among patients with diabetes (OR = 0.74, 95%CI: 0.64-0.87), possibly related to the anti-inflammatory properties of statins mediated by JAK inhibition.
Given the well-established benefits of aspirin in the prevention of colorectal cancer, many recent studies have investigated the use of aspirin for the prevention other cancers, including HCC. Early evidence suggests that aspirin may work to prevent liver disease progression and hepatocarcinogenesis via a range of mechanisms including inhibition of the proinflammatory cyclooxygenase-2 enzyme, the modulation of bioactive lipids, and the inhibition of platelet degranulation[155-159]. A recent New England Journal of Medicine populational study[160] aimed to quantify the association of aspirin with HCC and liver-related mortality. Using a national registry, the researchers identified all Swedish adults diagnosed with chronic hepatitis B or hepatitis C between 2005 to 2015 and compared patients without a history of aspirin use in this group (50,275 patients) to ones starting to take low-dose aspirin (14,205 patients). With a median follow-up of 7.9 years, the estimated cumulative incidence of HCC was 4.0% among aspirin users vs. 8.3% among non-aspirin users (adjusted HR = 0.69, 95%CI: 0.62-0.76). This effect appeared to be dose-dependent, with the adjusted hazard ratio being more significant with increased years of use; with short term use (3 months to 1 year) the adjusted HR was 0.90 (95%CI: 0.76-1.06), while with long term use (5 or more years) the adjusted HR was 0.57 (95%CI: 0.42-0.70) suggesting a causal link. Notably, the 10-year liver-related mortality was 11.0% in the aspirin user group versus 17.9% in the nonuser group [adjusted HR = 0.73 (95%CI: 0.67-0.81) without conferring an increased gastrointestinal bleeding risk aspirin users 7.8% risk vs. non-users 6.9%]. The strong correlation of aspirin use with a decreased HCC risk and overall liver mortality in patients with viral hepatitis warrants further work to assess aspirin’s role in HCCs of metabolic origin.
Several studies, particularly in Asia have investigated the effect of thiazolidinediones (TZDs) on the risk of HCC development among diabetes mellitus patients, and a growing body of evidence suggests that TZDs may in fact have an effective role. In a Taiwanese-based 2017 study[161], 76,349 newly diagnosed patients were identified between 2000 and 2010. Among diabetics who received TZDs, the incidence of HCC development was significantly lower (418.3 vs. 484.6 per 100,000 person-years) (aHR = 0.53, 95%CI: 0.38-0.77).
The use of metformin in diabetic subjects has been widely associated with a reduced incidence of HCC[162-166]. The mechanism underlying this effect is related to the activation of AMPK, the same mechanism implicated in the beneficial effects of physical exercise in HCC[167]. In a 2019 systematic review evaluating the use of metformin as a protective factor for HCC in diabetic patients[168], researchers selected 8 studies; 4 case-control and 4 cohort studies. All of the studies observed that metformin therapy was associated with a lower risk of HCC compared with non-metformin therapy. A meta-analysis of the case-control studies found odds ratio of 0.468 (95%CI: 0.275-0.799) for the association between the use of metformin and HCC development. Inversely, insulin was associated with an increased risk of HCC development.
Management of HCC
The therapeutic algorithm adopted for HCC patients in the context of MS does not differ from that used for other etiologies of HCC[1]. As with all HCC cases, the treatment allocation is based on the Barcelona Clinical Liver Cancer staging criteria[169].
Unfortunately, the majority of HCC patients do not qualify for curative treatment approaches, such as surgical resection, at the time of diagnosis given tumor extent or underlying liver dysfunction, although we have previously discussed that NAFLD-HCC patients often have better liver function due to the absence of underlying cirrhosis in a significant proportion of patients[170]. Of the patients that qualify, studies have reported significant perioperative morbidity (> 50%) and mortality (> 10%) in NAFLD-HCC patients undergoing curative resection[171]. For this reason, understanding the true survival benefit in these patients is crucial when recommending curative treatment. In a recent Singaporean study examining the outcomes of surgically resected NAFLD-HCC patients[34], researchers analyzed a 15-year prospective cohort of 996 HCC patients who underwent curative liver resection. Of these, 152 patients were identified as NAFLD-HCC based on a histological diagnosis. Compared to the 844 non-NAFLD HCC counterparts, a higher volume of complication rates was seen in the NAFLD-HCC group, which resulted in longer inpatient stay but lower 90-day mortality rates (NAFLD HCC 1.99% vs. non-NAFLD HCC 2.46%, P = 0.0355). In particular, NAFLD-HCC patients exhibited almost twice the proportion of minor complications (41.2% vs. 24.2%, P < 0.0001) and major complications (16.2% vs. 8.1%, P < 0.0001). However, after the perioperative period, long-term outcomes were significantly better in the NAFLD group in terms of overall survival (OS), with 5-year OS rates of 70.1% vs. 60.9% (P = 0.0355), although this result was limited in that the groups were not equally distributed on propensity score (P = 0.0411).
For patients with advanced stages of HCC, the multikinase inhibitor sorafenib was the first systemic agent to demonstrate a significant improvement in OS[172]. Approved worldwide in 2007, sorafenib remained the only available agent for HCC until 2016 as all phase III trials failed during this period. A recent international cohort study[173] examined the effect of sorafenib on OS in patients with NAFLD as compared to other etiologies. NAFLD-HCC patients comprised 3.6% of the 5201 HCC patients receiving sorafenib in this cohort. Despite NAFLD-associated patients being having a more advanced stage of HCC and starting at a lower dose of sorafenib, after adjusting for known prognostic factors, there was no difference in sorafenib-specific survival between NAFLD and other etiologies (HR = 0.99, 95%CI: 0.85-1.16, P = 0.92). There were also no observed differences in the OS of HCC patients with or without NAFLD (adjusted HR = 0.94, 95%CI: 0.76-1.16, P = 0.57). Interesting, emerging preclinical data[174] suggests sorafenib administered at approximately one-tenth the clinical dose for HCC, effectively prevents the progression of NASH in both mice and monkeys without any observed significant adverse effects. The benefit here was linked not to the kinase targets of sorafenib as seen in HCC, but rather the induction of mild mitochondrial uncoupling and subsequent AMPK activation.
In 2017, the HCC drug development pipeline saw a decisive shift as the first drugs other than sorafenib began showing promise. Regorafenib was approved in 2017[175] as a second-line therapy, lenvatinib as a first-line therapy in 2018[176], ramucirumab, and cabozantinib as second-line therapies in 2019[177,178], and the combination of nivolumab plus ipilimumab in 2020[39]; these are all approved for use in sorafenib-pretreated patients only. In 2020, the landmark IMbrave150 trial[179] (atezolizumab plus bevacizumab combination therapy) demonstrated an improvement in both overall survival and progression-free survival over sorafenib. The OS at 12-month was 67.2% (95%CI: 61.3-73.1) with atezolizumab-bevacizumab and 54.6% (95%CI: 45.2-64.0) with sorafenib, while the median progression-free survival was 6.8 months (95%CI: 5.7-8.3) and 4.3 months (95%CI: 4.0-5.6) in the respective groups (HR for disease progression or death 0.59, 95%CI: 0.47-0.76, P < 0.001). In a separate analysis of patient-reported outcomes from this trial[180], patients who received atezolizumab plus bevacizumab reported significantly longer delays in median time to deterioration of quality of life (median TTD 11.2 months vs. 3.6 months, HR = 0.63, 95%CI: 0.46-0.85), physical functioning (median TTD 13.1 months vs. 4.9 months, HR = 0.53, 95%CI: 0.39-0.73) and role functioning (median TTD 9.1 months vs. 3.6 months, HR = 0.62, 95%CI: 0.46-0.84). The median time to deterioration (TDD) of several disease-related symptoms (such as anorexia, diarrhea, fatigue, and pain) was also significantly longer after combined therapy. Largely based on this data, the combination therapy atezolizumab (anti-PD-L1) plus bevacizumab (anti-VEGF) was approved by the FDA and the EU and now represents a first-line therapy alternative for healthy patients, with no worse than Child-Pugh class A cirrhosis, a high performance status and with special attention to esophageal varices, where present[181]. Accordingly, a significant proportion of patients with NAFLD-related HCC will have become candidates for systemic front-line therapy with atezolizumab plus bevacizumab. Notably however, HCCs of non-viral etiology were poorly characterized and comprised only 30% of the total participants in this study.
As discussed earlier, NAFLD modifies the local immune microenvironment, causing a decrease of anti-tumoral CD4+ T cells while simultaneously promoting tumoral activity in CD8+ T cells, NK T cells, and Th17 cells[65,182,183], which has brought up questions about the potential implications for immunotherapy use in NAFLD-HCC patients[184]. Indeed, subgroup analyses of the recent CheckMate 459 phase III trial[185] revealed that nivolumab was less effective in terms of OS in HCC patients with non-viral etiologies (vs. sorafenib: HR = 0.95, 95%CI: 0.74-1.22). Subgroup analysis of the IMbrave150 trial[179] reported similar results in patients receiving atezolizumab plus bevacizumab with non-viral HCC (vs. sorafenib: HR = 0.91, 95%CI: 0.52-1.60). Notably, the studies did not differentiate between NAFLD and alcoholic liver disease in the non-viral HCC subgroup. Despite these limitations, recent data based on murine models[186] suggests that immunotherapy may be less effective for NAFLD-related HCC patients as related to a loss of CD4+ T-cells in the NASH liver and a more immunosuppressive immune cell phenotype in the tumor environment.
The presence of obesity, one of the principal features of the MS, may have implications on the efficacy of bevacizumab in HCC. Obesity has been shown to promote resistance to anti-VEGF therapy in breast cancer patients via the upregulation of IL-6, an inflammatory factor, and fibroblast growth factor 2, an angiogenic promoter. Similarly, in studies analyzing first-line bevacizumab use in metastatic colorectal cancer, visceral fat area, and obesity were associated with worse patient outcomes[187,188]. However, a pooled analysis of individual data from 2085 patients enrolled in eight first-line metastatic CRC trials between 1991 to 2013 found no interaction between obesity and bevacizumab in terms of survival[189]. Further research and more extensive subgroup analyses are needed to obtain a better understanding of the efficacy of combined atezolizumab plus bevacizumab therapy in the MS-related HCC subgroup of patients, along with further analysis of the non-cirrhotic patients in this group.
CONCLUSION
The worldwide increase in the prevalence of the MS is leading to an increase in liver disease and HCC incidence. Despite recent significant advances in the management of other etiologies of liver disease such as hepatitis C, the growing burden of NAFLD-HCC will continue to be a major health issue and will continue to increase the global burden of HCC. Additional research is clearly needed to improve risk stratification of HCC risk and to improve management strategies of patients with NAFLD-HCC. Although a lot has been learned about the molecular mechanisms underpinning hepatocarcinogenesis in these patients, much more has to be unraveled so that we can translate these findings into improved patient outcomes.
DECLARATIONS
Authors’ contributionsConception, drafting, and final review of the manuscript: Zampaglione L, Ferrari J, Pedica F, Goossens N
Availability of data and materialsNot applicable.
Financial support and sponsorshipGoossens N is supported by a grant of the Ligue Genevoise contre le cancer (ref number 1906).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Association for the Study of the Liver; Electronic address: easloffice@easloffice.eu; European Association for the Study of the Liver. EASL clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol 2018;69:182-236.
2. Liu Z, Jiang Y, Yuan H, et al. The trends in incidence of primary liver cancer caused by specific etiologies: results from the Global Burden of Disease Study 2016 and implications for liver cancer prevention. J Hepatol 2019;70:674-83.
3. Noureddin M, Rinella ME. Nonalcoholic fatty liver disease, diabetes, obesity, and hepatocellular carcinoma. Clin Liver Dis 2015;19:361-79.
6. Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J Hepatol 2016;64:1388-402.
7. Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: mayo clinic experiences with a hitherto unnamed disease. Mayo Clin Proc 1980;55:434-8.
9. Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology 2010;52:1836-46.
10. Eslam M, Sanyal AJ, George J. International Consensus Panel. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 2020;158:1999-2014.e1.
11. Eslam M, Newsome PN, Sarin SK, et al. A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J Hepatol 2020;73:202-9.
12. Younossi ZM, Stepanova M, Afendy M, et al. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin Gastroenterol Hepatol 2011;9:524-530.e1; quiz e60.
13. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016;64:73-84.
14. Foschi FG, Bedogni G, Domenicali M, et al. Prevalence of and risk factors for fatty liver in the general population of Northern Italy: the Bagnacavallo Study. BMC Gastroenterol 2018;18:177.
15. Soresi M, Noto D, Cefalù AB, et al. Metabolic Syndrome Study Group. Nonalcoholic fatty liver and metabolic syndrome in Italy: results from a multicentric study of the Italian Arteriosclerosis society. Acta Diabetol 2013;50:241-9.
16. Younossi ZM, Otgonsuren M, Henry L, et al. Association of nonalcoholic fatty liver disease (NAFLD) with hepatocellular carcinoma (HCC) in the United States from 2004 to 2009. Hepatology 2015;62:1723-30.
17. Cho EJ, Kwack MS, Jang ES, et al. Relative etiological role of prior hepatitis B virus infection and nonalcoholic fatty liver disease in the development of non-B non-C hepatocellular carcinoma in a hepatitis B-endemic area. Digestion 2011;84 Suppl 1:17-22.
18. Myers S, Neyroud-Caspar I, Spahr L, et al. NAFLD and MAFLD as emerging causes of HCC: a populational study. JHEP Rep 2021;3:100231.
19. Office fédéral de la statitique. Enquête Suisse Sur La Santé 2017. https://www.bfs.admin.ch/bfs/fr/home/statistiques/sante/determinants/exces-poids.html [Last accessed on 17 Jan 2021].
20. Younossi Z, Stepanova M, Ong JP, et al. Global Nonalcoholic Steatohepatitis Council. Nonalcoholic steatohepatitis is the fastest growing cause of hepatocellular carcinoma in liver transplant candidates. Clin Gastroenterol Hepatol 2019;17:748-755.e3.
21. Angulo P, Kleiner DE, Dam-Larsen S, et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015;149:389-97.e10.
22. Kanwal F, Kramer JR, Mapakshi S, et al. Risk of hepatocellular cancer in patients with non-alcoholic fatty liver disease. Gastroenterology 2018;155:1828-37.e2.
23. Amarapurkar DN, Dharod M, Gautam S, Patel N. Risk of development of hepatocellular carcinoma in patients with NASH-related cirrhosis. Trop Gastroenterol 2013;34:159-63.
24. Yatsuji S, Hashimoto E, Tobari M, Taniai M, Tokushige K, Shiratori K. Clinical features and outcomes of cirrhosis due to non-alcoholic steatohepatitis compared with cirrhosis caused by chronic hepatitis C. J Gastroenterol Hepatol 2009;24:248-54.
25. Hsiang JC, Bai WW, Raos Z, et al. Epidemiology, disease burden and outcomes of cirrhosis in a large secondary care hospital in South Auckland, New Zealand. Intern Med J 2015;45:160-9.
26. Estes C, Anstee QM, Arias-Loste MT, et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016-2030. J Hepatol 2018;69:896-904.
27. Goossens N, Bellentani S, Cerny A, et al. Nonalcoholic fatty liver disease burden - Switzerland 2018-2030. Swiss Med Wkly 2019;149:w20152.
28. Chalasani N, Younossi Z, Lavine JE, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018;67:328-57.
29. Kodama K, Kawaguchi T, Hyogo H, et al. Clinical features of hepatocellular carcinoma in nonalcoholic fatty liver disease patients without advanced fibrosis. J Gastroenterol Hepatol 2019;34:1626-32.
30. Mittal S, El-Serag HB, Sada YH, et al. Hepatocellular carcinoma in the absence of cirrhosis in united states veterans is associated with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol 2016;14:124-31.e1.
31. Tokushige K, Hyogo H, Nakajima T, et al. Hepatocellular carcinoma in Japanese patients with nonalcoholic fatty liver disease and alcoholic liver disease: multicenter survey. J Gastroenterol 2016;51:586-96.
32. Yasui K, Hashimoto E, Komorizono Y, et al. Japan NASH Study Group. Characteristics of patients with nonalcoholic steatohepatitis who develop hepatocellular carcinoma. Clin Gastroenterol Hepatol 2011;9:428-33; quiz e50.
33. Piscaglia F, Svegliati-Baroni G, Barchetti A, et al. HCC-NAFLD Italian Study Group. Clinical patterns of hepatocellular carcinoma in nonalcoholic fatty liver disease: a multicenter prospective study. Hepatology 2016;63:827-38.
34. Koh YX, Tan HJ, Liew YX, et al. Liver resection for nonalcoholic fatty liver disease-associated hepatocellular carcinoma. J Am Coll Surg 2019;229:467-478.e1.
35. Paradis V, Zalinski S, Chelbi E, et al. Hepatocellular carcinomas in patients with metabolic syndrome often develop without significant liver fibrosis: a pathological analysis. Hepatology 2009;49:851-9.
36. Bengtsson B, Stål P, Wahlin S, Björkström NK, Hagström H. Characteristics and outcome of hepatocellular carcinoma in patients with NAFLD without cirrhosis. Liver Int 2019;39:1098-108.
37. Tobari M, Hashimoto E, Taniai M, et al. The characteristics and risk factors of hepatocellular carcinoma in nonalcoholic fatty liver disease without cirrhosis. J Gastroenterol Hepatol 2020;35:862-9.
38. Singal AG, Pillai A, Tiro J. Early detection, curative treatment, and survival rates for hepatocellular carcinoma surveillance in patients with cirrhosis: a meta-analysis. PLoS Med 2014;11:e1001624.
39. Yau T, Kang YK, Kim TY, et al. Efficacy and safety of nivolumab plus ipilimumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib: the CheckMate 040 randomized clinical trial. JAMA Oncol 2020;6:e204564.
40. Romeo S, Sanyal A, Valenti L. Leveraging Human genetics to identify potential new treatments for fatty liver disease. Cell Metab 2020;31:35-45.
41. Trépo E, Valenti L. Update on NAFLD genetics: from new variants to the clinic. J Hepatol 2020;72:1196-209.
42. Bianco C, Jamialahmadi O, Pelusi S, et al. Non-invasive stratification of hepatocellular carcinoma risk in non-alcoholic fatty liver using polygenic risk scores. J Hepatol 2021;74:775-82.
43. Ioannou GN, Green P, Kerr KF, Berry K. Models estimating risk of hepatocellular carcinoma in patients with alcohol or NAFLD-related cirrhosis for risk stratification. J Hepatol 2019;71:523-33.
44. Adler M, Larocca L, Trovato FM, Marcinkowski H, Pasha Y, Taylor-Robinson SD. Evaluating the risk of hepatocellular carcinoma in patients with prominently elevated liver stiffness measurements by FibroScan: a multicentre study. HPB (Oxford) 2016;18:678-83.
45. Kanwal F, Singal AG. Surveillance for hepatocellular carcinoma: current best practice and future direction. Gastroenterology 2019;157:54-64.
46. Fujiwara N, Friedman SL, Goossens N, Hoshida Y. Risk factors and prevention of hepatocellular carcinoma in the era of precision medicine. J Hepatol 2018;68:526-49.
48. Marchesini G, Brizi M, Morselli-labate AM, et al. Association of nonalcoholic fatty liver disease with insulin resistance. Am J Med 1999;107:450-5.
50. Davila JA, Morgan RO, Shaib Y, McGlynn KA, El-Serag HB. Diabetes increases the risk of hepatocellular carcinoma in the United States: a population based case control study. Gut 2005;54:533-9.
51. Yang JD, Ahmed F, Mara KC, et al. Diabetes is associated with increased risk of hepatocellular carcinoma in patients with cirrhosis from nonalcoholic fatty liver disease. Hepatology 2020;71:907-16.
52. Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of US adults. N Engl J Med 2003;348:1625-38.
53. Park EJ, Lee JH, Yu GY, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010;140:197-208.
54. Healy ME, Lahiri S, Hargett SR, et al. Dietary sugar intake increases liver tumor incidence in female mice. Sci Rep 2016;6:22292.
55. Piguet AC, Saran U, Simillion C, et al. Regular exercise decreases liver tumors development in hepatocyte-specific PTEN-deficient mice independently of steatosis. J Hepatol 2015;62:1296-303.
56. Baumeister SE, Schlesinger S, Aleksandrova K, et al. Association between physical activity and risk of hepatobiliary cancers: a multinational cohort study. J Hepatol 2019;70:885-92.
57. Caussy C, Soni M, Cui J, et al. Familial NAFLD Cirrhosis Research Consortium. Nonalcoholic fatty liver disease with cirrhosis increases familial risk for advanced fibrosis. J Clin Invest 2017;127:2697-704.
58. Long MT, Gurary EB, Massaro JM, et al. Parental non-alcoholic fatty liver disease increases risk of non-alcoholic fatty liver disease in offspring. Liver Int 2019;39:740-7.
59. Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461-5.
60. BasuRay S, Smagris E, Cohen JC, Hobbs HH. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology 2017;66:1111-24.
61. Mazo DF, Malta FM, Stefano JT, et al. Validation of PNPLA3 polymorphisms as risk factor for NAFLD and liver fibrosis in an admixed population. Ann Hepatol 2019;18:466-71.
62. Liu YL, Patman GL, Leathart JB, et al. Carriage of the PNPLA3 rs738409 C >G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J Hepatol 2014;61:75-81.
63. Liu YL, Reeves HL, Burt AD, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat Commun 2014;5:4309.
64. Pelusi S, Baselli G, Pietrelli A, et al. Rare pathogenic variants predispose to hepatocellular carcinoma in nonalcoholic fatty liver disease. Sci Rep 2019;9:3682.
65. Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol 2019;16:411-28.
66. Singal AG, Manjunath H, Yopp AC, et al. The effect of PNPLA3 on fibrosis progression and development of hepatocellular carcinoma: a meta-analysis. Am J Gastroenterol 2014;109:325-34.
67. Yang J, Trépo E, Nahon P, et al. PNPLA3 and TM6SF2 variants as risk factors of hepatocellular carcinoma across various etiologies and severity of underlying liver diseases. Int J Cancer 2019;144:533-44.
68. Trépo E, Romeo S, Zucman-Rossi J, Nahon P. PNPLA3 gene in liver diseases. J Hepatol 2016;65:399-412.
69. Dongiovanni P, Petta S, Mannisto V, et al. Statin use and non-alcoholic steatohepatitis in at risk individuals. J Hepatol 2015;63:705-12.
70. Dongiovanni P, Donati B, Fares R, et al. PNPLA3 I148M polymorphism and progressive liver disease. World J Gastroenterol 2013;19:6969-78.
71. Pirazzi C, Adiels M, Burza MA, et al. Patatin-like phospholipase domain-containing 3 (PNPLA3) I148M (rs738409) affects hepatic VLDL secretion in humans and in vitro. J Hepatol 2012;57:1276-82.
72. Scorletti E, West AL, Bhatia L, et al. Treating liver fat and serum triglyceride levels in NAFLD, effects of PNPLA3 and TM6SF2 genotypes: results from the WELCOME trial. J Hepatol 2015;63:1476-83.
73. Liu Z, Que S, Zhou L, et al. The effect of the TM6SF2 E167K variant on liver steatosis and fibrosis in patients with chronic hepatitis C: a meta-analysis. Sci Rep 2017;7:9273.
74. Smagris E, Gilyard S, BasuRay S, Cohen JC, Hobbs HH. Inactivation of Tm6sf2, a gene defective in fatty liver disease, impairs lipidation but not secretion of very low density lipoproteins. J Biol Chem 2016;291:10659-76.
75. Mahdessian H, Taxiarchis A, Popov S, et al. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc Natl Acad Sci U S A 2014;111:8913-8.
76. Prill S, Caddeo A, Baselli G, et al. The TM6SF2 E167K genetic variant induces lipid biosynthesis and reduces apolipoprotein B secretion in human hepatic 3D spheroids. Sci Rep 2019;9:11585.
77. Kim DS, Jackson AU, Li YK, et al. Novel association of TM6SF2 rs58542926 genotype with increased serum tyrosine levels and decreased apoB-100 particles in Finns. J Lipid Res 2017;58:1471-81.
78. Dongiovanni P, Petta S, Maglio C, et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 2015;61:506-14.
79. Pirola CJ, Sookoian S. The dual and opposite role of the TM6SF2-rs58542926 variant in protecting against cardiovascular disease and conferring risk for nonalcoholic fatty liver: a meta-analysis. Hepatology 2015;62:1742-56.
80. Donati B, Dongiovanni P, Romeo S, et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci Rep 2017;7:4492.
81. Tang S, Zhang J, Mei TT, et al. Association of TM6SF2 rs58542926 T/C gene polymorphism with hepatocellular carcinoma: a meta-analysis. BMC Cancer 2019;19:1128.
82. Buch S, Stickel F, Trépo E, et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat Genet 2015;47:1443-8.
83. Mancina RM, Dongiovanni P, Petta S, et al. The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of european descent. Gastroenterology 2016;150:1219-1230.e6.
84. Gijón MA, Riekhof WR, Zarini S, Murphy RC, Voelker DR. Lysophospholipid acyltransferases and arachidonate recycling in human neutrophils. J Biol Chem 2008;283:30235-45.
85. Johansen A, Rosti RO, Musaev D, et al. Mutations in MBOAT7, encoding lysophosphatidylinositol acyltransferase I, lead to intellectual disability accompanied by epilepsy and autistic features. Am J Hum Genet 2016;99:912-6.
86. Teo K, Abeysekera KWM, Adams L, et al. EU-PNAFLD Investigators. , GOLD Consortium. rs641738C>T near MBOAT7 is associated with liver fat, ALT and fibrosis in NAFLD: a meta-analysis. J Hepatol 2021;74:20-30.
87. Willer CJ, Sanna S, Jackson AU, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet 2008;40:161-9.
88. Beer NL, Tribble ND, McCulloch LJ, et al. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum Mol Genet 2009;18:4081-8.
89. Barbara M, Scott A, Alkhouri N. New insights into genetic predisposition and novel therapeutic targets for nonalcoholic fatty liver disease. Hepatobiliary Surg Nutr 2018;7:372-81.
90. Raimondo A, Rees MG, Gloyn AL. Glucokinase regulatory protein: complexity at the crossroads of triglyceride and glucose metabolism. Curr Opin Lipidol 2015;26:88-95.
91. Kawaguchi T, Shima T, Mizuno M, et al. Risk estimation model for nonalcoholic fatty liver disease in the Japanese using multiple genetic markers. PLoS One 2018;13:e0185490.
92. Dewey FE, Murray MF, Overton JD, et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science 2016;354:aaf6814.
93. Abul-Husn NS, Cheng X, Li AH, et al. A protein-truncating HSD17B13 variant and protection from chronic liver disease. N Engl J Med 2018;378:1096-106.
94. Yang J, Trépo E, Nahon P, et al. A 17-beta-hydroxysteroid dehydrogenase 13 variant protects from hepatocellular carcinoma development in alcoholic liver disease. Hepatology 2019;70:231-40.
95. Wang P, Wu CX, Li Y, Shen N. HSD17B13 rs72613567 protects against liver diseases and histological progression of nonalcoholic fatty liver disease: a systematic review and meta-analysis. Eur Rev Med Pharmacol Sci 2020;24:8997-9007.
96. Moeller G, Adamski J. Integrated view on 17beta-hydroxysteroid dehydrogenases. Mol Cell Endocrinol 2009;301:7-19.
97. Tsachaki M, Odermatt A. Subcellular localization and membrane topology of 17β-hydroxysteroid dehydrogenases. Mol Cell Endocrinol 2019;489:98-106.
98. Saloniemi T, Jokela H, Strauss L, Pakarinen P, Poutanen M. The diversity of sex steroid action: novel functions of hydroxysteroid (17β) dehydrogenases as revealed by genetically modified mouse models. J Endocrinol 2012;212:27-40.
99. Luci C, Bourinet M, Leclère PS, Anty R, Gual P. Chronic inflammation in non-alcoholic steatohepatitis: molecular mechanisms and therapeutic strategies. Front Endocrinol (Lausanne) 2020;11:597648.
100. Gehrke N, Schattenberg JM. Metabolic inflammation-a role for hepatic inflammatory pathways as drivers of comorbidities in nonalcoholic fatty liver disease? Gastroenterology 2020;158:1929-1947.e6.
101. Cai J, Zhang XJ, Li H. The role of innate immune cells in nonalcoholic steatohepatitis. Hepatology 2019;70:1026-37.
103. Koo SY, Park EJ, Lee CW. Immunological distinctions between nonalcoholic steatohepatitis and hepatocellular carcinoma. Exp Mol Med 2020;52:1209-19.
104. Stiglund N, Strand K, Cornillet M, et al. Retained NK cell phenotype and functionality in non-alcoholic fatty liver disease. Front Immunol 2019;10:1255.
105. Narayanan S, Surette FA, Hahn YS. The Immune landscape in nonalcoholic steatohepatitis. Immune Netw 2016;16:147-58.
106. Cai L, Zhang Z, Zhou L, et al. Functional impairment in circulating and intrahepatic NK cells and relative mechanism in hepatocellular carcinoma patients. Clin Immunol 2008;129:428-37.
107. Hoechst B, Voigtlaender T, Ormandy L, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology 2009;50:799-807.
109. Arrese M, Cabrera D, Kalergis AM, Feldstein AE. Innate immunity and inflammation in NAFLD/NASH. Dig Dis Sci 2016;61:1294-303.
110. Szabo G, Petrasek J. Inflammasome activation and function in liver disease. Nat Rev Gastroenterol Hepatol 2015;12:387-400.
111. Aron-Wisnewsky J, Prifti E, Belda E, et al. Major microbiota dysbiosis in severe obesity: fate after bariatric surgery. Gut 2019;68:70-82.
112. Debédat J, Clément K, Aron-Wisnewsky J. Gut microbiota dysbiosis in human obesity: impact of bariatric surgery. Curr Obes Rep 2019;8:229-42.
113. Abenavoli L, Scarpellini E, Colica C, et al. Gut microbiota and obesity: a role for probiotics. Nutrients 2019;11:2690.
114. Aoun A, Darwish F, Hamod N. The influence of the gut microbiome on obesity in adults and the role of probiotics, prebiotics, and synbiotics for weight loss. Prev Nutr Food Sci 2020;25:113-23.
115. Gomes AC, Hoffmann C, Mota JF. The human gut microbiota: metabolism and perspective in obesity. Gut Microbes 2018;9:308-25.
116. Turnbaugh PJ, Bäckhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 2008;3:213-23.
117. Weiss GA, Hennet T. Mechanisms and consequences of intestinal dysbiosis. Cell Mol Life Sci 2017;74:2959-77.
118. Chatelier E, Nielsen T, Qin J, et al; MetaHIT consortium. Richness of human gut microbiome correlates with metabolic markers. Nature 2013;500:541-6.
119. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature 2006;444:1022-3.
121. Leung C, Rivera L, Furness JB, Angus PW. The role of the gut microbiota in NAFLD. Nat Rev Gastroenterol Hepatol 2016;13:412-25.
122. Brandi G, De Lorenzo S, Candela M, et al. Microbiota, NASH, HCC and the potential role of probiotics. Carcinogenesis 2017;38:231-40.
123. Yoshimoto S, Loo TM, Atarashi K, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013;499:97-101.
124. Wu L, Feng J, Li J, et al. The gut microbiome-bile acid axis in hepatocarcinogenesis. Biomed Pharmacother 2021;133:111036.
125. Ðanić M, Stanimirov B, Pavlović N, et al. Pharmacological applications of bile acids and their derivatives in the treatment of metabolic syndrome. Front Pharmacol 2018;9:1382.
127. Jiao Y, Lu Y, Li XY. Farnesoid X receptor: a master regulator of hepatic triglyceride and glucose homeostasis. Acta Pharmacol Sin 2015;36:44-50.
128. Ryan KK, Tremaroli V, Clemmensen C, et al. FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature 2014;509:183-8.
129. Su H, Ma C, Liu J, et al. Downregulation of nuclear receptor FXR is associated with multiple malignant clinicopathological characteristics in human hepatocellular carcinoma. Am J Physiol Gastrointest Liver Physiol 2012;303:G1245-53.
130. Wolfe A, Thomas A, Edwards G, Jaseja R, Guo GL, Apte U. Increased activation of the Wnt/β-catenin pathway in spontaneous hepatocellular carcinoma observed in farnesoid X receptor knockout mice. J Pharmacol Exp Ther 2011;338:12-21.
131. Takahashi S, Tanaka N, Fukami T, et al. Role of farnesoid X receptor and bile acids in hepatic tumor development. Hepatol Commun 2018;2:1567-82.
132. Bosman FT, Carneiro F, Hruban RH, Theise ND. . WHO classification of tumours of the digestive system. 4th ed. Geneva: World Health Organization; 2010.
133. Lokuhetty D. Organisation mondiale de la santé, Centre international de recherche sur le cancer. WHO classification of tumours. https://whobluebooks.iarc.fr/ [Last accessed on 17 Jan 2021].
134. Salomao M, Yu WM, Brown RS Jr, Emond JC, Lefkowitch JH. Steatohepatitic hepatocellular carcinoma (SH-HCC): a distinctive histological variant of HCC in hepatitis C virus-related cirrhosis with associated NAFLD/NASH. Am J Surg Pathol 2010;34:1630-6.
135. Salomao M, Remotti H, Vaughan R, Siegel AB, Lefkowitch JH, Moreira RK. The steatohepatitic variant of hepatocellular carcinoma and its association with underlying steatohepatitis. Hum Pathol 2012;43:737-46.
136. Shibahara J, Ando S, Sakamoto Y, Kokudo N, Fukayama M. Hepatocellular carcinoma with steatohepatitic features: a clinicopathological study of Japanese patients. Histopathology 2014;64:951-62.
137. Qin J, Higashi T, Nakagawa S, et al. Steatohepatitic variant of hepatocellular carcinoma is associated with both alcoholic steatohepatitis and nonalcoholic steatohepatitis: a study of 2 cohorts with molecular insights. Am J Surg Pathol 2020;44:1406-12.
138. Lee JS, Yoo JE, Kim H, et al. Tumor stroma with senescence-associated secretory phenotype in steatohepatitic hepatocellular carcinoma. PLoS One 2017;12:e0171922.
139. Calderaro J, Couchy G, Imbeaud S, et al. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J Hepatol 2017;67:727-38.
140. Ziol M, Poté N, Amaddeo G, et al. Macrotrabecular-massive hepatocellular carcinoma: a distinctive histological subtype with clinical relevance. Hepatology 2018;68:103-12.
141. Reddy SK, Steel JL, Chen HW, et al. Outcomes of curative treatment for hepatocellular cancer in nonalcoholic steatohepatitis versus hepatitis C and alcoholic liver disease. Hepatology 2012;55:1809-19.
142. Sanyal A, Poklepovic A, Moyneur E, Barghout V. Population-based risk factors and resource utilization for HCC: US perspective. Curr Med Res Opin 2010;26:2183-91.
143. Pelusi S, Cespiati A, Rametta R, et al. Prevalence and risk factors of significant fibrosis in patients with nonalcoholic fatty liver without steatohepatitis. Clin Gastroenterol Hepatol 2019;17:2310-9.e6.
144. Petersen KF, Dufour S, Befroy D, Lehrke M, Hendler RE, Shulman GI. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes 2005;54:603-8.
145. Promrat K, Kleiner DE, Niemeier HM, et al. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology 2010;51:121-9.
146. Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology 2015;149:367-78.e5; quiz e14.
147. Turati F, Trichopoulos D, Polesel J, et al. Mediterranean diet and hepatocellular carcinoma. J Hepatol 2014;60:606-11.
148. Gepner Y, Shelef I, Komy O, et al. The beneficial effects of Mediterranean diet over low-fat diet may be mediated by decreasing hepatic fat content. J Hepatol 2019;71:379-88.
149. EPIC - European Prospective Investigation into Cancer and Nutrition. https://epic.iarc.fr/ [Last accessed on 17 Jan 2021].
150. Lee Y, Doumouras AG, Yu J, et al. Complete resolution of nonalcoholic fatty liver disease after bariatric surgery: a systematic review and meta-analysis. Clin Gastroenterol Hepatol 2019;17:1040-60.e11.
151. Mattar SG, Velcu LM, Rabinovitz M, et al. Surgically-induced weight loss significantly improves nonalcoholic fatty liver disease and the metabolic syndrome. Ann Surg 2005;242:610-7; discussion 618.
152. Lassailly G, Caiazzo R, Buob D, et al. Bariatric surgery reduces features of nonalcoholic steatohepatitis in morbidly obese patients. Gastroenterology 2015;149:379-88; quiz e15.
153. Lassailly G, Caiazzo R, Ntandja-Wandji LC, et al. Bariatric surgery provides long-term resolution of nonalcoholic steatohepatitis and regression of fibrosis. Gastroenterology 2020;159:1290-301.e5.
154. Kwak M, Mehaffey JH, Hawkins RB, Hsu A, Schirmer B, Hallowell PT. Bariatric surgery is associated with reduction in non-alcoholic steatohepatitis and hepatocellular carcinoma: a propensity matched analysis. Am J Surg 2020;219:504-7.
155. Malehmir M, Pfister D, Gallage S, et al. Platelet GPIbα is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat Med 2019;25:641-55.
156. Beloribi-Djefaflia S, Vasseur S, Guillaumond F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016;5:e189.
157. Foderà D, D'Alessandro N, Cusimano A, et al. Induction of apoptosis and inhibition of cell growth in human hepatocellular carcinoma cells by COX-2 inhibitors. Ann N Y Acad Sci 2004;1028:440-9.
158. Kern MA, Schubert D, Sahi D, et al. Proapoptotic and antiproliferative potential of selective cyclooxygenase-2 inhibitors in human liver tumor cells. Hepatology 2002;36:885-94.
159. Chen H, Cai W, Chu ESH, et al. Hepatic cyclooxygenase-2 overexpression induced spontaneous hepatocellular carcinoma formation in mice. Oncogene 2017;36:4415-26.
160. Simon TG, Duberg AS, Aleman S, Chung RT, Chan AT, Ludvigsson JF. Association of aspirin with hepatocellular carcinoma and liver-related mortality. N Engl J Med 2020;382:1018-28.
161. Huang MY, Chung CH, Chang WK, et al. The role of thiazolidinediones in hepatocellular carcinoma risk reduction: a population-based cohort study in Taiwan. Am J Cancer Res 2017;7:1606-16.
162. Donadon V, Balbi M, Mas MD, Casarin P, Zanette G. Metformin and reduced risk of hepatocellular carcinoma in diabetic patients with chronic liver disease. Liver Int 2010;30:750-8.
163. Hassan M, Li D, Kaseb A, et al. Association of diabetes duration and diabetes treatment with the risk of hepatocellular carcinoma. Cancer 2010;116:1938-46.
164. Bosetti C, Franchi M, Nicotra F, et al. Insulin and other antidiabetic drugs and hepatocellular carcinoma risk: a nested case-control study based on Italian healthcare utilization databases. Pharmacoepidemiol Drug Saf 2015;24:771-8.
165. Kasmari AJ, Welch A, Liu G, Leslie D, McGarrity T, Riley T. Independent of cirrhosis, hepatocellular carcinoma risk is increased with diabetes and metabolic syndrome. Am J Med 2017;130:746.e1-7.
166. Miele L, Bosetti C, Turati F, et al. Diabetes and insulin therapy, but not metformin, are related to hepatocellular cancer risk. Gastroenterol Res Pract 2015;2015:570356.
167. Zheng L, Yang W, Wu F, et al. Prognostic significance of AMPK activation and therapeutic effects of metformin in hepatocellular carcinoma. Clin Cancer Res 2013;19:5372-80.
168. Cunha V, Cotrim HP, Rocha R, Carvalho K, Lins-Kusterer L. Metformin in the prevention of hepatocellular carcinoma in diabetic patients: a systematic review. Ann Hepatol 2020;19:232-7.
169. Association For The Study Of The Liver; European Organisation For Research And Treatment Of Cancer. EASL-EORTC clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol 2012;56:908-43.
171. Cauchy F, Zalinski S, Dokmak S, et al. Surgical treatment of hepatocellular carcinoma associated with the metabolic syndrome. Br J Surg 2013;100:113-21.
172. Llovet JM, Ricci S, Mazzaferro V, et al. SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359:378-90.
173. Howell J, Samani A, Mannan B, Hajiev S, sharma R. Impact of NAFLD on clinical outcomes in hepatocellular carcinoma treated with sorafenib: an international cohort study. JCO 2021;39:289.
174. Jian C, Fu J, Cheng X, et al. Low-dose sorafenib acts as a mitochondrial uncoupler and ameliorates nonalcoholic steatohepatitis. Cell Metab 2020;31:892-908.e11.
175. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017;389:56-66.
176. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet 2018;391:1163-73.
177. Zhu AX, Kang Y, Yen C, et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2019;20:282-96.
178. Abou-Alfa GK, Meyer T, Cheng AL, et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N Engl J Med 2018;379:54-63.
179. Finn RS, Qin S, Ikeda M, et al. IMbrave150 Investigators. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med 2020;382:1894-905.
180. Galle PR, Finn RS, Qin S, et al. Patient-reported outcomes (PROs) from the Phase III IMbrave150 trial of atezolizumab (atezo) + bevacizumab (bev) vs sorafenib (sor) as first-line treatment (tx) for patients (pts) with unresectable hepatocellular carcinoma (HCC). JCO 2020;38:476.
181. Gordan JD, Kennedy EB, Abou-Alfa GK, et al. Systemic therapy for advanced hepatocellular carcinoma: ASCO guideline. J Clin Oncol 2020;38:4317-45.
182. Rakhra K, Bachireddy P, Zabuawala T, et al. CD4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell 2010;18:485-98.
183. Ma C, Kesarwala AH, Eggert T, et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016;531:253-7.
184. Pinter M, Scheiner B, Peck-Radosavljevic M. Immunotherapy for advanced hepatocellular carcinoma: a focus on special subgroups. Gut 2021;70:204-14.
185. Yau T, Park J, Finn R, et al. CheckMate 459: a randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Ann Oncol 2019;30:v874-5.
186. Heinrich B, Brown Z, Vormehr M, et al. Abstract 1728: Nonalcoholic steatohepatitis (NASH) impairs treatment of intrahepatic metastases with CD4 + T cell dependent RNA vaccine. Cancer Res 2018;78:1728.
187. Artaç M, Korkmaz L, Coşkun HŞ, et al. Bevacuzimab may be less effective in obese metastatic colorectal cancer patients. J Gastrointest Cancer 2019;50:214-20.
188. Guiu B, Petit JM, Bonnetain F, et al. Visceral fat area is an independent predictive biomarker of outcome after first-line bevacizumab-based treatment in metastatic colorectal cancer. Gut 2010;59:341-7.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Zampaglione L, Ferrari J, Pedica F, Goossens N. HCC in metabolic syndrome: current concepts and future directions. Hepatoma Res 2021;7:55. http://dx.doi.org/10.20517/2394-5079.2021.22
AMA Style
Zampaglione L, Ferrari J, Pedica F, Goossens N. HCC in metabolic syndrome: current concepts and future directions. Hepatoma Research. 2021; 7: 55. http://dx.doi.org/10.20517/2394-5079.2021.22
Chicago/Turabian Style
Zampaglione, Lucia, Jacopo Ferrari, Federica Pedica, Nicolas Goossens. 2021. "HCC in metabolic syndrome: current concepts and future directions" Hepatoma Research. 7: 55. http://dx.doi.org/10.20517/2394-5079.2021.22
ACS Style
Zampaglione, L.; Ferrari J.; Pedica F.; Goossens N. HCC in metabolic syndrome: current concepts and future directions. Hepatoma. Res. 2021, 7, 55. http://dx.doi.org/10.20517/2394-5079.2021.22
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 22 clicks
Cite This Article 22 clicks

 Like This Article 5
likes
Like This Article 5
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.