
Bridging molecular basis, prognosis, and treatment of pediatric liver tumors
 0
0Abstract
A deeper understanding of the genetic and molecular basis of hepatoblastoma (HB) has fueled the hope to help in identifying genes and signaling pathways that are amenable to therapeutic intervention. However, it has become clear that HB is a genetically very simple cancer and that rather alterations of the transcriptome or epigenome will facilitate a more stratified and rationalized approach to current therapeutics. In this review, we discuss recent findings on genomic, transcriptomic, and epigenomic data and their potential to serve as biomarkers and predictors of patient’s outcome. We also describe the state of the art in HB experimental biology, the in vitro and in vivo HB models that are currently available, and their use to improve our understanding of this disease and identify new treatment options.
Keywords
INTRODUCTION
Hepatoblastoma (HB) is the predominant liver tumor in childhood; however, it is a rare tumor with an incidence of only approximately one case per million children per year[1]. In contrast to HB, pediatric hepatocellular carcinoma (HCC) is much more uncommon and has a worse prognosis[2]. The difficulty of accessing biological samples from childhood liver cancers has prevented deeper understanding of its molecular nature, thereby challenging translational research. Because of this, there is an urgent need for centralized biorepositories of human samples, cell lines, and murine models to be able to move translational research of childhood liver cancer forward[3]. Up to now, studies focused on deciphering the molecular factors driving the oncogenesis and tumor progression of HB have been performed on a restricted number of in vitro and in vivo models as well as in limited cohorts of patients, who, in some cases, had received heterogeneous treatments. Nevertheless, despite these limitations, the scientific community has improved the molecular knowledge of HB and identified its main molecular driver, β-catenin, as well as molecular prognostic subtypes, all of which provide the basis for precision medicine in the future. The main research findings in HB research, ranging from its genetics, transcriptomics, and methylomics to the potential therapeutic strategies, are summarized below.
GENETIC AND GENOMIC STUDIES
In contrast to HCC, the most frequent liver tumor in adults, which develops on a cirrhotic liver background usually caused by chronic viral hepatitis B or C infection as well as by alcohol consumption[4], liver tumors in children, adolescents, and young adults typically occur on apparently normal liver. In young children, most primary liver tumors are HB, whereas, in adolescents and young adults, the main histologic subtypes are fibrolamellar carcinoma and HCC[5-7]. Hepatic liver tumors with HB and HCC histological features currently indicated as hepatocellular malignant neoplasms not otherwise specified (HCN-NOS), previously called transitional liver cell tumors (TLCTs), also occur on normal liver and are typically diagnosed in older children and young adolescents[8-10]. Several studies have explored patients’ genetic profiles in search of the genomic hallmarks of hepatoblastoma pathogenesis. The high mutation rate (60%-92%) found in CTNNB1, which encodes β-catenin, ranks HB among the human tumors with the most frequent constitutive activation of Wnt/β-catenin/TCF signaling[11-16]. Evidence for this pathway as the genetic driver in HB is also supported by the increased risk to develop HB for children affected by familial adenomatous polyposis, a disorder caused by germline mutation of the APC gene involved in β-catenin degradation[17], and by the identification of mutations in AXIN1 and AXIN2, two important Wnt pathway-related players[18,19]. Additional evidence for the genetic/epigenetic origin of this tumor is provided by increased risk associated with congenital anomalies, such as Beckwith-Wiedemann syndrome[20], and by the increased risk for children exposed to perinatal and maternal factors such as very low weight at birth and eclampsia during pregnancy[21,22]. Recent whole-exome sequencing studies have explored the genetic landscape of human HB and found the lowest mutation burden (2.9 mutations per tumor) among pediatric cancers[14,16,23]. Deciphering the genetics of HB is strongly contributing to our understanding of tumor biology and improving patient stratification through the identification of diagnostic, prognostic, and theranostic biomarkers. At the diagnostic level, evaluation of INI1/SMARCB1 gene status by immunohistochemistry and/or genetic analysis in pediatric primary liver tumors with low serum alpha-fetoprotein (AFP) allows discriminating HB, which are all INI1 wildtype, from malignant rhabdoid liver tumors, where this gene is deleted[24]. Telomerase reverse-transcriptase (TERT) promoter mutation and increased expression have been frequently found in HCN-NOS/TLCTs and could be used as diagnostic markers to identify such tumors[14,25]. Furthermore, nuclear factor and erythroid 2 like 2 gene (NFE2L2, also called NRF2), a key regulator of antioxidant stress-response, has been found mutated in 9%-10% of HBs and associated with poor prognosis[14,26,27]. As alteration of the NFE2L2-related pathway is strongly involved in chemoresistance[28], NFE2L2 mutation should be evaluated as a chemotherapy efficacy predictive biomarker. Comprehensive genomic profiling analyses also revealed additional genes affected by mutations, amplification, or loss at very low frequency[29,30]. Recently, a global dysregulation of RNA editing, an epigenetic mechanism that confers specific nucleotide changes on RNA transcripts without altering the sequence of genomic DNA, has been reported in HB accompanied by a specific hyperediting of BLCAP (bladder cancer-associated protein), a gene with tumor suppressor functions[27]. These genes represent potentially actionable targets to be further evaluated in view of a precision medicine approach for HB patients.
TRANSCRIPTOMIC AND EPIGENOMIC STUDIES
Besides activating CTNNB1 mutation, which is found in the majority of HB cases, these tumors have a low mutation burden, suggesting that the molecular determinants of the clinical heterogeneity of HB should not be sought among their genetic aberrations. Instead, several papers have associated the different clinical behavior of HB with differences in their transcriptome. Using gene expression arrays, a transcriptomic study of 24 HBs clearly revealed two main prognostic subclasses of tumors, which were named C1 and C2[11]. Both transcriptomic subtypes had similar percentages of CTNNB1 mutation; however, they displayed a strong difference in their gene expression profile in terms of stem-cell/progenitor-like and proliferating markers as well as in hepatic function and Wnt/β-catenin target related genes. The differences in the transcriptome of C1 and C2 tumors were also linked to tumor histology, because, when a simplified pathological parameter called “main epithelial component” was taken into account, C2 tumors displayed predominantly immature histotypes (i.e., embryonal and crowded fetal), whereas C1 tumors showed a predominantly well-differentiated fetal histotype, which resembled early and late stages of liver development, respectively. More importantly, the two transcriptomic subtypes were shown to be associated with clinical prognostic factors (i.e., metastasis and vascular invasion) and patient outcome. Specifically, children harboring C2 tumors showed a significantly reduced probability of event-free survival as compared with patients with C1 tumors. Out of these findings, a 16-gene signature discriminating C1 and C2 subclasses was defined in an attempt to translate the molecular findings to the clinical setting[11]. Of note, the dichotomy of HB samples was also found in a subsequent microRNA (miR) array analysis of 49 HB samples that identified Cm1 and Cm2 subgroups by determining a specific four-miR signature[31]. In a recent study, using a large tri-national cohort of surgical samples from 174 HB patients, the prognostic prediction of the 16-gene signature was validated and proposed for inclusion in a new stratification system in combination with clinical factors with the aim of improving risk-adapted management of HB patients[26]. Interestingly, another recent study has demonstrated the prognostic prediction of the 16-gene signature in 35 pre-treated HB samples from diagnostic biopsies[15].
Almost 10 years after the identification of the C1 and C2 subclasses[11], three independent studies suggested the presence of a third transcriptomic subclass of HBs. Using gene expression arrays, Sumazin et al.[16] defined HB1, HB2, and HB3 with distinct NFE2L2 and Wnt-signaling activity and LIN28B, HMGA2, SALL4, and AFP gene expression. By performing RNA-sequencing in 25 HBs, besides C1 tumors,
The study of DNA methylation has revealed a global genome-wide DNA hypomethylation of HB[33-35]. In addition, Carrillo-Reixach et al.[27] reported two epigenomic subtypes of HB called Epi-CA and Epi-CB that overlapped with C1/C2B and C2-Pure tumors, respectively. The epigenetic differences between Epi-CA and Epi-CB tumors were found in the degree of DNA hypomethylation and CpG island hypermethylation. In particular, Epi-CB tumors showed a more pronounced DNA hypomethylation and specific CpG-island hypermethylation than Epi-CA tumors did. In line with the transcriptomic studies, the differences in the DNA methylation of the tumors were also associated with tumor histology, specifically the degree of immaturity of the main epithelial component, and patient clinical outcome. Therefore, patients harboring tumors with an Epi-CB subtype, which had predominantly a C2 transcriptome, showed a worse prognosis than patients with tumors of the Epi-CA subtype with a similar C1 or C2B transcriptome. The assessment of percentage of unmethylated Alu regions measured using the QUAlu method[36] was proposed as a tool to discriminate Epi-CA and Epi-CB tumors taking into account the global degree of DNA hypomethylation[27]. Later, the finding of two main groups of HBs based on the DNA methylation data, significantly associated with C1/C2 tumors, was also reported by Sekiguchi et al.[15] who named them F and E from their correlation with fetal and embryonic histology, respectively. In this study, the authors found differences in the methylation level of the HNF4/CEBPA-binding regions in gene bodies, which were more highly hypermethylated in tumors of the E cluster as compared with tumors of the F cluster.
BIOMARKERS TO IMPROVE FUTURE CLINICAL MANAGEMENT OF HB
Altogether genomic, transcriptomic, and epigenomic studies have offered new clues about HB pathogenesis and identified biomarkers with strong prognostic predictions. Currently, the treatment of HB patients is based on the ongoing Pediatric Hepatic International Tumor Trial (PHITT), which uses a highly refined risk stratification system of the Children’s Hepatic tumors International Collaboration (CHIC)[37]. This stratification relies on the main clinical prognostic factors such as the pretreatment extent of disease (PRETEXT) stage, metastasis, patient age, AFP serum levels, and annotation factors related to vein, intrahepatic, and extrahepatic tumor dissemination. Up to now, no molecular data have been incorporated into clinical risk-adapted systems, despite that fact that they have proven effective for prognostic prediction when combined with CHIC stratification[11,26,27]. First, the 16-gene signature identified to discriminate C1 and C2 tumors was identified as an independent prognostic factor when compared to clinical variables including PRETEXT, vascular invasion, and metastasis[11]. This signature was widely validated in a recent study, using a large cohort of 174 patients, that identified the C2 subtype as the only independent factor contributing significant additional prognostic information to the clinical parameters compared with three other biomarkers studied (CTNNB1, NFE2L2, and TERT mutations)[26]. Finally, a recent study proposed a first molecular risk stratification of HB by integrating novel transcriptomic and epigenomic biomarkers such as the C2-Pure subtype, the 14q32-signature of the DLK1/DIO3 locus, and the Epi-CB tumors[27]. In this study, the combination of biomarkers performed better when stratifying patients according to their prognosis than using individual biomarkers did. Therefore, in a similar way to the CHIC clinical stratification, the integration of multiple molecular prognostic factors has a cumulative effect in terms of risk prediction. Finally, the combination of clinical and molecular risk-staging systems (CHIC and MRS) resulted in better patient risk prediction and highlighted the importance of incorporating molecular factors to the clinical setting [Figure 1].
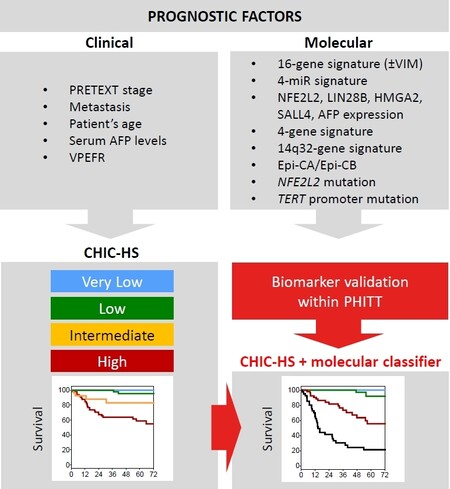
Figure 1. Incorporation of molecular factors into the risk stratification system of childhood liver cancer. The current CHIC-HS (Children’s Hepatic tumors International Collaboration-Hepatoblastoma Stratification) staging system[37] is based on clinical characteristics, such as PRETEXT (pretreatment extent of disease), alpha-fetoprotein (AFP), and the PRETEXT annotation factors vascular involvement (V: hepatic vein/inferior vena cava; P: portal vein), extrahepatic tumor extension (E), multifocality (F), and tumor rupture (R). New molecular risk stratification systems are based on specific gene/miR expression signatures[11,16,27,31,32], epigenetic patterns[27], or mutations in nuclear factor and erythroid 2 like 2 (NFE2L2) and the telomerase reverse-transcriptase (TERT) promoter[26], which are currently validated within the Pediatric Hepatic International Tumor Trial (PHITT). The combination of both clinical and molecular factors will improve clinical management and prediction of outcome in childhood liver cancer patients in the future.
In summary, the implementation of a combined clinical and molecular risk staging system is key for moving precision medicine of childhood liver cancer forward and to improve the current patient morbidity and mortality. The prospective cohort of patients enrolled in the ongoing PHITT (NCT03017326) provides a unique opportunity to perform a biomarker validation study using a large patient cohort as a pre-step to the incorporation of a highly validated biomarker panel in the next clinical trials.
PRECLINICAL TESTING OF NEW THERAPIES IN “CLASSICAL” TUMOR CELL LINES
The first pediatric liver tumor models for testing new drugs in the preclinical setting were based on cell lines that had been established from freshly dissected tumors and grown on regular plastic dishes. The most prominent models used are the HB cell lines HUH6[38], HepT1[39], HepT3[40], and HepG2[41,42], as well as the pediatric HCC cell line Hep3B[43]. As these models closely recapitulate the genetic and transcriptional repertoire of their original tumors (i.e., all HB cell lines display the characteristic HB mutations in CTNNB1, whereas Hep3B carries an AXIN1 mutation), they were used in many studies to functionally validate new candidate genes and signaling pathways and inhibit their aberrant activation by genetic and therapeutical approaches [Table 1].
Reported preclinical drug testing studies using pediatric liver tumor models
| Target | Agent | In vitro models | Dosage | Response | In vivo models | Ref. |
| PI3K | LY294002 | HUH6, HepT1 | 0-50 µM | 10-30 µM | - | [43] |
| mTOR | rapamycin | HUH6, HepT1, HepG2 | 0-100 nM | 100 nM | HUH6# | [47] |
| Smoothened | cyclopamine | HUH6, HepT1, HepT3, HepG2 | 0-20 µM | 7.5 µM | - | [50] |
| Bcl-2, Bcl-xL | ABT-737 | HUH6, HepT1 | 0-100 µM | 10 µM | HUH6# | [53,54] |
| NK1R | aprepitant | HUH6, HepT1, HepG2 | 0-100 µM | 30 µM | HUH6# | [56] |
| proteasome | bortezomib | HUH6, HepG2 | - | 7-62 nM | HUH6# | [31] |
| MYCN | MLN8237, JQ1 | HUH6, HepT1, HepG2 | 0-10 µM | 1 µM | - | [57] |
| SP8 | mithramycin A | HUH6, HepT1, HepG2, Hep3B | 0-100 nM | 10-30 nm | - | [58] |
| CHKA | MN58b, TCD-717 | HUH6, HepG2 | 0-8 µM | 6 µM | MRS-3 PDX* | [27] |
| MEK1 | trametinib | B6-2 | 0-100 nM | 1 nM | B6-2 PDX* | [61] |
| PLK1 | volasertib | HB-214, HB-243, HB-279, HB-282, HB-284, HB-295 | 0-100 µM | 1.7 µM | HB-214 PDX* | [63] |
| - | chloroquine | HB-243, HB-279, HB-295, HB-282, HB-284, HB-303 | 0-10 µM | 10 µM | - | [65] |
| MDM4 | NSC207895, ATSP-7041 | B6-2, HUH6, HepT1, HepG2 | 0-100 µM | 1-7 µM | HepT1§ | [66] |
One first example is the activation of the phosphatidylinositol-3-kinase (PI3K)/AKT pathway, which can be detected in 79% of HB cases[44]. The trigger for the constitutive activation of this pathway is overexpression of the insulin-like growth factor gene, which is mainly caused by aberrant methylation of its P3 promoter[45]. In addition, rare mutations of the PI3KCA gene might also contribute to PI3K/AKT activity[44]. Preclinical testing of the synthetic PI3K inhibitor LY294002 effectively suppressed growth of tumor cells and induced apoptosis[44]. Of note, knockdown of PI3KCA mimicked the LY294002-mediated inhibition of PI3K in these models[44].
It is known that the tumor growth promoting effects of the activated PI3K/AKT pathway are mainly mediated over the mammalian target of rapamycin (mTOR)[46]. The discovery that the immunosuppressant rapamycin (also known as sirolimus) is able to inhibit mTOR and thereby elicit a strong anti-tumorigenic effect in primary and metastatic cancers in mice[47] has paved the way for many preclinical and clinical studies. Consequently, it has been shown that rapamycin efficiently blocked PI3K/AKT signaling also in HB cells by inhibiting proliferation and inducing apoptosis, both in vitro and in vivo[48]. Of note, the tumor-suppressive effects of rapamycin on HB cells were achieved without the feared positive feedback activation of PI3K/AKT signaling through mTOR inhibition, as reported for rhabdomyosarcoma[49]. As a first promising effect of rapamycin in a single HB patient after liver transplantation has already been reported[50], it is tempting to speculate that rapamycin might be a hopeful new agent to treat HB patients, especially those undergoing liver transplantation.
Another example of the successful use of “classical” tumor cell lines was the proof that Hedgehog (Hh) signaling is active in approximately half of all HB cell lines and primary tumors[51]. Consequently, blockade of the Hh pathway by the smoothened inhibitor cyclopamine resulted in a significant growth retardation and induction of cell death of the Hh-activated models HepT3 and HUH6. As previous studies have also found efficacy of cyclopamine in the HCC model HUH7[52], targeting the Hh pathway especially with clinically approved second-generation smoothened inhibitors such as Vismodegib seems to be a potential therapeutic option for liver cancer patients in general.
Disruption of pathways leading to programmed cell death plays a major role in many cancer types, and elevated expression of the anti-apoptotic Bcl-2 family members Bcl-2 and Bcl-xL are frequently found in therapy resistant tumors[53]. ABT-737, a small-molecule inhibitor of Bcl-2 and Bcl-xL, was shown not only to reduce cell viability[54] but also to enhance the cytotoxic effect of paclitaxel when used in combination in HB models[55]. As the combination of ABT-737 and paclitaxel allows a tenfold reduction of paclitaxel to achieve a similar therapeutic effect, inhibition of anti-apoptotic mediators might display a therapeutic option to reduce toxic side effects of current treatments.
The neurokinin-1 receptor (NK1R) has been described as an integral part of cancer cells that can be targeted by its antagonist aprepitant, a clinically approved drug used for chemotherapy-induced nausea and vomiting[56]. In high doses, aprepitant has been reported to suppress growth of HB cells propagated on plastic as well as in vivo[57]. As high expression of NK1R, especially of the truncated splice variant, can be found in almost all HB cases[57], NK1R may serve as a novel therapeutic target in HB that warrants further exploration.
Genetic permutation analysis of transcriptomic data revealed breast cancer 1 (BRCA1) and Fanconi anemia (FA) complex genes (FANCI and FANCD2) to be upregulated in the HUH6 and HepG2 models, which belong to the C2A group of the 4-gene signature[32]. Although well-known FA/BRCA pathway inhibitors such as GW7647, ML323, pimozide, and MLN4924 had very high effect concentrations in viability assays, the proteasome inhibitor bortezomib was effective in the low nanomolar range, rendering it compatible for clinical applications[32]. Consequently, testing bortezomib in the preclinical HUH6 xenograft model significantly reduced tumor volume.
Another gene with a significantly high expression in C2 tumors of the 16-gene signature is MYCN, which maps to 2p24.1, a chromosomal region known to be frequently duplicated in HB[11]. Eberherr et al.[58] found that MYCN is generally upregulated in pediatric liver tumors, and that treating HB and HCC cell lines with the known MYCN inhibitors MLN8237 and JQ1 induced dose-dependent growth arrest by trapping cells in the cell cycle.
Recent transcriptomic data reveal choline kinase alpha (CHKA), a key enzyme of the biosynthesis of phosphatidylcholine via the CDP/choline pathway, to be the most widely overexpressed coding gene in high-risk and intermediate-risk tumors[27]. Treatment of the “classical” models HUH6 and HepG2 with the CHKA inhibitors MN58b and TCD-717 showed a dose-dependent reduction of cell viability and colony formation, and knockdown of CHKA expression in HUH6 cells phenocopied these effects[27].
Integrative analysis of transcriptomic and methylomic data of metastasized and non-metastasized HB samples identified the Sp8 transcription factor (SP8) as a highly upregulated gene in metastatic tumors[59]. Interestingly, HB patients with high SP8 expression levels were also prognostic for poor survival. SP8-mediated aggressive traits such as cell motility and clonogenic growth could be abrogated in SP8-silenced cell models using RNA interference. Of clinical relevance, application of the FDA-approved pan SP transcription factor mithramycin A abolished SP8-induced effects[59].
PATIENT-DERIVED XENOGRAFT MODELS
Several drugs have been identified as potentially effective against HB by cell line screening. Drug efficacy validation in preclinical studies with animal models is usually required prior to clinical trials. Several genetically engineered mouse models have been described that recapitulate HB features[60], including the Cited1-Ctnnb1 mouse, which provided evidence that activating Ctnnb1 mutation is sufficient to drive tumorigenesis in the liver for the first time[61]. While these models are very useful tools to shed light on HB biology, the lack of the human genetic background limits the use of transgenic mouse models for the validation of new anticancer treatments. Patient-derived xenografts (PDXs) have recently become reference models for the preclinical evaluation of anti-cancer therapy and are gaining a central tool in preclinical pediatric oncology programs worldwide (http://www.ncipptc.org,https://www.itccp4.eu). During the last years, several HB PDX models have been generated, by either orthotopic or heterotopic (interscapular fat pad) implantation of patient’s tumor fragments[62,63]. These models preserve the histological and genetic features of the patients they have been derived from; therefore, they represent powerful tools to discover and validate new therapies for HB patients. In particular, as the majority of HB PDXs are developed from surgical samples that received previous therapies or at relapse, they represent a valuable preclinical tool to identify second-line treatments. The combination of duparlisib (a pan-PI3K inhibitor) and trametinib (a MEK-1/2 inhibitor) was shown to inhibit tumor growth of a NRAS-mutated orthotopic PDX[62], and evaluation of several potential second-line drugs such as irinotecan, temozolomide, temsirolimus, sirolimus, sorafenib, crizotinib, paclitaxel, and volasertib on heterotopically implanted PDXs allowed the identification of irinotecan alone or in combination with temozolomide or volasertib as promising anti-cancer treatment for HB patients[63,64]. Moreover, the inhibition of CHKA using MN58b fully abrogated tumor growth by lowering the proliferation rate and reverting the progenitor-like phenotype in a PDX model established from a high-risk HB[27]. The availability of a large HB PDX panel fully characterized at the molecular level is also particularly relevant as it allows the investigation of the molecular markers associated to PDX response, large-scale pre-clinical assays, and the development of companion biomarkers that are becoming an important requirement to grant the authorization from the medicines regulatory authorities to run clinical trials.
TUMOR CELL LINES DERIVED FOM PATIENT-DERIVED XENOGRAFTS
Recent advances have allowed the generation of cell lines derived from PDX models of pediatric liver cancers by growing them under stem cell culturing conditions[62,63]. These second-generation models are molecularly robust and reflect a variety of histological subtypes[64]. As drugs identified in these models can be easily validated in the corresponding PDX in mice [Table 1], this allows for a timely preclinical testing of new drugs in a representative spectrum of pediatric liver tumors.
The first study using this kind of model applied trametinib, an inhibitor of the mitogen-activated protein kinase kinase enzymes MEK1 and MEK2 to the B6 cell line, which has been derived from a xenograft from a HB with HCC-like features with a mutation in the neuroblastoma ras viral oncogene homolog gene
Expression profiling analyses have identified the polo-like kinase 1 (PLK1) gene to be overexpressed in a set of 74 HB samples[65]. Of note, high PLK1 expression was associated with a significantly poorer outcome. Kats et al.[64] tested the PLK1 inhibitor volasertib in six cell lines derived from PDX models and could show a strong tumor-suppressive effect in half of the cell lines, as well as in HB-214 when transplanted into athymic nude mice. Most interestingly, in vitro activity of volasertib was achieved at concentrations known to be safe in phase I trials in children and adults[64].
To more accurately mimic the architecture and organization of solid tumors, the same PDX cell lines have recently been further developed to be grown as three-dimensional spheroids[66]. As a proof-of-concept, Eloranta et al.[66] assayed the well-documented anti-neoplastic activity of chloroquine in these models and could show a reduced viability and induction of apoptosis. These organized cell culture models will bridge the gap between cell lines grown on plastic and in vivo models and substantially further anticancer therapeutic development in pediatric liver cancers.
Recently, a new study has been published reporting on a novel therapeutic strategy that reactivates p53 by the inhibition of the p53 regulator murine double minute 4 (MDM4), which is highly expressed in HB patients[67]. Inhibition of MDM4 by its inhibitors NSC207895 and ATSP-7041 as well as short hairpin RNA-mediated knockdown of MDM4 expression led to upregulation of p53 activity and a subsequent inhibition of tumor cell growth. The in vivo efficacy of MDM4 inhibition was proven with the orthotopic HepT1 xenograft model in mice using NSC207895[67].
FUTURE ASPECTS
In recent decades, great advances in the comprehension of the molecular pathogenesis of HB have been made. Currently, there is a need to better understand rare and aggressive forms of childhood liver cancer (i.e., metastatic/recurrent tumors, HB with HCC-like features, and HB with AFP < 100 ng/mL), all of which have been little studied due to the difficulty of obtaining biological specimens. Additionally, there is also a pressing need to incorporate molecular data into clinical risk stratification of patients to improve clinical management of childhood liver cancer. To address these shortcomings, emerging artificial intelligence tools can be applied to study the public datasets obtained from the molecular profiling of tumors (i.e., genome, transcriptome, and methylome), which have been generated in recent years thanks to the advances and the decreasing costs of molecular measurement technologies. It is clear that the application of big data in the field of cancer has a huge potential, and, in the case of rare cancers, it is useful not only to analyze molecular data associated with clinical and pathological parameters in depth but also to increase the sample size of the studies. Moreover, it is vital to promote the establishment of centralized biorepositories of human samples, including living tumors (i.e., cell lines, organoids, and murine models), to validate biomarkers and test new therapies to move personalized medicine for pediatric patients with liver cancer forward.
The ongoing international clinical trial PHITT is designed to meet the above-mentioned needs. In parallel to an improvement in treatments for patients, it is intended to establish one of the largest and most complete biorepositories in the world, which will include tissue, blood, and urine samples at different times of treatment as well as living tumors (i.e., patient-derived xenografts and organoids). The samples are already being collected and molecularly profiled, and the omics databases will be exploited through sophisticated computational and artificial intelligence tools with the aim of identifying new biomarkers and signaling pathways involved in highly aggressive pediatric liver malignances, so as to be able to provide a comprehensive and highly-validated panel of diagnostic and prognostic biomarkers. One of the important objectives of the PHITT is to improve the current molecular knowledge of childhood liver cancer and its clinical management by integrating the use of biomarkers into clinical practice with the final aim of advancing the improvement of quality of life and survival of children suffering from primary liver cancers.
DECLARATIONS
Authors’ contributionsReview conception and design, draft manuscript preparation and approval of the final version of the manuscript: Armengol C, Cairo S, Kappler R
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis article was possible thanks to the inputs from AGAUR (2017-SGR-490), the European Union's Horizon 2020 research and innovation programme under grant agreement No 668596 (ChiLTERN) and grant agreement No 826121 (iPC). CA was supported by Ramón y Cajal (RYC-2010-07249) program of the Ministry of Science and Innovation of Spain and RK by the Bettina Bräu foundation Munich and the Gänseblümchen foundation Voerde.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Kehm RD, Osypuk TL, Poynter JN, Vock DM, Spector LG. Do pregnancy characteristics contribute to rising childhood cancer incidence rates in the United States? Pediatr Blood Cancer 2018:65.
2. Schmid I, von Schweinitz D. Pediatric hepatocellular carcinoma: challenges and solutions. J Hepatocell Carcinoma 2017;4:15-21.
3. Rikhi RR, Spady KK, Hoffman RI, Bateman MS, Bateman M, Howard LE. Hepatoblastoma: a need for cell lines and tissue banks to develop targeted drug therapies. Front Pediatr 2016;4:22.
4. Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Primers 2021;7:6.
5. Czauderna P. Adult type vs. Childhood hepatocellular carcinoma--are they the same or different lesions? Med Pediatr Oncol 2002;39:519-23.
6. Czauderna P, Mackinlay G, Perilongo G, et al. Hepatocellular carcinoma in children: results of the first prospective study of the International Society of Pediatric Oncology group. J Clin Oncol 2002;20:2798-804.
7. Katzenstein HM, Krailo MD, Malogolowkin MH, et al. Hepatocellular carcinoma in children and adolescents: results from the Pediatric Oncology Group and the Children's Cancer Group intergroup study. J Clin Oncol 2002;20:2789-97.
8. Lopez-Terrada D, Alaggio R, de Davila MT, et al. Towards an international pediatric liver tumor consensus classification: proceedings of the Los Angeles COG liver tumors symposium. Mod Pathol 2014;27:472-91.
9. Prokurat A, Kluge P, Kosciesza A, Perek D, Kappeler A, Zimmermann A. Transitional liver cell tumors (TLCT) in older children and adolescents: a novel group of aggressive hepatic tumors expressing beta-catenin. Med Pediatr Oncol 2002;39:510-8.
10. Zimmermann A. The emerging family of hepatoblastoma tumours: from ontogenesis to oncogenesis. Eur J Cancer 2005;41:1503-14.
11. Cairo S, Armengol C, De Reynies A, et al. Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell 2008;14:471-84.
12. Koch A, Denkhaus D, Albrecht S, Leuschner I, von Schweinitz D, Pietsch T. Childhood hepatoblastomas frequently carry a mutated degradation targeting box of the beta-catenin gene. Cancer Res 1999;59:269-73.
13. Wei Y, Fabre M, Branchereau S, Gauthier F, Perilongo G, Buendia MA. Activation of beta-catenin in epithelial and mesenchymal hepatoblastomas. Oncogene 2000;19:498-504.
14. Eichenmüller M, Trippel F, Kreuder M, et al. The genomic landscape of hepatoblastoma and their progenies with HCC-like features. J Hepatol 2014;61:1312-20.
15. Sekiguchi M, Seki M, Kawai T, et al. Integrated multiomics analysis of hepatoblastoma unravels its heterogeneity and provides novel druggable targets. NPJ Precis Oncol 2020;4:20.
16. Sumazin P, Chen Y, Trevino LR, et al. Genomic analysis of hepatoblastoma identifies distinct molecular and prognostic subgroups. Hepatology 2017;65:104-21.
17. Hirschman BA, Pollock BH, Tomlinson GE. The spectrum of APC mutations in children with hepatoblastoma from familial adenomatous polyposis kindreds. J Pediatr 2005;147:263-6.
18. Koch A, Weber N, Waha A, et al. Mutations and elevated transcriptional activity of conductin (AXIN2) in hepatoblastomas. J Pathol 2004;204:546-54.
19. Taniguchi K, Roberts LR, Aderca IN, et al. Mutational spectrum of beta-catenin, AXIN1, and AXIN2 in hepatocellular carcinomas and hepatoblastomas. Oncogene 2002;21:4863-71.
20. Trobaugh-Lotrario AD, Venkatramani R, Feusner JH. Hepatoblastoma in children with Beckwith-Wiedemann syndrome: does it warrant different treatment? J Pediatr Hematol Oncol 2014;36:369-73.
21. Ansell P, Mitchell CD, Roman E, Simpson J, Birch JM, Eden TO. Relationships between perinatal and maternal characteristics and hepatoblastoma: a report from the UKCCS. Eur J Cancer 2005;41:741-8.
22. McLaughlin CC, Baptiste MS, Schymura MJ, Nasca PC, Zdeb MS. Maternal and infant birth characteristics and hepatoblastoma. Am J Epidemiol 2006;163:818-28.
23. Grobner SN, Worst BC, Weischenfeldt J, et al. The landscape of genomic alterations across childhood cancers. Nature 2018;555:321-7.
24. Vokuhl C, Oyen F, Haberle B, von Schweinitz D, Schneppenheim R, Leuschner I. Small cell undifferentiated (SCUD) hepatoblastomas: all malignant rhabdoid tumors? Genes Chromosomes Cancer 2016;55:925-31.
25. Zhou S, Venkatramani R, Gupta S, et al. Hepatocellular malignant neoplasm, NOS: a clinicopathological study of 11 cases from a single institution. Histopathology 2017;71:813-22.
26. Cairo S, Armengol C, Maibach R, et al. A combined clinical and biological risk classification improves prediction of outcome in hepatoblastoma patients. Eur J Cancer 2020;141:30-9.
27. Carrillo-Reixach J, Torrens L, Simon-Coma M, et al. Epigenetic footprint enables molecular risk stratification of hepatoblastoma with clinical implications. J Hepatol 2020;73:328-41.
28. Jeong Y, Hellyer JA, Stehr H, et al. Role of KEAP1/NFE2L2 Mutations in the chemotherapeutic response of patients with non-small cell lung cancer. Clin Cancer Res 2020;26:274-81.
29. Jia D, Dong R, Jing Y, et al. Exome sequencing of hepatoblastoma reveals novel mutations and cancer genes in the Wnt pathway and ubiquitin ligase complex. Hepatology 2014;60:1686-96.
30. Lee H, El Jabbour T, Ainechi S, et al. General paucity of genomic alteration and low tumor mutation burden in refractory and metastatic hepatoblastoma: comprehensive genomic profiling study. Hum Pathol 2017;70:84-91.
31. Cairo S, Wang Y, de Reynies A, et al. Stem cell-like micro-RNA signature driven by Myc in aggressive liver cancer. Proc Natl Acad Sci U S A 2010;107:20471-6.
32. Hooks KB, Audoux J, Fazli H, et al. New insights into diagnosis and therapeutic options for proliferative hepatoblastoma. Hepatology 2018;68:89-102.
33. Cui X, Liu B, Zheng S, Dong K, Dong R. Genome-wide analysis of DNA methylation in hepatoblastoma tissues. Oncol Lett 2016;12:1529-34.
34. Honda S, Minato M, Suzuki H, et al. Clinical prognostic value of DNA methylation in hepatoblastoma: four novel tumor suppressor candidates. Cancer Sci 2016;107:812-9.
35. Maschietto M, Rodrigues TC, Kashiwabara AY, et al. DNA methylation landscape of hepatoblastomas reveals arrest at early stages of liver differentiation and cancer-related alterations. Oncotarget 2017;8:97871-89.
36. Buj R, Mallona I, Diez-Villanueva A, et al. Quantification of unmethylated Alu (QUAlu): a tool to assess global hypomethylation in routine clinical samples. Oncotarget 2016;7:10536-46.
37. Meyers RL, Maibach R, Hiyama E, et al. Risk-stratified staging in paediatric hepatoblastoma: a unified analysis from the Children's Hepatic tumors International Collaboration. Lancet Oncol 2017;18:122-31.
38. Doi I. Establishment of a cell line and its clonal sublines from a patient with hepatoblastoma. Gann 1976;67:1-10.
39. Pietsch T, Fonatsch C, Albrecht S, Maschek H, Wolf HK, von Schweinitz D. Characterization of the continuous cell line HepT1 derived from a human hepatoblastoma. Lab Invest 1996;74:809-18.
40. Hartmann W, Waha A, Koch A, et al. p57(KIP2) is not mutated in hepatoblastoma but shows increased transcriptional activity in a comparative analysis of the three imprinted genes p57(KIP2), IGF2, and H19. Am J Pathol 2000;157:1393-403.
41. Aden DP, Fogel A, Plotkin S, Damjanov I, Knowles BB. Controlled synthesis of HBsAg in a differentiated human liver carcinoma-derived cell line. Nature 1979;282:615-6.
42. Lopez-Terrada D, Cheung SW, Finegold MJ, Knowles BB. Hep G2 is a hepatoblastoma-derived cell line. Hum Pathol 2009;40:1512-5.
43. Knowles BB, Howe CC, Aden DP. Human hepatocellular carcinoma cell lines secrete the major plasma proteins and hepatitis B surface antigen. Science 1980;209:497-9.
44. Hartmann W, Kuchler J, Koch A, et al. Activation of phosphatidylinositol-3'-kinase/AKT signaling is essential in hepatoblastoma survival. Clin Cancer Res 2009;15:4538-45.
45. Eriksson T, Frisk T, Gray SG, et al. Methylation changes in the human IGF2 p3 promoter parallel IGF2 expression in the primary tumor, established cell line, and xenograft of a human hepatoblastoma. Exp Cell Res 2001;270:88-95.
46. Seeliger H, Guba M, Kleespies A, Jauch KW, Bruns CJ. Role of mTOR in solid tumor systems: a therapeutical target against primary tumor growth, metastases, and angiogenesis. Cancer Metastasis Rev 2007;26:611-21.
47. Guba M, von Breitenbuch P, Steinbauer M, et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat Med 2002;8:128-35.
48. Wagner F, Henningsen B, Lederer C, et al. Rapamycin blocks hepatoblastoma growth in vitro and in vivo implicating new treatment options in high-risk patients. Eur J Cancer 2012;48:2442-50.
49. Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 2007;26:1932-40.
50. Nielsen D, Briem-Richter A, Sornsakrin M, Fischer L, Nashan B, Ganschow R. The use of everolimus in pediatric liver transplant recipients: first experience in a single center. Pediatr Transplant 2011;15:510-4.
51. Eichenmüller M, Gruner I, Hagl B, et al. Blocking the hedgehog pathway inhibits hepatoblastoma growth. Hepatology 2009;49:482-90.
52. Huang S, He J, Zhang X, et al. Activation of the hedgehog pathway in human hepatocellular carcinomas. Carcinogenesis 2006;27:1334-40.
53. Cory S, Adams JM. Killing cancer cells by flipping the Bcl-2/Bax switch. Cancer Cell 2005;8:5-6.
54. Lieber J, Kirchner B, Eicher C, et al. Inhibition of Bcl-2 and Bcl-X enhances chemotherapy sensitivity in hepatoblastoma cells. Pediatr Blood Cancer 2010;55:1089-95.
55. Lieber J, Eicher C, Wenz J, et al. The BH3 mimetic ABT-737 increases treatment efficiency of paclitaxel against hepatoblastoma. BMC Cancer 2011;11:362.
56. Munoz M, Covenas R. The Neurokinin-1 receptor antagonist aprepitant: an intelligent bullet against cancer? Cancers (Basel) 2020;12:2682.
57. Berger M, Neth O, Ilmer M, et al. Hepatoblastoma cells express truncated neurokinin-1 receptor and can be growth inhibited by aprepitant in vitro and in vivo. J Hepatol 2014;60:985-94.
58. Eberherr C, Beck A, Vokuhl C, et al. Targeting excessive MYCN expression using MLN8237 and JQ1 impairs the growth of hepatoblastoma cells. Int J Oncol 2019;54:1853-63.
59. Wagner AE, Schwarzmayr T, Haberle B, et al. SP8 promotes an aggressive phenotype in hepatoblastoma via FGF8 activation. Cancers (Basel) 2020;12:2294.
60. Whitlock RS, Yang T, Vasudevan SA, Woodfield SE. Animal modeling of pediatric liver cancer. Cancers (Basel) 2020;12:273.
61. Mokkapati S, Niopek K, Huang L, et al. beta-catenin activation in a novel liver progenitor cell type is sufficient to cause hepatocellular carcinoma and hepatoblastoma. Cancer Res 2014;74:4515-25.
62. Bissig-Choisat B, Kettlun-Leyton C, Legras XD, et al. Novel patient-derived xenograft and cell line models for therapeutic testing of pediatric liver cancer. J Hepatol 2016;65:325-33.
63. Nicolle D, Fabre M, Simon-Coma M, et al. Patient-derived mouse xenografts from pediatric liver cancer predict tumor recurrence and advise clinical management. Hepatology 2016;64:1121-35.
64. Kats D, Ricker CA, Berlow NE, et al. Volasertib preclinical activity in high-risk hepatoblastoma. Oncotarget 2019;10:6403-17.
65. Yamada S, Ohira M, Horie H, et al. Expression profiling and differential screening between hepatoblastomas and the corresponding normal livers: identification of high expression of the PLK1 oncogene as a poor-prognostic indicator of hepatoblastomas. Oncogene 2004;23:5901-11.
66. Eloranta K, Cairo S, Liljestrom E, et al. Chloroquine triggers cell death and inhibits PARPs in cell models of aggressive hepatoblastoma. Front Oncol 2020;10:1138.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Armengol C, Cairo S, Kappler R. Bridging molecular basis, prognosis, and treatment of pediatric liver tumors. Hepatoma Res 2021;7:50. http://dx.doi.org/10.20517/2394-5079.2021.19
AMA Style
Armengol C, Cairo S, Kappler R. Bridging molecular basis, prognosis, and treatment of pediatric liver tumors. Hepatoma Research. 2021; 7: 50. http://dx.doi.org/10.20517/2394-5079.2021.19
Chicago/Turabian Style
Armengol, Carolina, Stefano Cairo, Roland Kappler. 2021. "Bridging molecular basis, prognosis, and treatment of pediatric liver tumors" Hepatoma Research. 7: 50. http://dx.doi.org/10.20517/2394-5079.2021.19
ACS Style
Armengol, C.; Cairo S.; Kappler R. Bridging molecular basis, prognosis, and treatment of pediatric liver tumors. Hepatoma. Res. 2021, 7, 50. http://dx.doi.org/10.20517/2394-5079.2021.19
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 14 clicks
Cite This Article 14 clicks

 Like This Article 11
likes
Like This Article 11
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.