
Clinical implications of hepatic progenitor cell activation in non-alcoholic fatty liver disease
 0
0Abstract
Non-alcoholic fatty liver disease (NAFLD) is one of the most prominent causes of liver-related morbidity in the Western world. NAFLD is a chronic disease characterised by accumulation of triglycerides in hepatocytes. Upon damage, hepatocytes drive regeneration to sustain homeostasis of the liver. However, 30-40 years of ongoing replication induced by chronic lipid damage and oxidative stress increase senescence of the hepatocytes. At this stage, activation of a reserve compartment is seen, known as the hepatic progenitor cells (HPCs). HPCs are bipotent cells which can differentiate into hepatocytes or cholangiocytes depending on the underlying aetiology in order to facilitate liver regeneration. Activation of HPCs is observed as ductular reaction (DR), comprising an expansion of transit amplifying cells of the terminal branches of the biliary tree. DR is usually observed in advanced NAFLD but is also associated with histological severity and distinct molecular profiles. In this context, information about HPCs and their activation in the form of DR may add a both diagnostic and prognostic values when assessing NAFLD patients. In this review, we analyse HPCs characteristics and development, and the clinical impact of their activation in subjects with NAFLD.
Keywords
Introduction
Non-alcoholic fatty liver disease (NAFLD) is the leading cause of chronic liver disease worldwide[1]. The natural history of NAFLD includes a wide range of phenotypes, from completely asymptomatic conditions to a relatively fast progression to its necroinflammatory form [non-alcoholic steatohepatitis(NASH)] with or without fibrosis and eventually liver cirrhosis and/or hepatocellular carcinoma (HCC)[2,3]. NAFLD is commonly considered the hepatic manifestation of the metabolic syndrome and frequently develops in patients with an altered metabolic environment and who often present comorbidities such as obesity, diabetes, arterial hypertension, or dyslipidaemia[4]. However, it should be kept in mind that these characteristics, and hence NAFLD, may also be found in non-obese individuals[5]. Sedentary lifestyle and overnutrition are classically considered as drivers of NAFLD, though recent studies suggest that diets with high fructose intake increase the risk of NAFLD and NASH[6]. Consequently, NAFLD is considered a complex disease, which occurs as a result of the interaction between environmental risk factors and a susceptible polygenic background[7]. Recent advances in the field have made it increasingly clear that genetic factors play an important role in determining response to high-calorie, high fat diets and subsequent metabolic stress in NAFLD patients. Several genetic modifiers have now been identified, including single nucleotide polymorphisms in PNPLA3 (patatin-like phospholipase domain-containing protein 3), TM6SF2 (transmembrane 6 superfamily member 2), and HSD17B13 (hydroxysteroid 17-beta dehydrogenase 13)[8-10].
At present, the clinical diagnosis of NAFLD is ultimately confirmed by histological assessment of tissue following liver biopsy that remains the “gold standard” method for disease classification[11]. Although invasive, liver biopsy rarely results in complications and allows defining the histological hallmarks of the disease (steatosis, hepatocyte ballooning, lobular inflammation, and fibrosis stage)[12] and accurately excludes other chronic liver diseases which may have similar presentations[13]. It is therefore clinically important to distinguish the features of patients who may develop severe disease in order to optimise treatment and follow up. In this context, information about hepatic progenitor cells (HPCs) and their activation in the form of ductular reaction (DR) may add both diagnostic and prognostic values when assessing NAFLD patients. As there are currently no approved treatments for NASH, lifestyle modification and diet represent the only available standard of care. In addition, drugs with a potential benefit for patients with NAFLD (e.g., vitamin E or pioglitazone) are limited by the burden of their side effects and the impracticality to be administered to all individuals with the disease[14]. Hence, the efforts of the scientific community are towards the discovery of biomarkers able to stratify patients and identify a target population at increased risk of disease progression. In this review, we analyse the characteristics and development of HPCs, and the clinical impact of HPC activation in subjects with NAFLD.
Hepatic progenitor cell-mediated regeneration
Upon injury, the liver can self-renew by proliferation of hepatocytes and cholangiocytes followed by proliferation of mesenchymal and endothelial cells[15]. However, it has been shown that in many chronic liver diseases, including NAFLD, hepatocytes and cholangiocytes become increasingly senescent[16-18]. This may be due, in part, to ongoing replication over a 30-40 year period of chronic damage, as well as a range of other factors such as oxidative stress, cholestasis, or hypoxia[19]. At this point, activation and proliferation of a reserve compartment known as HPCs, can be seen in the smallest terminal branches of the biliary tree, the canals of Hering, where they are normally quiescent [Figure 1][20-22]. In human disease, activation of HPCs presents itself in the form of DR, a transient amplification of HPCs, which can be seen at the portal-parenchymal interface[23-25]. However, in rodents, HPCs are termed “oval cells” due to their characteristic shape and are seen to expand more as single cells rather than the DR typically seen in humans[26].
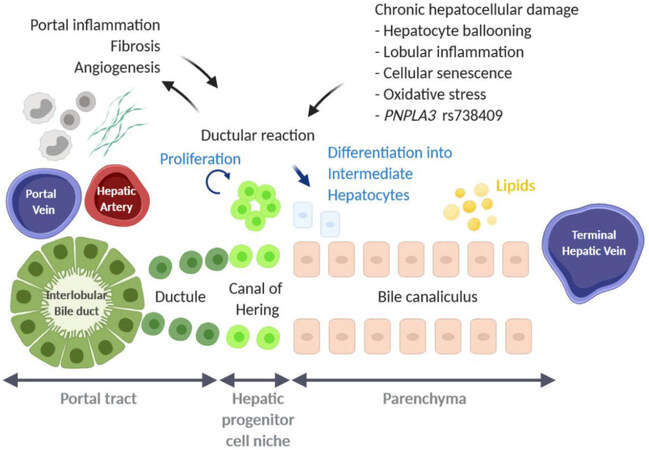
Figure 1. Schematic overview of hepatic progenitor cell activation, observed as ductular reaction, in human non-alcoholic fatty liver disease. Image has been created using BioRender
HPCs are bipotent cells which possess the ability to differentiate into cholangiocytes or hepatocytes depending on the underlying aetiology and on which cells are most damaged[27]. The strong differentiation potential of HPCs was recently demonstrated by Lu et al.[28] using targeted deletion of Mdm2 (E3 ubiquitin-protein ligase) in murine hepatocytes to induce cell senescence. In these mice, there was significant activation of HPCs followed by complete liver regeneration. Furthermore, isolation and transplantation of HPCs from wild-type mice confirmed a substantial contribution of HPCs towards regeneration of hepatocytes and cholangiocytes as well as restoration of the liver parenchyma. Using lineage tracing mouse models, Raven et al.[29] showed that concurrent inhibition of hepatocyte proliferation by p21 overexpression directly triggered ductular reaction. Upon differentiation towards hepatocytes, HPCs have been shown to gradually lose their biliary features[27,30], including markers such as keratin 19 (K19) and keratin 7 (K7). Loss of K19 is seen early in differentiation, with expression of K7 and epithelial cell adhesion molecule (EPCAM) lost later on in hepatocyte differentiation [Figure 2][30].
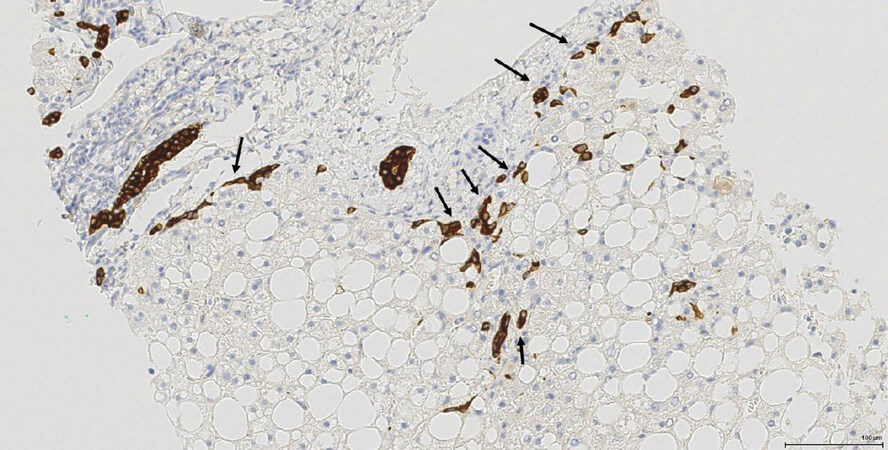
Figure 2. HPCs in NASH with moderate fibrosis (F2 stage) (needle biopsy sample). Keratin 7 immunostaining decorates the interlobular bile ducts, ductular reaction/HPCs (arrows). HPCs: hepatic progenitor cells; NASH: non-alcoholic steatohepatitis
HPCs reside in a specialised micro-environment, their so-called niche. Upon HPC activation and differentiation, the composition of the niche changes during chronic disease which is aetiology dependent and is characterised by infiltrating inflammatory cells, specific extracellular matrix composition, vascularisation, innervation, and even growth factors[31,32]. In addition to the changes in biomarker expression, commitment to a specific cell fate of HPCs is further defined by alternate activation of different pathways through interaction with the different cells in their niche. For example, expression of the Notch ligand Jagged 1 during biliary regeneration promotes Notch signalling in HPCs and, consequently, their differentiation to cholangiocytes[33]. Conversely, an increase in Wnt3a expression by macrophages gives rise to canonical Wnt signalling in HPCs, resulting in maintenance of Numb expression, an inhibitor of Notch signalling, and commitment to a hepatocyte cell fate[33]. Similarly, YAP and HIPPO signalling have an opposite function in regulating liver cell fate[34]. The extracellular matrix also plays an important role in determining the cell fate of HPCs. Laminins in particular have been reported to sustain biliary features and differentiate induced-pluripotent stem cells into cholangiocytes[35-37], whereas the inhibition of laminin deposition in a mouse model of chronic parenchymal damage has been described to result in enhanced differentiation of HPCs/oval cells into hepatocytes[38]. Using mouse models of chronic biliary injury, Peng et al.[39] showed that the disruption of the integrin receptor α5β6, an adhesion receptor that interacts with fibronectin and transforming growth factor beta 1, not only inhibited the response of oval cells to tissue damage but also reduced fibrosis. This suggests that HPCs and their niche can have, besides a role in liver regeneration, a pro-fibrotic effect. DR and the presence of intermediate hepatocytes is a phenome of older age and advanced disease[40]. Promotion of the appropriate pathways therefore plays a key role in parenchymal regeneration and resolution of liver injury.
Portal inflammation and ductular reaction in NAFLD
Portal chronic inflammation has been associated with features of metabolic syndrome in NAFLD patients. The Nonalcoholic Steatohepatitis Clinical Research Network (NASH CRN) has shown that patients with mild or moderate portal inflammation were more likely to present higher BMI, higher insulin values, and insulin resistance when compared to patients with no signs of portal inflammation[41]. In addition, a significantly higher rate of patients with portal inflammation took medication for the treatment of diabetes and/or cardiovascular and hypertensive conditions. Histologically, patients with portal inflammation had significantly increased ballooning and advanced fibrosis, together with a higher likelihood of a definite NASH diagnosis[41]. Another study investigated the clinical features of 160 biopsy-proven NAFLD patients and the correlation with portal inflammation[42]. They confirmed patients with a higher degree of portal inflammation presented higher age, BMI, cholesterol, alanine aminotransferase, gamma-glutamyl transpeptidase, and ferritin as well as arterial hypertension and diabetes. In a cohort of 619 NAFLD patients with a median follow-up duration 12.6 years, Angulo et al.[43] showed using univariate analysis that there was a significantly different survival free of liver transplantation and survival free of liver-related events according to ballooning grade, portal inflammation grade, the NASH categories, and fibrosis stage, but not among steatosis grade, lobular inflammation grade, or NAFLD Activity Score (NAS) categories[43]. Moreover, they found a significant correlation between fibrosis and portal inflammation, although in the multivariate analysis only fibrosis was associated with long-term outcome for patients.
The extent of DR in adult and paediatric NAFLD patients has been strongly associated with the diagnosis of NASH, disease severity, including histological features such as hepatocyte ballooning, lobular inflammation, and fibrosis, but also portal inflammation[17,44]. In this context, the distinct micro-environment of DR could play a pivotal function in the homing of immune cells into the portal tract. In particular, portal leukocytes expressing CD3, CD8, or CD68, exhibit a significant and incremental variation across NAFLD stages[45]. Although these leukocytes were also observed in control livers and at early disease stages, all types of portal leukocytes significantly increased with the progression of NASH in concomitant with DR and fibrosis deposition. Interestingly, the increase in portal macrophages in patients with mild NAFLD has been reported to precede the expression of proinflammatory cytokines such as IL1B and TNF[45]. Numerous cytokines link inflammation with HPC proliferation and differentiation, such as TNF-like weak inducer of apoptosis (TWEAK), transforming growth factor beta, interferon-gamma, and interleukin 17[46-49]. Many of these key modulators of liver inflammation and regeneration can be tracked back to portal infiltration. Infiltrating T helper (Th)17 lymphocytes have been implicated in several types of liver diseases including NAFLD and alcoholic liver disease[45,50], while TWEAK has been reported to originate from infiltrating macrophages[51].
Furthermore, the degree of DR also reflects the amount of parenchymal damage. Patients carrying the PNPLA3 I148M variant have been characterized by a prominent activation of HPC niche that was associated with a more aggressive histological pattern and portal fibrogenesis, and increased oxidative stress[52]. Interestingly, these patients displayed increased portal infiltration of pro-inflammatory S100A9-positive macrophages. The influx of S100A9-positive cells in advanced NAFLD has been correlated with an increase of monocyte-derived C-C motif chemokine receptor 2 (CCR2)-positive macrophages in the portal tract[53]. There is compelling data in both mouse models and humans which show that macrophage infiltration into the liver is predominantly controlled by CCR2 and its ligand C-C motif chemokine ligand (CCL2)[54-56]. It has been shown through CCR2-/- mice that CCR2 plays an important role in the recruitment of monocyte-derived macrophages and stellate cells[57]. Earlier studies in high fat diet-fed mice also demonstrated that CCL2 and CCR2 were crucial in development of steatosis, insulin resistance, and obesity[58,59]. More recent studies have now confirmed that monocyte-derived macrophages primarily express CCR2 and the inflammatory phenotype associated with steatosis, fibrosis, and angiogenesis, rather than Kupffer cells[53]. This supports a potentially key role of recruited macrophages in the development and progression of NAFLD. Therapeutic inhibition of CCR2-positive monocyte-derived macrophages using cenicriviroc has been reported to reduce fibrosis in human disease[60]. Although the effect on NASH amelioration was limited, patients on the cenicriviroc arm achieved significant improvement in fibrosis stage by ≥ 1 stage and no worsening of steatohepatitis when compared to those on placebo [20% vs. 10%; OR: 2.20 (95%CI: 1.11-4.35)]. These results were more prominent in patients with higher histological NASH activity and were equally represented in all fibrosis subgroups[60]. If we consider that fibrosis is the main driver of outcomes in NASH patients[43], we can still consider these results as relevant. Interestingly, mild or no portal inflammation at baseline were among factors predicting cenicriviroc response, in terms of fibrosis regression[60]. In other words, although cenicriviroc targets specificreceptors, the complexity of the inflammatory portal milieu may predict treatment response in patients with NAFLD. As mentioned, HPCs were linked to increased portal fibrogenesis, hence they might be considered as therapeutic target in future design.
Nevertheless, portal chronic inflammation acquires even more significance since it may not disappear after NASH pharmacological or bariatric surgical treatment, as highlighted in studies from clinical trials[61]. Indeed, increased portal inflammation in post-treatment liver biopsies has been related to NASH resolution[62]. Despite considerable evidence of the importance of macrophage infiltration in disease progression, current research into the mechanisms involved in their recruitment during NAFLD is very limited and remains incompletely understood.
Hepatic progenitor cell secretome
HPCs are not only associated with portal inflammation but they themselves can affect the recruitment, differentiation, and functional properties of immune cells by secreting a range of proteins[63]. HPCs have been reported to be a source of the chemokine CCL2 which can, apart from attracting CCR2-positive monocytes, induce a distinct metabolic shift in hepatocytes[64-66]. In vitro, CCL2 can induce lipid accumulation in primary mouse hepatocytes and in human HCC cell lines, while overexpression of Ccl2 in mice induces hepatic steatosis, decreases AMP-activated protein kinase activity and alters the hepatic mitochondrial dynamics[65,66]. In paediatric NAFLD, the expression of resistin and glucagon Like Peptide 1 (GLP-1) in HPCs has been correlated with advanced features of NAFLD, fibrosis and steatosis, respectively, while the expression of adiponectin in HPCs was inversely correlated with the NASH Activity Score[44]. Serum protein and hepatic mRNA levels of the adipokine resistin have been reported to be increased in adult patients with NASH[67], while adiponectin plasma levels are markedly diminished in patients with visceral obesity, type 2 diabetes mellitus, and NASH[68,69]. Resistin can increase the uptake of oxidised low-density lipoprotein by macrophages through their scavenger receptors, thereby promoting foamy cell formation in vitro, suggesting that HPCs influence macrophage function[70-72]. Recently, our group showed that the formation of foamy macrophages in human NAFLD is mediated by the macrophage scavenger receptor 1, resulting in the release of pro-inflammatory cytokines, such as TNFa, but also inducing metabolic changes in the liver[73,74]. Adiponectin, on the other hand, is regarded as an anti-inflammatory modulator in NAFLD and is protective against HCC formation[69,75]. Though HPCs in NAFLD show a decreased expression of adiponectin, indicative for a shift towards a pro-inflammatory phenotype, they also secrete GLP-1, which can decrease transaminases and reduce intrahepatic triglycerides and macrophage infiltration in a NASH mouse model[44,76]. In a double blinded, randomised trial, NASH patients treated with the GLP-1 analogue liraglutide showed reduced metabolic dysfunction, insulin resistance, and lipotoxicity compared to the placebo group[77]. This suggests that some of the factors released by HPCs can also be anti-inflammatory. The dual pro- and anti-inflammatory phenotype, and the pleiotropic metabolic effects of the HPC secretome on other cells in the liver highlights the complexity of the progenitor cell-mediated regeneration during NAFLD progression.
Gene signatures of advanced NAFLD
Over recent years, high-through transcriptomic analyses have been used in efforts to examine changes that occur as NAFLD progresses [Table 1]. Though some studies are limited to dichotomous comparisons between mild and advanced disease, the expression of HPC markers has been found to be enriched in more advanced stage of the disease[78-84]. In a small cohort of 22 patients with NAFLD or alcoholic liver disease, Starmann et al.[78] reported the expression of KRT7, KRT23, and p62 to be enriched in steatohepatitis when compared to steatosis. K23 has been described as a stress-related DR marker associated with prominent inflammation and fibrosis in chronic liver disease, that can be induced in vitro by treatment with TWEAK and the type I acute phase inducer interleukin (IL)-1β[85]. Interestingly, other transcriptomics studies using larger NAFLD cohorts, have reported that the expression of TNF receptor superfamily member 12A (TNFRSF12A), the receptor for TWEAK, is increasingly expressed in advanced NAFLD[83,84]. Immunopositivity for TNFRSF12A in NASH has been described to be found in DR, endothelial cells and myofibroblasts[86]. This means that TWEAK not only induces HPC proliferation but could also induce pathways protective against cell stress.
| Comparison | NAFLD patients | Platform | Dif exp genes | Selected HPC/stem cell markers | Reference |
|---|---|---|---|---|---|
| Steatosis vs. (N)ASH | n = 22 | Microarray | 2543 | KRT7, KRT23, p62 | Starmann et al.[78] 2012 |
| NAFLD F0-1 vs. F3-4 | n = 72 | Microarray | 64 | EPCAM, JAG1, SOX9, CD24, GLI2 | Moylan et al.[79] 2014 |
| sSmple steatosis vs. NASH | n = 39 | Microarray | 22 | SPP1 | Arendt et al.[80] 2015 |
| NAFL vs. NASH F0-2 | n = 31 | RNAseq | 132 | THY1, LAMA1 | Suppli et al.[81] 2019 |
| Lobular inflammation vs. fibrosis | n = 118 | RNAseq | 34 | SPP1 | Gerhard et al.[82] 2018 |
| Regression gene-level NAS and fibrosis | n = 72 | RNAseq | 20 | TNFRSF12A | Hoang et al.[83] 2019 |
| NAFL or NASH F0/1 vs. NASH F2 or F3 or F4 | n = 206 | RNAseq | 25 | THY1, TNFRSF12A | Govaere et al.[84] 2020 |
| NAS ≥ 4 vs. NAS < 4 | n = 206 | RNAseq | 369 | CD24, EPCAM, GLI2, SPP1, TNFRSF12A | Govaere et al.[84] 2020 |
| Unsupervised clustering of NAFLD patients | n = 206 | RNAseq | Top 250 | CD24, EPCAM, JAG1, KRT7, KRT19, LAMC2, SOX9, TACSTD2, THY1, TNFRSF12A | Govaere et al.[84] 2020 |
Hepatocytes can activate the p62-KEAP1-NRF2 pathway to cope with oxidative stress[87]. Phosphorylation of p62 disrupts the cytoplasmic binding of KEAP1 to NRF2, facilitating the transport of NRF2 to the nucleus where it regulates autophagy and “stemness” pathways[88,89]. In NASH, p62-positive Mallory-Denk bodies, cytoplasmic inclusions commonly observed in ballooned hepatocytes, correlate with increased parenchymal necroinflammation[90]. Similarly, the expression of the sonic hedgehog signaling molecule, found in ballooned hepatocytes, correlates with the grade of hepatocyte ballooning and fibrosis stage[91]. Interestingly, expression of GLI2, the transcription factor downstream of the sonic hedgehog pathway, has been found to be expressed in DR and was associated with portal inflammation and fibrosis stage in a cohort of 30 NAFLD patients[91]. In chronic liver disease, including biliary fibrosis and viral hepatitis, sonic hedgehog signaling is believed necessary to generate, maintain, and expand certain populations of HPCs[92,93]. It is still not fully known whether ballooned hepatocytes can directly activate HPCs, though activation of similar pathways would suggest so. This would explain the association between DR and the histological severity in NAFLD. Recently, our group showed that the expression of HPC markers was associated with a high NAFLD Activity Score using RNA sequencing performed on 206 NAFLD biopsy samples[84]. Unsupervised clustering stratified the 206 NAFLD patients based on disease activity, fibrosis stage, age, serum aspartate aminotransferase, type 2 diabetes mellitus, and carriage of the PNPLA3 rs738409 variant[84]. Intriguingly, this was correlated with a high gene expression of HPC markers including CD24, EPCAM, JAG1, KRT7, KRT19, LAMC2, SOX9, TACSTD2, THY1, and TNFRSF12A[Table 1].
In conclusion, HPC activation in NAFLD, observed as DR, has been associated with histological severity and distinct molecular profiles, and may confer both diagnostic and prognostic values when assessing NAFLD patients.
Declarations
Authors’ contributionsContributed to the manuscript drafting and critically revised the manuscript for intellectual content: Younes R, Burgoyne R, Tiniakos D, Govaere O
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work has been supported by the EPoS (Elucidating Pathways of Steatohepatitis) consortium funded by the Horizon 2020 Framework Program of the European Union under Grant Agreement 634413.
Conflicts of interestRamy Younes is currently employed by Boehringer Ingelheim International. Dina Tiniakos reports consultation fees from Intercept Pharmaceuticals Inc, Allergan, Cirius Therapeutics and an educational grant from Histoindex Pte Ltd. All other authors declare that they have no competing interests.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2021.
REFERENCES
1. Younossi Z, Anstee QM, Marietti M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 2018;15:11-20.
2. Marengo A, Jouness RI, Bugianesi E. Progression and natural history of nonalcoholic fatty liver disease in adults. Clin Liver Dis 2016;20:313-24.
3. Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol 2019;16:411-28.
4. Bugianesi E, McCullough AJ, Marchesini G. Insulin resistance: a metabolic pathway to chronic liver disease. Hepatology 2005;42:987-1000.
6. Jensen T, Abdelmalek MF, Sullivan S, et al. Fructose and sugar: a major mediator of non-alcoholic fatty liver disease. J Hepatol 2018;68:1063-75.
7. Anstee QM, Darlay R, Cockell S, et al. Genome-wide association study of non-alcoholic fatty liver and steatohepatitis in a histologically-characterised cohort. J Hepatol 2020;73:505-15.
8. Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461-5.
9. Kozlitina J, Smagris E, Stender S, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2014;46:352-6.
10. Abul-Husn NS, Cheng X, Li AH, et al. A protein-truncating HSD17B13 variant and protection from chronic liver disease. N Engl J Med 2018;378:1096-106.
11. European Association for the Study of the L, European Association for the Study of D, European Association for the Study of O. EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J Hepatol 2016;64:1388-402.
12. Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313-21.
13. Younes R, Govaere O, Petta S, et al. Presence of serum antinuclear antibodies does not impact long-term outcomes in nonalcoholic fatty liver disease. Am J Gastroenterol 2020;115:1289-92.
14. Leoni S, Tovoli F, Napoli L, Serio I, Ferri S, Bolondi L. Current guidelines for the management of non-alcoholic fatty liver disease: A systematic review with comparative analysis. World J Gastroenterol 2018;24:3361-73.
15. Libbrecht L, Roskams T. Hepatic progenitor cells in human liver diseases. Semin Cell Dev Biol 2002;13:389-96.
16. Aravinthan A, Scarpini C, Tachtatzis P, et al. Hepatocyte senescence predicts progression in non-alcohol-related fatty liver disease. J Hepatol 2013;58:549-56.
17. Richardson MM, Jonsson JR, Powell EE, et al. Progressive fibrosis in nonalcoholic steatohepatitis: association with altered regeneration and a ductular reaction. Gastroenterology 2007;133:80-90.
18. Sasaki M, Ikeda H, Yamaguchi J, Miyakoshi M, Sato Y, Nakanuma Y. Bile ductular cells undergoing cellular senescence increase in chronic liver diseases along with fibrous progression. Am J Clin Pathol 2010;133:212-23.
19. Roskams T, Yang SQ, Koteish A, et al. Oxidative stress and oval cell accumulation in mice and humans with alcoholic and nonalcoholic fatty liver disease. Am J Pathol 2003;163:1301-11.
20. Roskams T. Liver stem cells and their implication in hepatocellular and cholangiocarcinoma. Oncogene 2006;25:3818-22.
21. Van Haele M, Roskams T. Hepatic progenitor cells: an update. Gastroenterol Clin North Am 2017;46:409-20.
22. Roskams TA, Theise ND, Balabaud C, et al. Nomenclature of the finer branches of the biliary tree: canals, ductules, and ductular reactions in human livers. Hepatology 2004;39:1739-45.
23. Desmet VJ. Ductal plates in hepatic ductular reactions. Hypothesis and implications. I. Types of ductular reaction reconsidered. Virchows Arch 2011;458:251-9.
24. Desmet VJ. Ductal plates in hepatic ductular reactions. Hypothesis and implications. II. Ontogenic liver growth in childhood. Virchows Arch 2011;458:261-70.
25. Desmet VJ. Ductal plates in hepatic ductular reactions. Hypothesis and implications. III. Implications for liver pathology. Virchows Arch 2011;458:271-9.
27. Ko S, Russell JO, Molina LM, Monga SP. Liver progenitors and adult cell plasticity in hepatic injury and repair: knowns and unknowns. Annu Rev Pathol 2020;15:23-50.
28. Lu WY, Bird TG, Boulter L, et al. Hepatic progenitor cells of biliary origin with liver repopulation capacity. Nat Cell Biol 2015;17:971-83.
29. Raven A, Lu WY, Man TY, et al. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature 2017;547:350-4.
30. Govaere O, Roskams T. Pathogenesis and prognosis of hepatocellular carcinoma at the cellular and molecular levels. Clin Liver Dis 2015;19:261-76.
31. Govaere O, Cockell S, Van Haele M, et al. High-throughput sequencing identifies aetiology-dependent differences in ductular reaction in human chronic liver disease. J Pathol 2019;248:66-76.
32. Zanchi A, Reidy J, Feldman HJ, et al. Innervation of the proximal human biliary tree. Virchows Arch 2020;477:385-92.
33. Boulter L, Govaere O, Bird TG, et al. Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat Med 2012;18:572-9.
34. Yimlamai D, Christodoulou C, Galli GG, et al. Hippo pathway activity influences liver cell fate. Cell 2014;157:1324-38.
35. Govaere O, Wouters J, Petz M, et al. Laminin-332 sustains chemoresistance and quiescence as part of the human hepatic cancer stem cell niche. J Hepatol 2016;64:609-17.
36. Govaere O, Petz M, Wouters J, et al. The PDGFRalpha-laminin B1-keratin 19 cascade drives tumor progression at the invasive front of human hepatocellular carcinoma. Oncogene 2017;36:6605-16.
37. Takayama K, Mitani S, Nagamoto Y, et al. Laminin 411 and 511 promote the cholangiocyte differentiation of human induced pluripotent stem cells. Biochem Biophys Res Commu 2016;474:91-6.
38. Espanol-Suner R, Carpentier R, Van Hul N, et al. Liver progenitor cells yield functional hepatocytes in response to chronic liver injury in mice. Gastroenterology 2012;143:1564-75 e7.
39. Peng ZW, Ikenaga N, Liu SB, et al. Integrin alphavbeta6 critically regulates hepatic progenitor cell function and promotes ductular reaction, fibrosis, and tumorigenesis. Hepatology 2016;63:217-32.
40. Delladetsima J, Alexandrou P, Giaslakiotis K, et al. Hepatic progenitor cells in chronic hepatitis C: a phenomenon of older age and advanced liver disease. Virchows Arch 2010;457:457-66.
41. Brunt EM, Kleiner DE, Wilson LA, et al. Portal chronic inflammation in nonalcoholic fatty liver disease (NAFLD): a histologic marker of advanced NAFLD-Clinicopathologic correlations from the nonalcoholic steatohepatitis clinical research network. Hepatology 2009;49:809-20.
42. Rakha EA, Adamson L, Bell E, et al. Portal inflammation is associated with advanced histological changes in alcoholic and non-alcoholic fatty liver disease. J Clin Pathol 2010;63:790-5.
43. Angulo P, Kleiner DE, Dam-Larsen S, et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015;149:389-97 e10.
44. Nobili V, Carpino G, Alisi A, et al. Hepatic progenitor cells activation, fibrosis, and adipokines production in pediatric nonalcoholic fatty liver disease. Hepatology 2012;56:2142-53.
45. Gadd VL, Skoien R, Powell EE, et al. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014;59:1393-405.
46. Tirnitz-Parker JE, Viebahn CS, Jakubowski A, et al. Tumor necrosis factor-like weak inducer of apoptosis is a mitogen for liver progenitor cells. Hepatology 2010;52:291-302.
47. Nguyen LN, Furuya MH, Wolfraim LA, et al. Transforming growth factor-beta differentially regulates oval cell and hepatocyte proliferation. Hepatology 2007;45:31-41.
48. Knight B, Lim R, Yeoh GC, Olynyk JK. Interferon-gamma exacerbates liver damage, the hepatic progenitor cell response and fibrosis in a mouse model of chronic liver injury. J Hepatol 2007;47:826-33.
49. Guillot A, Gasmi I, Brouillet A, et al. Interleukins-17 and 27 promote liver regeneration by sequentially inducing progenitor cell expansion and differentiation. Hepatol Commun 2018;2:329-43.
50. Ceulemans A, Verhulst S, Van Haele M, et al. RNA-sequencing-based comparative analysis of human hepatic progenitor cells and their niche from alcoholic steatohepatitis livers. Cell Death Dis 2017;8:e3164.
51. Bird TG, Lu WY, Boulter L, et al. Bone marrow injection stimulates hepatic ductular reactions in the absence of injury via macrophage-mediated TWEAK signaling. Proc Natl Acad Sci U S A 2013;110:6542-7.
52. Carpino G, Pastori D, Baratta F, et al. PNPLA3 variant and portal/periportal histological pattern in patients with biopsy-proven non-alcoholic fatty liver disease: a possible role for oxidative stress. Sci Rep 2017;7:15756.
53. Krenkel O, Puengel T, Govaere O, et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018;67:1270-83.
54. Mitchell C, Couton D, Couty JP, et al. Dual role of CCR2 in the constitution and the resolution of liver fibrosis in mice. Am J Pathol 2009;174:1766-75.
55. Seki E, de Minicis S, Inokuchi S, et al. CCR2 promotes hepatic fibrosis in mice. Hepatology 2009;50:185-97.
56. Zimmermann HW, Seidler S, Nattermann J, et al. Functional contribution of elevated circulating and hepatic non-classical CD14CD16 monocytes to inflammation and human liver fibrosis. PLoS One 2010;5:e11049.
57. Miura K, Yang L, van Rooijen N, Ohnishi H, Seki E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am J Physiol Gastrointest Liver Physiol 2012;302:G1310-21.
58. Kanda H, Tateya S, Tamori Y, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest 2006;116:1494-505.
59. Weisberg SP, Hunter D, Huber R, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest 2006;116:115-24.
60. Friedman SL, Ratziu V, Harrison SA, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018;67:1754-67.
61. Neuschwander-Tetri BA, Brunt EM, Wehmeier KR, Oliver D, Bacon BR. Improved nonalcoholic steatohepatitis after 48 weeks of treatment with the PPAR-gamma ligand rosiglitazone. Hepatology 2003;38:1008-17.
62. Tiniakos DG AQ, Burt AD. Fatty liver disease. In: Burt AD FL, Hubscher SG, editors. MacSween’s Pathology of the Liver: Philadelphia: Elsevier; 2017. pp. 308-71.
63. Lukacs-Kornek V, Lammert F. The progenitor cell dilemma: Cellular and functional heterogeneity in assistance or escalation of liver injury. J Hepatol 2017;66:619-30.
64. Chen L, Luo M, Sun X, et al. DJ-1 deficiency attenuates expansion of liver progenitor cells through modulating the inflammatory and fibrogenic niches. Cell Death Dis 2016;7:e2257.
65. Clement S, Juge-Aubry C, Sgroi A, et al. Monocyte chemoattractant protein-1 secreted by adipose tissue induces direct lipid accumulation in hepatocytes. Hepatology 2008;48:799-807.
66. Luciano-Mateo F, Cabre N, Fernandez-Arroyo S, et al. Chemokine C-C motif ligand 2 overexpression drives tissue-specific metabolic responses in the liver and muscle of mice. Sci Rep 2020;10:11954.
67. Shen C, Zhao CY, Wang W, et al. The relationship between hepatic resistin overexpression and inflammation in patients with nonalcoholic steatohepatitis. BMC Gastroenterol 2014;14:39.
68. Hui JM, Hodge A, Farrell GC, Kench JG, Kriketos A, George J. Beyond insulin resistance in NASH: TNF-alpha or adiponectin? Hepatology 2004;40:46-54.
69. Adolph TE, Grander C, Grabherr F, Tilg H. Adipokines and Non-Alcoholic Fatty Liver Disease: Multiple Interactions. Int J Mol Sci 2017;18.
70. Xu W, Yu L, Zhou W, Luo M. Resistin increases lipid accumulation and CD36 expression in human macrophages. Biochem Biophys Res Commun 2006;351:376-82.
71. Lee TS, Lin CY, Tsai JY, et al. Resistin increases lipid accumulation by affecting class A scavenger receptor, CD36 and ATP-binding cassette transporter-A1 in macrophages. Life Sci 2009;84:97-104.
72. Jamaluddin MS, Weakley SM, Yao Q, Chen C. Resistin: functional roles and therapeutic considerations for cardiovascular disease. Br J Pharmacol 2012;165:622-32.
73. Govaere O, Martinez-Lopez N, Petersen S, et al. Macrophage Scavenger Receptor 1 mediates lipid-induced inflammation in non-alcoholic fatty liver disease. 2020. Available from: https://www.biorxiv.org/content/10.1101/2020.02.01.930115v1.abstract. [Last accessed on 21 Dec 2020].
74. Kragh Petersen S, Bilkei-Gorzo O, Govaere O, Hartlova A. Macrophages and scavenger receptors in obesity-associated non-alcoholic liver fatty disease (NAFLD). Scand J Immunol 2020;92:e12971.
75. Walker S, Wankell M, Ho V, et al. Targeting mTOR and Src restricts hepatocellular carcinoma growth in a novel murine liver cancer model. PLoS One 2019;14:e0212860.
76. Valdecantos MP, Ruiz L, Pardo V, et al. Differential effects of a glucagon-like peptide 1 receptor agonist in non-alcoholic fatty liver disease and in response to hepatectomy. Sci Rep 2018;8:16461.
77. Armstrong MJ, Hull D, Guo K, et al. Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. J Hepatol 2016;64:399-408.
78. Starmann J, Falth M, Spindelbock W, et al. Gene expression profiling unravels cancer-related hepatic molecular signatures in steatohepatitis but not in steatosis. PLoS One 2012;7:e46584.
79. Moylan CA, Pang H, Dellinger A, et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 2014;59:471-82.
80. Arendt BM, Comelli EM, Ma DW, et al. Altered hepatic gene expression in nonalcoholic fatty liver disease is associated with lower hepatic n-3 and n-6 polyunsaturated fatty acids. Hepatology 2015;61:1565-78.
81. Suppli MP, Rigbolt KTG, Veidal SS, et al. Hepatic transcriptome signatures in patients with varying degrees of nonalcoholic fatty liver disease compared with healthy normal-weight individuals. Am J Physiol Gastrointest Liver Physiol 2019;316:G462-G72.
82. Gerhard GS, Legendre C, Still CD, Chu X, Petrick A, DiStefano JK. Transcriptomic profiling of obesity-related nonalcoholic steatohepatitis reveals a core set of fibrosis-specific genes. J Endocr Soc 2018;2:710-26.
83. Hoang SA, Oseini A, Feaver RE, et al. Gene expression predicts histological severity and reveals distinct molecular profiles of nonalcoholic fatty liver disease. Sci Rep 2019;9:12541.
84. Govaere O, Cockell S, Tiniakos D, et al. Transcriptomic profiling across the non-alcoholic fatty liver disease spectrum reveals gene signatures for steatohepatitis and fibrosis. Science Translational Medicine (accepted for publication) 2020; doi: 10.1126/scitranslmed.aba4448.
85. Guldiken N, Kobazi Ensari G, Lahiri P, et al. Keratin 23 is a stress-inducible marker of mouse and human ductular reaction in liver disease. J Hepatol 2016;65:552-9.
86. Wilhelm A, Shepherd EL, Amatucci A, et al. Interaction of TWEAK with Fn14 leads to the progression of fibrotic liver disease by directly modulating hepatic stellate cell proliferation. J Pathol 2016;239:109-21.
87. Taniguchi K, Yamachika S, He F, Karin M. p62/SQSTM1-Dr. Jekyll and Mr. Hyde that prevents oxidative stress but promotes liver cancer. FEBS Lett 2016;590:2375-97.
88. Ichimura Y, Waguri S, Sou YS, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell 2013;51:618-31.
89. Dai X, Yan X, Wintergerst KA, Cai L, Keller BB, Tan Y. Nrf2: redox and metabolic regulator of stem cell state and function. Trends Mol Med 2020;26:185-200.
90. Brunt EM, Tiniakos DG. Histopathology of nonalcoholic fatty liver disease. World J Gastroenterol 2010;16:5286-96.
91. Guy CD, Suzuki A, Zdanowicz M, et al. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology 2012;55:1711-21.
92. Omenetti A, Diehl AM. Hedgehog signaling in cholangiocytes. Curr Opin Gastroenterol 2011;27:268-75.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Younes R, Burgoyne R, Tiniakos D, Govaere O. Clinical implications of hepatic progenitor cell activation in non-alcoholic fatty liver disease. Hepatoma Res 2021;7:15. http://dx.doi.org/10.20517/2394-5079.2020.119
AMA Style
Younes R, Burgoyne R, Tiniakos D, Govaere O. Clinical implications of hepatic progenitor cell activation in non-alcoholic fatty liver disease. Hepatoma Research. 2021; 7: 15. http://dx.doi.org/10.20517/2394-5079.2020.119
Chicago/Turabian Style
Younes, Ramy, Rachel Burgoyne, Dina Tiniakos, Olivier Govaere. 2021. "Clinical implications of hepatic progenitor cell activation in non-alcoholic fatty liver disease" Hepatoma Research. 7: 15. http://dx.doi.org/10.20517/2394-5079.2020.119
ACS Style
Younes, R.; Burgoyne R.; Tiniakos D.; Govaere O. Clinical implications of hepatic progenitor cell activation in non-alcoholic fatty liver disease. Hepatoma. Res. 2021, 7, 15. http://dx.doi.org/10.20517/2394-5079.2020.119
About This Article
Special Issue
Copyright

Data & Comments
Data
0

 Cite This Article 12 clicks
Cite This Article 12 clicks

 Like This Article 26
likes
Like This Article 26
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.