
First cases of MPV17 related mitochondrial DNA depletion syndrome with compound heterozygous mutations in p.R50Q/p.R50W: a case report

Abstract
Mutations in MPV17 lead to severe mitochondrial DNA depletion syndrome (MTDPS). All known p.R50W variants in MPV17 are lethal. The homozygous variant p.R50Q in MPV17 among patients with Navajo neurohepatopathy is known to allow longer survival, although heterozygous variants p.R50Q have not been reported. This is the first clinical report in compound heterozygosity MPV17 mutation (p.R50W/p.R50Q). Three siblings were admitted due to multiple hepatic nodules; none presented neurological abnormalities. However, they suffered from severe hypoglycemia and cyclic vomiting. The diagnosis of MPV17-related MTDPS was confirmed by detection of a compound heterozygous MPV17 mutation (p.R50W/p.R50Q), and striking reduction of hepatic mitochondrial DNA. One patient developed pediatric-onset of hepatocellular carcinoma. Notably, all patients survived for extended periods, including two patients who received liver transplantation, which contrasted the high mortality rate associated with p.R50W mutations, as previously reported. The p.R50Q mutation might be associated with longer survival and improved liver transplantation outcomes.
Keywords
Introduction
The causes of mitochondrial disorders are defects in mitochondrial DNA (mtDNA) or in nuclear genes that affect mtDNA biogenesis and maintenance. Defects in nuclear genes can result in the accumulation of mtDNA deletions, or with mtDNA depletion syndrome (MTDPS)[1-3]. The latter is an autosomal recessive disease associated with decreased mtDNA copy number in clinically affected tissues[4-6]. The disease has three known phenotypes: hepatocerebral, myopathic, and encephalomyopathy. Hepatocerebral MTDPS is linked to pathogenic variants in DNA polymerase gamma[7], Twinkle (PEO1)[8], deoxyguanosine kinase[9], and mtDNA maintenance protein MPV17[4].
MPV17-related MTDPS is a very rare disease. To date, MPV17-related hepatocerebral MTDPS has been reported in 96 patients[4,5,10-30]. Disease prognosis is severe, given that ~80% of patients die from liver failure during early childhood[30]. Hepatic cirrhosis has been diagnosed in 20 patients, while three patients had hepatocellular carcinoma (HCC)[10,15,25]. Neurological manifestations were also reported in 91% of patients with developmental delays, and 74% of patients with generalized hypotonia. These patients also experienced MR imaging (MRI) abnormalities; metabolic manifestations, including hypoglycemia and lactic acidosis; failure to thrive; feeding difficulties; and retinaltubulopathy.
Currently, 48 pathogenic variants of MPV17 are known, occurring exclusively in a few families. Five cases with p.R50W mutations (three homozygous and two heterozygous) died of liver failure during early childhood[4,12,25,30]. Previous case studies identified homozygous variant p.R50Q in MPV17 among patients with Navajo neurohepatopathy (NNH)[10,15,29]. One specific homozygous mutation, p.R50Q, has been reported in several Navajo individuals from the southwestern United States and is the cause of NNH. This condition clinically manifests as severe sensory and motor neuropathy, corneal anesthesia, ulcers, cerebral leukoencephalopathy, failure to thrive, and metabolic acidosis. To date, compound heterozygous variant p.R50Q with other mutations has not been reported in MPV17-related MTDPS.
In this case report, we describe for the first time three patients with MTDPS who possessed the compound heterozygous MPV17 variant p.R50Q/p.R50W. Unlike deceased outcomes of p.R50W variants as previously reported, our three cases with p.R50Q/R50W mutations survived without signs of typical neurological manifestations.
Case report
Patients
The proband was a 6-year-old male (Case 1) and the first child of healthy, nonconsanguineous parents with no hereditary history of the disease (for family pedigree, see Figure 1A). The child was delivered via caesarian section after a dinochorionic and diamniotic pregnancy. Case 2 was the first girl of a twin of Case 1. A second girl (Case 3) was born three years after Cases 1 and 2, delivered without any complication. Cases 1-3 showed similar clinical manifestation: all patients suffered from severe hypoglycemia and cyclic vomiting. During infancy, Cases 1 and 3 presented with liver failure; although there was a mild spontaneous remission, acute liver failure was repeated through episodes of febrile infection. Case 2 presented with liver dysfunction only.
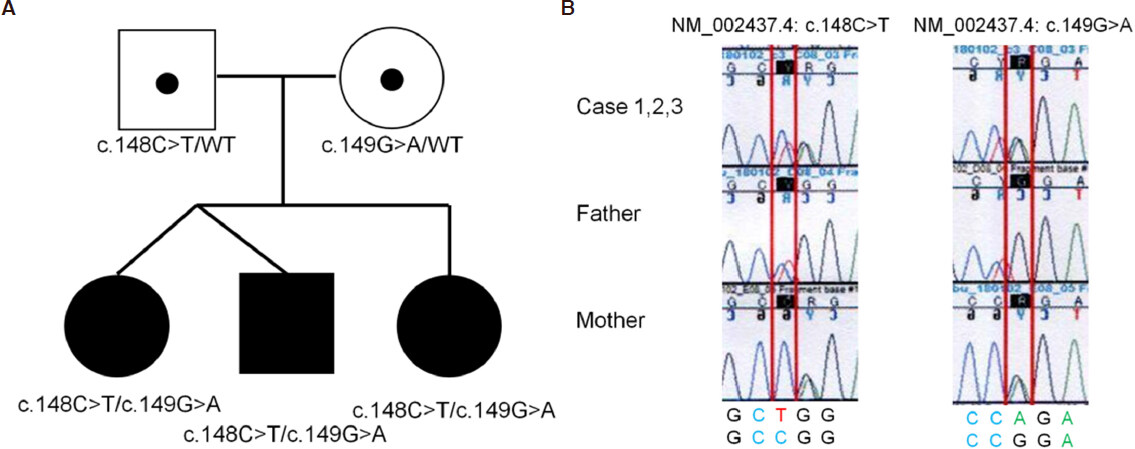
Figure 1. Family pedigree (A). Whole-exome and Sanger sequencing revealed compound heterozygous missense mutations with NM_002437.4:c.148C>T (p.R50W) and c.149G>A (R50Q) in MPV17, inherited from each of their parents (B)
In Case 1, the patient developed recurrent vomiting with hypoglycemia at the age of six, and abdominal ultrasound found multiple liver masses. Each patient was admitted to our hospital due to multiple hepatic nodules, detected by abdominal ultrasound at age six in Case 2, and at age three in Case 3 [Figure 2A].
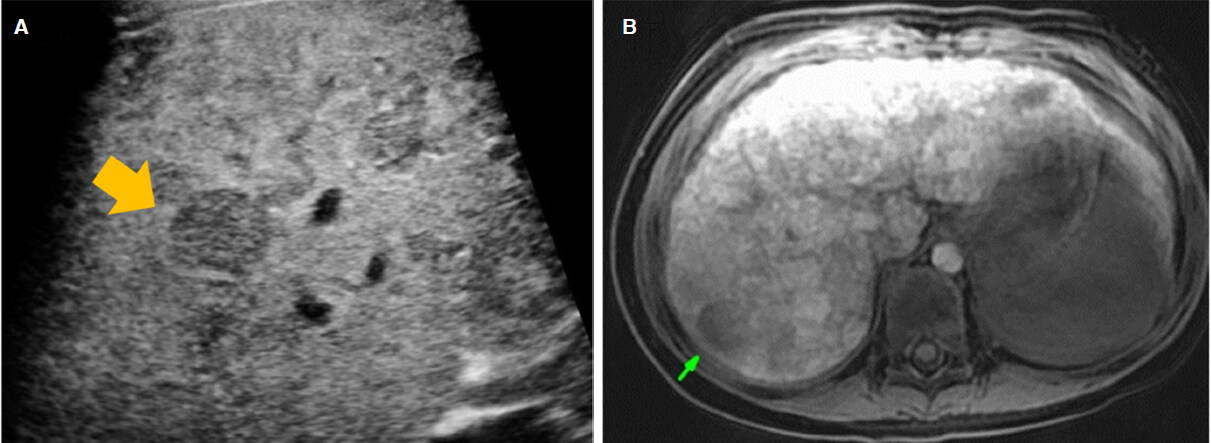
Figure 2. Abdominal ultrasound showed the mass in the liver (A). EOB-enhanced magnetic resonance images indicated the washed-out portal-venous phase and liver-cell-enhance phase in S7 hepatic mass (B)
Table 1 shows the clinical findings of three patients at first admission to our hospital. Case 2 and 3 showed no manifestations of neurological abnormality, but Case 1 exhibited mild intellectual disability (IQ 68). Serological tests for viral hepatitis A, B, and C, cytomegalovirus, and Epstein-Barr virus excluded the presence of these infections. Wilson’s disease was excluded by measuring ceruloplasmin, serum copper, and urinary copper.
Clinical findings of three patients with MPV17-related mitochondria hepatopathy at the first visit to our hospital
| Case1 | Case2 | Case3 | |
|---|---|---|---|
| Sex | Male | Female | Female |
| Height (cm) (SD) | 109.9 (-1.3 SD) | 110 (-0.9 SD) | 88.3 (-1.8 SD) |
| Weight (kg) (SD) | 17.3 (-1.1 SD) | 19.4 (-0.3 SD) | 13.6 (-0.1 SD) |
| Neurology | Normal | Normal | Normal |
| IQ | 68 | 92 | 113 |
| Metabolism | Hypoglycemia | Hypoglycemia | Hypoglycemia |
| AST (U/l) | 53 | 47 | 82 |
| ALT (U/l) | 30 | 28 | 36 |
| GGT (U/l) | 201 | 99 | 143 |
| Alb (mg/dL) | 2.7 | 3.1 | 3.1 |
| Plt (104/μL) | 21.3 | 12.3 | 28.1 |
| PT (%) | 67.4 | 61.5 | 55.3 |
| Lactic acid (mg/dL) | 22.0 | 19.2 | 18.6 |
| Pyruvic acid (mg/dL) | 1.2 | 1.0 | 1.0 |
| AFP (ng/mL) | 413 | 1332 | 3078 |
| Abdominal MRI | Multifocal masses | Multifocal masses | Multifocal masses |
| Liver histology | Cirrhosis/steatosis | Cirrhosis/steatosis | Not done |
| Head MRI | Normal | Normal | Normal |
| Child-Pugh score | 6 | 6 | 6 |
All three patients had multiple masses with hyper-echogenic occupied lesions and low-echogenic occupied lesions measuring between 0.5 and 1.0 cm, which were negatively detected by enhanced computed tomography. Transverse T2-weighted magnetic resonance (MR) image shows numerous well-defined hypointense masses in the liver, while transverse EOB-enhanced MRI demonstrated negative arterial enhancement, portal-venous phase, and liver-cell-enhance phase studies showed high intensity with masses in all three patients.
Liver biopsies revealed advanced fibrosis with regenerative nodules and mild steatosis in Cases 1 and 2 [Figure 3A]. In Case 3, liver biopsy was avoided due to hemorrhagic tendency. Electron microscopy revealed that these two children had giant mitochondria with increased inclusion-body count in hepatocytes [Figure 3B].
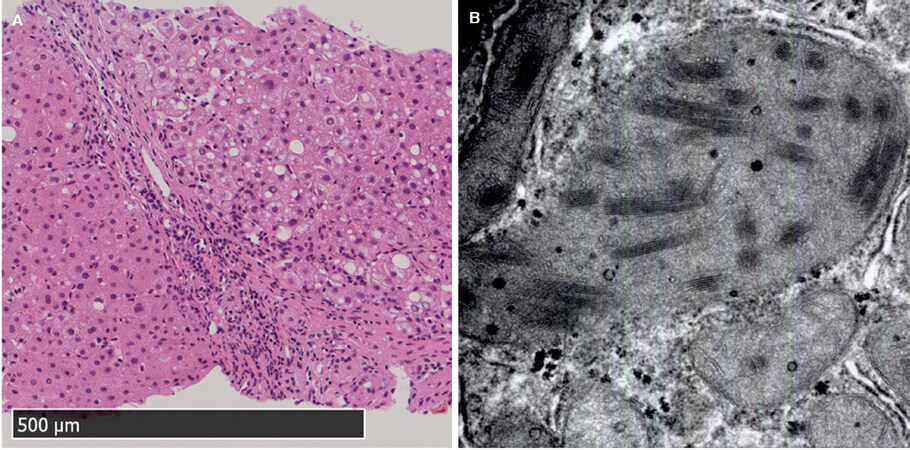
Figure 3. Liver biopsies at age six showed advanced fibrosis with regenerative nodules and mild steatosis: (A) hematoxylin-eosin staining (magnification, low power field); and (B) electron microscopy (magnification, ×25,000) revealed giant mitochondria with increased inclusion bodies in hepatocytes
After obtaining approval from the institutional review board of Saiseikai Yokohama-shi Tobu Hospital and informed consent from parents, biochemical examination and molecular studies were performed.
A biochemical analysis was performed at the Department of Metabolism, Chiba Prefectural Children’s Hospital. Case 1 and 2 livers were analyzed for tissue-specific enzyme activity and mtDNA levels. Hepatic respiratory chain complex I, II, III, and IV (CI-CIV) activities were evaluated as described previously[31]. The results of this analysis showed that Case 1 had decreased CI and CIII activities, and increased citrate synthase (CS) activity (CI, 3.5%; CIII, 28.4% of CS activity; CS, 213.6% of total protein). Similarly, Case 2 exhibited decreased CI, CIII, and CIV activities, and increased CS activity (CI, 9.8%; CIII, 17.0%; CIV, 20.0% of CS activity; CS, 263.7% of total protein). Next, a quantitative PCR was performed to evaluate the mtDNA copy number using previously reported methods[32], the analysis of which revealed a striking reduction of hepatic mtDNA in both patients (Case 1, 0.7%; Case 2, 0.6%; normal range 40%-150%).
DNA was extracted from peripheral blood samples of the three patients and their parents. A whole-exome analysis using Hiseq 2500 (Illumina, Sana Clara, USA) identified compound heterozygous missense mutations with NM_002437.4:c.148C>T (p.R50W) and c.149G>A (p.R50Q) in MPV17, respectively, inherited from each of their parents [Figure 1B]. Sanger sequencing confirmed these results.
These results confirmed a diagnosis of MPV17-related MTDPS.
Clinical treatment
Patients were given ubiquinone carnitine as medication for mitochondrial rescue supplementation. A lipid-rich diet efficiently controlled hypoglycemia and normalized liver function for a previous case study[14]. However, a lipid-rich, carbohydrate-poor diet (lipid 45%, carbohydrate 45%, and protein 10%) caused severe hypoglycemia with vomiting symptoms in Cases 1-3. Episodic hypoglycemia and vomiting were dramatically reduced with the avoidance of fasting and uncooked cornstarch, along with the provision of a carbohydrate-rich diet with medium-chain triglyceride powder (lipid 30%, carbohydrate 60%, and protein 10%) six times per day.
Six months later, Case 2 was readmitted to our hospital because of an increasing mass measuring 2.9 cm located in segment 7, as revealed by abdominal ultrasound imaging. It showed no evidence of vascular invasion or metastasis. EOB-enhanced MR images revealed early arterial enhancement, as well as washed-out portal-venous and liver-cell-enhance phases [Figure 2B]. We diagnosed HCC. At age 7, Case 2 underwent living-donor-liver transplantation from her father. The resected liver was completely cirrhotic, and histology indicated well-differentiated HCC [Figure 4]. After liver transplantation, Case 2 did not experience post-transplant complications. Case 1 also developed end-stage liver disease and obtained a liver transplant from his grandfather five months after Case 2’s operation. Case 1’s resected liver was also completely cirrhotic but without neoplasm stigma. As with Case 2, Case 1 did not experience post-surgery complications. We also observed Case 3 to detect any increases in hepatic masses. At the time of writing, all three patients exhibited normal cognition and neurological function.
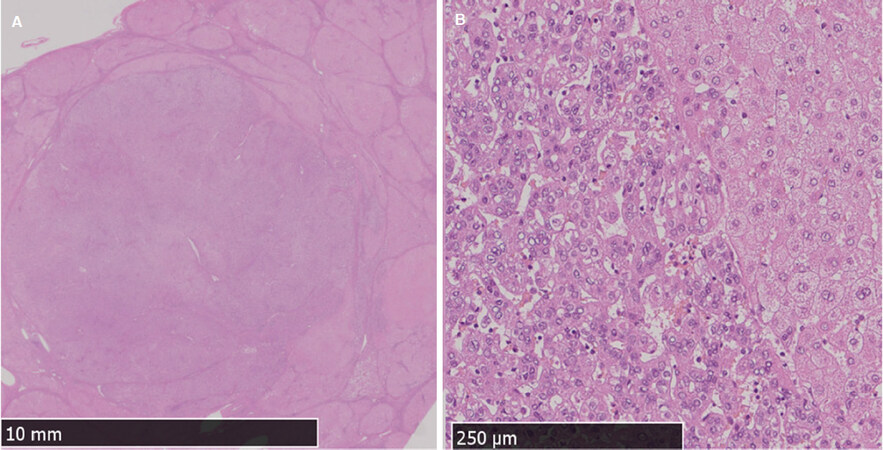
Figure 4. Explanted Case 2 liver with multiple large nodules. A histological analysis (hematoxylin-eosin staining) of the Case 2 S7 mass revealed: (A) a high nucleo-to-cytoplasm ratio (magnification, low power field); and (B) sharply delineated small aggregates of highly pleomorphic small hepatocytes, which are typical of well-differentiated hepatocellular carcinoma (magnification, high power field)
Discussion
This is the first clinical report in compound heterozygous mutation (p.R50Q/p.R50W) of MPV17, and three cases were diagnosed with hepatocerebral MTDPS. In contrast with deceased outcomes of p.R50W variants previously reported, our three cases with p.R50Q/R50W mutations survived for a more extended period without neurological manifestations. One case developed HCC. Two of our cases underwent liver transplantation, and both showed positive post-transplant outcomes at the time of writing.
Table 2 shows previous patients who had p.R50Q homozygous mutations, and p.R50W homozygous or heterozygous mutations, and compared with our cases. All previous cases with p.R50W mutations (three homozygous and two heterozygous) died prematurely during early childhood (age of death: infancy; 1, 1, and 19 months old; and 10 years old)[4,12,25,30]. Although all our patients survived, one p.R50W homozygous patient died at 10 years old after liver transplantation and showed longer survival. However, while the patient progressed to end-stage liver disease, she was affected by severe ascites, malnutrition, and jaundice during pre-liver transplantation period[25]. In comparison to this case, our patients lacked severe hepatic manifestations. Regarding p.R50Q mutation, currently there are 11 known cases of homozygous p.R50Q mutation[4,10,11,25,29]; eight of them presented with NNH. It is known that patients possessing the p.R50Q homozygous mutation have higher survival rate (45%; 5 out of 11 patients), although some cases resulted in death by infantile-onset liver failure (age of death: 7 months old and 2 and 5 years old). It was surprising that none of the three cases with p.R50Q/R50W mutations died during early childhood (age of follow-up: 8, 8, and 5 years old, respectively), in contrast to 100% mortality observed in cases with p.R50W variants (age of death: infancy; 1, 1, and 19 months old; and 10 years old, respectively) and 55% in cases with p.R50Q homozygous variants (age of death: 6 months old and 2, 5, 15, 16, and 20 years old, respectively).
Clinical phenotype of patients with MPV-17-related mitochondria hepatopathy with p.R50Q, or p.R50W mutations
| p.R50Q/p.R50W n = 3 | p.R50W reported n = 5 | p.R50Q reported n = 11 | |||||
|---|---|---|---|---|---|---|---|
| Sex | Female | 2 | (67) | 4 | (80) | 4 | (36) |
| Outcome | Alive | 3 | (100) | 0 | (0) | 5 | (46) |
| Liver transplantation | 2 | (66) | 1 | (20) | 5 | (46) | |
| Hepatic symptoms | 3 | (100) | 5 | (100) | 11 | (100) | |
| HCC | 1 | (33) | 1 | (20) | 1 | (9) | |
| Hepatomegaly | 3 | (100) | 3 | (60) | 3 | (27) | |
| Cirrhosis | 2 | (67) | 1 | (20) | 6 | (55) | |
| Liver dysfunction | 3 | (100) | 5 | (100) | 11 | (100) | |
| Liver failure | 2 | (67) | 5 | (100) | 9 | (82) | |
| Cholestasis | 0 | (0) | 3 | (60) | 8 | (73) | |
| Steatosis | 0 | (0) | 4 | (80) | 7 | (64) | |
| Neurological symptoms | 1 | (33) | 4 | (80) | 10 | (91) | |
| Ataxia | 0 | (0) | 0 | (0) | 2 | (18) | |
| Corneal ulcers | 0 | (0) | 0 | (0) | 3 | (27) | |
| Developmental delay | 1 | (33) | 2 | (40) | 9 | (82) | |
| Dystonia | 0 | (0) | 2 | (40) | 0 | (0) | |
| Hypotonia | 0 | (0) | 2 | (40) | 3 | (27) | |
| Peripheral neuropathy | 0 | (0) | 0 | (0) | 8 | (73) | |
| Seizures | 0 | (0) | 1 | (20) | 1 | (9) | |
| MRI abnormality | |||||||
| Basal ganglia | 0 | (0) | 1 | (20) | 0 | (0) | |
| White matter | 0 | (0) | 2 | (40) | 5 | (46) | |
| Metabolic symptoms | 3 | (100) | 3 | (60) | 10 | (91) | |
| Lactic acidemia | 0 | (0) | 3 | (60) | 8 | (73) | |
| Hypoglycemia | 3 | (100) | 2 | (40) | 7 | (64) | |
| GI symptoms | 3 | (100) | 4 | (80) | 8 | (73) | |
| Feeding difficulties | 0 | (0) | 2 | (40) | 0 | (0) | |
| Failure to thrive | 3 | (100) | 4 | (80) | 8 | (73) | |
Additionally, our cases with a compound heterozygous p.R50Q/p.R50W mutation were free from neurological manifestations compared with high occurrence of neurological manifestations in p.R50W and p.R50Q mutations previously reported. Regarding neurological manifestations, in previous cases, 90% of p.R50Q homozygous variants and 80% of p.R50W homozygous variants had neurological symptoms such as ataxia, corneal ulcers, developmental delay, dystonia, hypotonia, peripheral neuropathy, and seizures. MRI abnormality was also observed in 40% patients who had p.R50Q homo and p.R50W cases, respectively. Compared with earlier cases, p.R50Q/p.R50W lacked neurological symptoms except mild intellectual disability observed in Case 1. Taken together, the fact that none of our cases showed mortality even though they had heterozygous p.R50W mutation and that they had no visible neurological manifestations suggests the p.R50Q mutation might be associated with longer survival compared with other mutations linked to severe outcomes of MPV17-related MTDPS, such as p.R50W.
Interestingly, one of our cases developed pediatric-onset HCC, increasing the number to four known patients with MPV17-related MTDPS who had this cancer [Table 3]. Out of four patients with HCC in MPV17-related MTDPS, no compound heterozygous mutation was reported. In contrast, two patients had homozygous mutations (another patient was not identified in one MPV17 mutation). Regarding treatment, all four patients with HCC had liver transplantation (LT), although post-LT course differed widely according to the types of genotypes. p.R50W homozygous patients died at 10 years old, one year after she had liver transplantation, while three patients showed extended survival with good-post-LT course.
Clinical manifestations of patients with MPV17-related hepatocerebral mitochondrial DNA depletion syndrome who developed hepatocellular carcinoma
| Case 2 | Patient 1 | Patient 2 | Patient 3 | |
|---|---|---|---|---|
| Sex | Female | Male | Male | Female |
| Ethnicity | Japan | Navajo | Caucasian | India |
| MPV17 | ||||
| Allele 1 | p.R50Q | p.R50Q | c.22insC | p.R50W |
| Allele 2 | p.R50W | p.R50Q | NA | p.R50W |
| Onset age | 4 years | Infancy | Infancy | 5 years |
| LT (age) | Done (7 years) | Done (11 years) | Done (NA) | Done (9 years) |
| Outcome (age) | Alive (8 years) | Alive (21 years) | Alive (9 years) | Died (10 years) |
| Hepatic manifestation | Cirrhosis, HCC | LF, Cirrhosis, Steatosis, HCC | LF, Cirrhosis, HCC | LF, Cirrhosis, Steatosis, HCC |
| Other manifestation | Hypoglycemia, FTT | DD, Peripheral neuropathy, MRI abnormalities, Hypoglycemia, FTT | DD, Hypotonia, Seizures, FTT | Seizures, Dystonia, MRI abnormalities, FTT |
| Ref. | This report | [10] | [15] | [25] |
Liver transplantation is one of the best treatments for HCC-induced cirrhotic liver. Although a recent study showed that liver transplantation for pediatric HCC patients with inherited metabolic disease has better chances of survival compared with pediatric HCC patients with non-inherited metabolic disease[33], its efficacy in MPV17-MTDPS remains controversial. The reason behind this is that the organ’s systemic complexity results in high morbidity during post-transplantation. Of the 17 known MPV17-related MTDPS patients who received liver transplantation, seven (41%) died during the post-transplantation period[30]. Of the five known MPV17-related MTDPS patients with the p.R50Q homozygous mutation who received liver transplantation, three (60%) survived[4,10,11,29]. In contrast, four patients with other mutations out of 12 (33%) survived liver transplantation[12-15,20,25,30]. Here, Case 1 received liver transplantation to treat end-stage liver disease, and Case 2 to treat hepatic cell carcinoma. At the time of writing this report, their post-transplant outcome was good. None of the patients had serious systemic organ complications. LT indication for MPV17-related MTDPS is still controversial, but LT may be an optional treatment for HCC with MPV17 related MTDPS with p.R50W mutation. In such cases, careful surveillance for systemic organ involvement should be applied, because long-term outcomes of post-LT course are still unknown.
As with previous studies of MTDPS, this case report describes a small sample size and short follow-up period. Therefore, we need to perform further studies on large patient cohorts, to determine the effectiveness and outcomes of liver transplantation in MPV17-related MTDPS. Long-term follow up should be performed, including regular neurological assessment.
This case study reported the first compound heterozygous p.R50Q/p.R50W mutation of MPV17. All the three siblings survived without neurological manifestations, in contrast with total mortality accompanied by systemic organ involvements in previous MPV17 mutation p.R50W. Two of our cases underwent liver transplantation, and both showed positive post-transplant outcomes at the time of writing. The p.R50Q mutation might be associated with more prolonged survival including liver transplantation outcomes, as compared with other previously described mutations linked to severe outcomes of MPV17-related MTDPS. Screens of the MPV17 gene should be performed in all cases of unknown, severe hepatic dysfunction in children.
Declarations
AcknowledgmentsWe thank Dr. Notohara Kenji at Kurashiki Central Hospital for advice on evaluating liver histopathology from adult hepatic neoplasms.
Authors’ contributionsDrafted the manuscript: Umetsu S
Made a special support from the metabolic evaluation: Murayama K, Shimura M
Made a special support from genetic evaluation: Uehara T, Kosaki K
Made a special support from Liver transplantation: Uchida H, Kasahara M
Assisted with liver histopathology: Irie R, Yoshioka T
Supervised diagnosis and treatment: Kobayashi S, Sogo T, Komatsu K, Inui A, Fujisawa T
Read and approved the final manuscript: Umetsu S, Inui A, Kobayashi S, Shimura M, Uehara T, Uchida H, Irie R, Sogo T, Komatsu H, Yoshioka T, Murayama K, Kosaki K, Kasahara M, Fujisawa T
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis study was supported by the following grants. Kei Murayama was supported by the Practical Research Project (19ek0109273, 18ek0109177) for Rare/Intractable Diseases from the Japan Agency for Medical Research and Development.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateThe institutional review board of Saiseikai Yokohama-shi Tobu Hospital approved, and informed consent was obtained from parent.
Consent to participateWritten informed consent was obtained from the guardian of the patients for publication of this Case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Copyright© The Author(s) 2020.
REFERENCES
1. Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain 2003;126:1905-12.
2. Spinazzola A, Invernizzi F, Carrara F, Lamantea E, Donati A, et al. Clinical and molecular features of mitochondrial DNA depletion syndromes. J Inherit Metab Dis 2009;32:143-58.
4. Spinazzola A, Viscomi C, Fernandez-Vizarra E, Carrara F, D’Adamo P, et al. MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat Genet 2006;38:570-5.
5. Spinazzola A, Santer R, Akman OH, Tsiakas K, Schaefer H, et al. Hepatocerebral form of mitochondrial DNA depletion syndrome: novel MPV17 mutations. Arch Neurol 2008;65:1108-13.
6. Munnich A, Rustin P. Clinical spectrum and diagnosis of mitochondrial disorders. Am J Med Genet 2001;106:4-17.
7. Ferrari G, Lamantea E, Donati A, Filosto M, Briem E, et al. Infantile hepatocerebral syndromes associated with mutations in the mitochondrial DNA polymerase-gammaA. Brain 2005;128:723-31.
8. Hakonen AH, Isohanni P, Paetau A, Herva R, Suomalainen A, et al. Recessive Twinkle mutations in early onset encephalopathy with mtDNA depletion. Brain 2007;130:3032-40.
9. Mandel H, Szargel R, Labay V, Elpeleg O, Saada A, et al. The deoxyguanosine kinase gene is mutated in individuals with depleted hepatocerebral mitochondrial DNA. Nat Genet 2001;29:337-41.
10. Karadimas CL, Vu TH, Holve SA, Chronopoulou P, Quinzii C, et al. Navajo neurohepatopathy is caused by a mutation in the MPV17 gene. Am J Hum Genet 2006;79:544-8.
11. Parini R, Furlan F, Notarangelo L, Spinazzola A, Uziel G, et al. Glucose metabolism and diet-based prevention of liver dysfunction in MPV17 mutant patients. J Hepatol 2009;50:215-21.
12. Wong LJ, Brunetti-Pierri N, Zhang Q, Yazigi N, Bove KE, et al. Mutations in the MPV17 gene are responsible for rapidly progressive liver failure in infancy. Hepatology 2007;46:1218-27.
13. Navarro-Sastre A, Martin-Hernandez E, Campos Y, Quintana E, Medina E, et al. Lethal hepatopathy and leukodystrophy caused by a novel mutation in MPV17 gene: description of an alternative MPV17 spliced form. Mol Genet Metab 2008;94:234-9.
14. Kaji S, Murayama K, Nagata I, Nagasaka H, Takayanagi M, et al. Fluctuating liver functions in siblings with MPV17 mutations and possible improvement associated with dietary and pharmaceutical treatments targeting respiratory chain complex II. Mol Genet Metab 2009;97:292-6.
15. El-Hattab AW, Li FY, Schmitt E, Zhang S, Craigen WJ, et al. MPV17-associated hepatocerebral mitochondrial DNA depletion syndrome: new patients and novel mutations. Mol Genet Metab 2010;99:300-8.
16. Merkle AN, Nascene DR, McKinney AM. MR imaging findings in the reticular formation in siblings with MPV17-related mitochondrial depletion syndrome. AJNR Am J Neuroradiol 2012;33:E34-5.
17. Al-Jasmi F, Penefsky HS, Souid AK. The phosphorescence oxygen analyzer as a screening tool for disorders with impaired lymphocyte bioenergetics. Mol Genet Metab 2011;104:529-36.
18. AlSaman A, Tomoum H, Invernizzi F, Zeviani M. Hepatocerebral form of mitochondrial DNA depletion syndrome due to mutation in MPV17 gene. Saudi J Gastroenterol 2012;18:285-9.
19. Nogueira C, de Souza CF, Husny A, Derks TG, Santorelli FM, et al. MPV17: fatal hepatocerebral presentation in a Brazilian infant. Mol Genet Metab 2012;107:764.
20. Uusimaa J, Evans J, Smith C, Butterworth A, Craig K, et al. Clinical, biochemical, cellular and molecular characterization of mitochondrial DNA depletion syndrome due to novel mutations in the MPV17 gene. Eur J Hum Genet 2014;22:184-91.
21. Piekutowska-Abramczuk D, Pronicki M, Strawa K, Karkucinska-Wieckowska A, Szymanska-Debinska T, et al. Novel c.191C>G (p.Pro64Arg) MPV17 mutation identified in two pairs of unrelated Polish siblings with mitochondrial hepatoencephalopathy. Clin Genet 2014;85:573-7.
22. Mendelsohn BA, Mehta N, Hameed B, Pekmezci M, Packman S, et al. Adult-onset fatal neurohepatopathy in a woman caused by MPV17 mutation. JIMD Rep 2014;13:37-41.
23. Al-Hussaini A, Faqeih E, El-Hattab AW, Alfadhel M, Asery A, et al. Clinical and molecular characteristics of mitochondrial DNA depletion syndrome associated with neonatal cholestasis and liver failure. J Pediatr 2014;164:553-9.e1-2.
24. Sarkhy AA, Al-Sunaid A, Abdullah A, AlFadhel M, Eiyad W. A novel MPV17 gene mutation in a Saudi infant causing fatal progressive liver failure. Ann Saudi Med 2014;34:175-8.
25. Vilarinho S, Choi M, Jain D, Malhotra A, Kulkarni S, et al. Individual exome analysis in diagnosis and management of paediatric liver failure of indeterminate aetiology. J Hepatol 2014;61:1056-63.
26. Bijarnia-Mahay S, Mohan N, Goyal D, Verma IC. Mitochondrial DNA depletion syndrome causing liver failure. Indian Pediatr 2014;51:666-8.
27. McKiernan P, Ball S, Santra S, Foster K, Fratter C, et al. Incidence of primary mitochondrial disease in children younger than 2 years presenting with acute liver failure. J Pediatr Gastroenterol Nutr 2016;63:592-7.
28. Kim J, Kang E, Kim Y, Kim JM, Lee BH, et al. MPV17 mutations in patients with hepatocerebral mitochondrial DNA depletion syndrome. Mol Genet Metab Rep 2016;8:74-6.
29. Bitting CP, Hanson JA. Navajo neurohepatopathy: a case report and literature review emphasizing clinicopathologic diagnosis. Acta Gastroenterol Belg 2016;79:463-9.
30. El-Hattab AW, Wang J, Dai H, Almannai M, Staufner C, et al. MPV17-related mitochondrial DNA maintenance defect: new cases and review of clinical, biochemical, and molecular aspects. Hum Mutat 2018;39:461-70.
31. Kirby DM, Crawford M, Cleary MA, Dahl HH, Dennett X, et al. Respiratory chain complex I deficiency: an underdiagnosed energy generation disorder. Neurology 1999;52:1255-64.
32. He L, Chinnery PF, Durham SE, Blakely EL, Wardell TM, et al. Detection and quantification of mitochondrial DNA deletions in individual cells by real-time PCR. Nucleic Acids Res 2002;30:e68.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Umetsu S, Inui A, Kobayashi S, Shimura M, Uehara T, Uchida H, Irie R, Sogo T, Komatsu H, Yoshioka T, Murayama K, Kosaki K, Kasahara M, Fujisawa T. First cases of MPV17 related mitochondrial DNA depletion syndrome with compound heterozygous mutations in p.R50Q/p.R50W: a case report. Hepatoma Res 2020;6:1. http://dx.doi.org/10.20517/2394-5079.2019.030
AMA Style
Umetsu S, Inui A, Kobayashi S, Shimura M, Uehara T, Uchida H, Irie R, Sogo T, Komatsu H, Yoshioka T, Murayama K, Kosaki K, Kasahara M, Fujisawa T. First cases of MPV17 related mitochondrial DNA depletion syndrome with compound heterozygous mutations in p.R50Q/p.R50W: a case report. Hepatoma Research. 2020; 6: 1. http://dx.doi.org/10.20517/2394-5079.2019.030
Chicago/Turabian Style
Umetsu, Shuichiro, Ayano Inui, Sohya Kobayashi, Masaru Shimura, Tomoko Uehara, Hajime Uchida, Rie Irie, Tsuyoshi Sogo, Haruki Komatsu, Takako Yoshioka, Kei Murayama, Kenjiro Kosaki, Mureo Kasahara, Tomoo Fujisawa. 2020. "First cases of MPV17 related mitochondrial DNA depletion syndrome with compound heterozygous mutations in p.R50Q/p.R50W: a case report" Hepatoma Research. 6: 1. http://dx.doi.org/10.20517/2394-5079.2019.030
ACS Style
Umetsu, S.; Inui A.; Kobayashi S.; Shimura M.; Uehara T.; Uchida H.; Irie R.; Sogo T.; Komatsu H.; Yoshioka T.; Murayama K.; Kosaki K.; Kasahara M.; Fujisawa T. First cases of MPV17 related mitochondrial DNA depletion syndrome with compound heterozygous mutations in p.R50Q/p.R50W: a case report. Hepatoma. Res. 2020, 6, 1. http://dx.doi.org/10.20517/2394-5079.2019.030
About This Article
Copyright

Data & Comments
Data


 Cite This Article 5 clicks
Cite This Article 5 clicks

 Like This Article 30
likes
Like This Article 30
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.