
A dimethylbromobenzene-cysteine stapled peptide dual inhibitor of the p53-MDM2/MDMX interactions

Abstract
Aim: Hepatocellular carcinoma (HCC) has emerged as one of the most commonly diagnosed forms of human cancer; yet, the current treatment for HCC is less effective than those used against other cancers. Transcription factor p53 induces cell cycle arrest and apoptosis in response to DNA damage and cellular stress, thereby playing a critical role in protecting cells from malignant transformation. The oncoproteins MDM2 and MDMX negatively regulate the activity and stability of the tumor suppressor protein p53, conferring tumor development and survival.
Methods: In this work, we firstly explored the feasibility of antagonists targeting the p53-binding domains of MDM2 and MDMX as a potential method for HCC therapy via the survival rate analysis in The Cancer Genome Atlas. Moreover, we developed a novel stapling strategy for peptide drug design using the reaction between mercapto group and bromine to crosslink the side chains of the two Cys at (i, i+4) positions, and apply it to a series of peptides derived from a dodecameric peptide antagonist of both MDM2 and MDMX, termed p53-MDM2/MDMX inhibitor (PMI).
Results: Notably, all of these stapled peptides can compete with p53 for MDM2 or MDMX binding as the similar affinity as PMI. More importantly, this stapling functionally rescued PMI that, on its own, failed to activate p53 because of its poor membrane permeability and susceptibility to proteolytic degradation.
Conclusion: Taken together, this work not only illustrates that the restoration of p53 is a potentially feasible program for HCC therapy, but promises an important new tool for peptide drug discovery and development for a variety of human diseases.
Keywords
Introduction
p53 acts as a tumor suppressor by initiating cell-cycle arrest, apoptosis, and senescence in response to cellular stress to maintain the integrity of the genome[1]. In a substantial proportion of cancers, p53 is wild type but the protein is inactivated; this offers an attractive strategy for cancer therapy based on p53 reactivation[2,3]. Recent studies in cancer patients have provided proof-of-concept for this approach[2]. Such activators are the product of basic research conducted over the past 20 years that has led to the appreciation of MDM2 and MDMX as the two major negative regulators of p53, which now seem to be “druggable” using a variety of strategies[4].
Of all human cancers, hepatocellular carcinoma (HCC) is the fifth most frequently diagnosed cancer worldwide and is the third leading cause of cancer death globally[5]. Yet, the current most common treatment for HCC is surgical resection, which is less effective than those used against other cancers[1]. Because the loss of p53 function plays a critical role in multistage hepatocarcinogenetic, the p53 gene has been regarded as a good candidate for modulating HCC risk[6]. Furthermore, the top two risk factors of HCC are metabolic disease (such as fatty liver) and viral infection (such as hepatitis B and C), both of which cause cirrhosis before HCC[7,8]. As one of the hallmarks of cancer, the changes observed in cancer cell metabolism and bioenergetics are also regulated by p53[9,10]. Therefore, the connection between p53 stress response and the disordered metabolic process leading to HCC is a potential avenue for HCC therapies.
Several classes of molecules that inhibit this interaction between p53/MDM2 (MDMX) have been developed (e.g., Nutlin and MI-219)[11,12]. They mimic the conserved residues from a region of the p53 N-terminal that are functional for the interaction with the N-terminal p53 binding domain of MDMX or MDMX[4]. This region forms an α-helix upon binding, enabling the three conserved hydrophobic residues of the MDM2 binding motif (F19, W23, and L26) to optimally embed into the hydrophobic binding groove located on MDM2 and its homologous MDMX protein[13,14]. Except for small molecules, it has been proved that the p53 peptide is appropriate as a biological tool and prototype therapeutic by enforcing its R-helical structure while preserving the key interacting residues that enable specific MDM2 and MDMX engagement[15].
As the wild-type p53 peptide (ETFSDLWKLLPE) has a low affinity for MDM2/MDMX and comes from a region of p53 that interacts with many other proteins[15], we explored the effects of stapling a peptide derived from phage selection experiments[14]. Phage display and rational design methods have been used to isolate linear peptides that bind MDM2 with high affinity[13]. The most avid of these published peptides, described by Pazgier et al.[14], named p53-MDM2/MDMX inhibitor (PMI), was used as the template for this study. Besides, when PMI helices are taken out of protein context and placed into aqueous buffer in isolation, it usually adopts random coil conformations, leading to a drastic reduction in biological activity and thus diminishing therapeutic potential[14,16]. To overcome it, numerous strategies have been developed to stabilize or mimic peptide helices[17-19]. Among these, the most straightforward, yet effective, strategy is sidechain cross-linking (‘‘peptide stapling’’)[16]. Since peptide stapling necessitates macrocyclization, an entropically unfavorable process, very few reactions are known to date that give rise to good yields along with the reinforced structures. These include disulfide bond formation[20], lactam formation[21], ruthenium-catalyzed ring closing metathesis[15], and copper-catalyzed azide-acetylene cycloaddition[22]. While these reactions have enabled the synthesis of stapled peptide helices, the development of additional stapling reactions with high yields and predictable structural effect is still highly desirable. Herein, we report the first synthesis of stapled PMI helices using 1,2(1,3 or 1,4)-dimethylbromobenzene reacting with the sulfydryl of cycstine and the subsequent structural, protein chemistry and in vitro anticancer activity studies of the stapled PMI.
Methods
Patient data
The data of p53, MDM2 and MDMX expression at mRNA level in HCC patients were collected and obtained from The Cancer Genome Atlas (TCGA) project[23,24] via the data portal on 03/24/2018.
General remarks
All synthetic peptide sources were obtained from CS Bio (Shanghai) Ltd. All other chemicals used in this study were purchased from Sigma-Aldrich unless otherwise specified. Acetonitrile and water (HPLC grade) were purchased from Fisher Scientific Ltd. All products were used as received without further purification.
Synthesis of peptides
All peptides were synthesized on appropriate resins on an CS bio 336X automated peptide synthesizer using the optimized HBTU activation/DIEA in situ neutralization protocol developed by an HBTU/HOBt protocol for Fmoc-chemistry SPPS.2 After cleavage and deprotection in a reagent cocktail containing 88% TFA, 5% phenol, 5% H2O and 2% TIPS, crude products were precipitated with cold ether and purified to homogeneity by preparative C18 reversed-phase HPLC. The molecular masses were ascertained by electrospray ionization mass spectrometry (ESI-MS).
Reversed phase analytical and preparative HPLC
Analytical HPLC was run on a Waters instrument using an analytical C18 column purchased from Waters at a flow rate of 1.0 mL/min. Solution A was ultrapure water containing 0.1% trifluoroacetic acid (TFA), and solution B was acetonitrile containing 0.1% TFA. The gradient is liner from 5% B to 65% B in 30 min. Preparative HPLC was run on a Preparative Waters instrument using an analytical C4 column purchased from Waters at a flow rate of 15.0 mL/min. Eluent A and B were same as the solution used in analytical HPLC. The gradient is liner from 25% B to 50% B in 60 min.
Preparation of stapled PMI
To prepare stapled PMI, the peptide was firstly dissolved in reaction buffer [80% 10 mmol/L PBS (pH 7.4) and 20% acetonitrile] at a concentration of 100 μmol/L, meanwhile 1,2(1,3 or 1,4)-dimethylbromobenzene were dissolved in DMSO at a concentration of 10 mmol/L. After the preparation of reaction fluid, 10 mL peptide buffer were magnetic stirred in a beaker at room temperature, and then 50 μL 1,2 (1,3 or 1,4)-dimethylbromobenzene buffers were mixed into the buffer in four times every 10 min. After the reaction, pure stapled PMI can be collected by preparative HPLC.
CD spectroscopy
CD spectra of variants at a concentration of 20 μmol/L in 10 mmol/L phosphate buffer (pH 7.4) were obtained at room temperature on a J-810 spectropolorimeter (Jasco, Easton, MD) using a 1-mm quartz cuvette as previous reports[25-27]. Scanned area was from 250 nm to 190 nm, and the scanning speed was 50 nm/min. Every curve was the average of three independent detections.
Fluorescence polarization-based competitive binding assay
As for fluorescence polarization assay, Fluorescein (FITC) was conjugated to 15-29p53 via its N-terminal amino group in DMF, and the resultant product 15-29p53-FITC were HPLC-purified and lyophilized. The Fluorescence polarization-based competitive binding assays were performed in Microfluor® 2, 96-well black plates (Thermo Fisher Scientific) and readings were taken using a Tecan Infinite M2000 fluorescence plate reader. Serially diluted Lupbin or corresponding peptide were prepared in Tris-HCl buffered saline (10 mmol/L Tris, 150 mmol/L NaCl, 1 mmol/L EDTA, pH 7.0) and incubated with 200 nmol/L 15-29p53-FITC/MDM2 or 50 nmol/L 15-29p53-FITC/MDMX in a total volume of 150 μL per well. After 2 h incubation at room temperature, fluorescence polarization was measured at λex = 470 nm and λem = 530 nm. Nonlinear regression analyses were performed to give rise to IC50 values.
Cell culture and cell viability analysis
Human colon cancer cell line HCT116+/+ (wild-type p53) was purchased from ATCC, and maintained in McCoy’s 5A medium with 10% FBS. The isogeneic HCT116-/- (p53 deletion) cells were presented by Prof. Bert Vogelstein of Johns Hopkins University (Baltimore, MD), and maintained in McCoy’s 5A medium with 10% FBS. human hepatoma cell line SK-Hep-1 was also purchased by ATCC, and maintained in DMEM with 10% FBS. All cells were maintained at 37 °C in an atmosphere of 5% CO2. For cell viability test, three cell lines were plated in 96-well plates at a density of 2,500 cells/well (100 μL). After 24 h, cells were treated with drug sample at the indicated concentrations and times in FBS-free mediums, respectively. The in vitro cytotoxicity was then measured by using a standard MTT (Thermo Fisher scientific) assay after 72 h drug treatment.
Apoptosis analysis
Necrosis/apoptosis was evaluated by flow cytometric analysis using the FITC Annexin V Apoptosis Detection Kit (BD Biosciences). Briefly, cells were treated with samples for 48 h. Cells were then harvested, washed twice with cold PBS, and re-suspended in 1× binding buffer at a concentration of 1 × 106 cells/mL. One hundred microliters of the solution (1 × 105 cells) was transferred to a 5 mL culture tube, followed by addition of 5 µL of FITC Annexin V and 5 µL of PI. After gentle vortexing and a 15-min incubation in the dark at room temperature, 400 µL of 1× binding buffer was added to the tube, and cells were analyzed by fluorescence-activated cell sorting (FACS).
Results
Wild-type p53 is a feasible target for HCC therapy
Research has shown that the tumor suppressor p53 has an important role in tumor progression, and that it is mutated or functionally inactivated in most human cancers[2]. As for HCC, p53 was nonsynonymously mutated in 259 (29.9%) of 867 hepatoma cases in TCGA [Figure 1A], suggesting that in a substantial proportion of HCC, TP53 (which encodes p53) is wild type but the protein is inactivated. To explore the importance of p53 and its two agonists- MDM2 and MDMX- in the HCC process, we analyzed the relationship between these protein expression and survival of HCC patients carried wild-type p53. As shown in Figure 1B and C, decreased expression of p53 was significantly associated with poor patient overall survival (P = 0.043) and disease-free survival (P = 0.037). It is well-known that the tumor suppressor activity and in vivo stability of p53 are abrogated by regulatory molecules such as the E3 ubiquitin ligase MDM2 and its homologue MDMX (also known as HDMX and MDM4) [Figure 1D][28,29]. This offers an attractive strategy for cancer therapy based on p53 reactivation by blocking the interaction between p53 and MDM2 (MDMX). In this case, the two major negative regulators of p53 now seem to be “druggable”, and recent studies in cancer patients have provided proof-of-concept for this approach[2]. To further verify the feasibility of p53 restoration via MDM2 and MDMX blocking for HCC therapy, we attempted to evaluate the association of MDM2 and MDMX expression with survival in 209 HCC patients carried wild-type p53. As expected, the 5-year overall survival [Figure 1E] and disease-free survival [Figure 1F] rates of MDM2 high-expressed cases are significantly higher than that of MDM2 low-expressed cases. Meanwhile, MDMX showed the same tendency as MDM2 [Figure 1G and H]. Collectively, all these results demonstrated that high-level p53 is beneficial to the survival of HCC patient, thus p53 restoration was a potentially feasible program for HCC therapy in p53-wild-type patients.
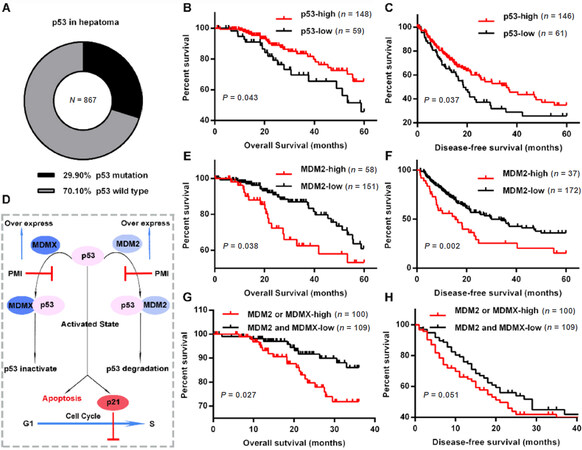
Figure 1. Wild-type p53 is a potential target for HCC therapy. A: The percentage of wild-type p53 in 867 HCC patients in The Cancer Genome Atlas; B and C: the Kaplan-Meier survival curves of overall survival and disease-free survival duration based on p53 expression in the mRNA level; D: the Schematic diagram for the mechanism of p53 function and its connection with MDM2 and MDMX; E-H: the Kaplan-Meier survival curves of overall survival and disease-free duration based on MDM2 and MDMX expression in the mRNA level. The receiver operating characteristic curve was used to define the cutoff, and log-rank analysis was used to test for significance. HCC: Hepatocellular carcinoma; PMI: p53-MDM2/MDMX inhibitor
Preparation of dimethylbromobenzene-cysteine stapled peptide
In this study, we firstly used PMI-a potent dodecameric peptide antagonist of MDM2 and MDMX that, despite its high affinity for both proteins[14], fails to activate p53 and kill p53+/+ tumor cells due presumably to its inability to traverse the cell membrane and susceptibility to proteolytic degradation[30]. Our new chemistry for stapling peptide entails an efficient click reaction between the bromine in dimethylbromobenzene and the mercapto group in peptide Cys. Due to the fact that the effective concentration in the molecule was much higher than the intermolecular concentration, dimethylbromobenzene would specifically be conjugated to the two Cys in one peptide rather than the two intermolecular Cys [Figure 2A]. Previous structural and functional studies of PMI (TSFAEYWNLLSP) identified Phe3, Tyr6, Trp7 and Leu10 as the most critical residues for MDM2/MDMX binding[14]. Thus, we maintained those four residues in the design of stapled peptides and introduced Cys-Cys pairs into (5,9) positions of PMI (Figure 2A, SP0 TSFACYWNCLSP). This N-acetylated and C-amidated peptides were synthesized using Fmoc-chemistry for solid phase peptide synthesis as our previous reports[31,32], and purified by HPLC to homogeneity. Crosslinking two Cys side chains was readily accomplished in 2 h in PBS/acetonitrile (4:1) buffer containing 100 μmol/L SP0 peptide and 150 μmol/L dimethylbromobenzene, as verified by ESI-MS [Figure 2B], resulting in 3 stapled constructs termed SP1, SP2 and SP3 [Figure 1A and B]. Not surprisingly, SP1, SP2 and SP3 partially adopted an α-helical structure in aqueous solution according to CD analyses, whereas SP0 and PMI showed very limited topological structure [Figure 2B and C], suggesting that crosslinking Cys-Cys side chains stabilized peptide conformation productive for targets binding.
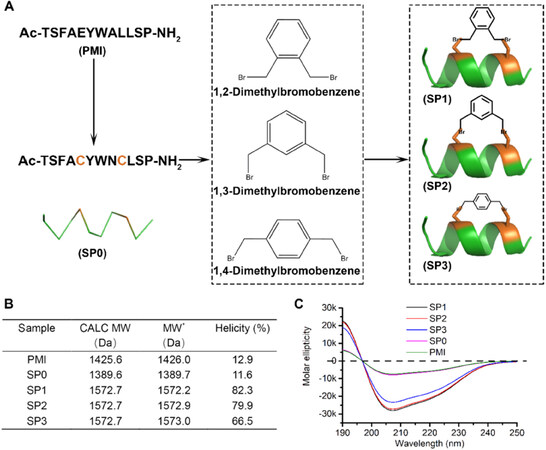
Figure 2. Characterization of dimethylbromobenzene-cysteine stapled peptide. A: Schematic diagram for the preparation of dimethylbromobenzene-cysteine stapled peptide; B: the table for the molecular weight and helicity of the p53-MDM2/MDMX inhibitor (PMI) and stapled peptides. CALC MW stands for the theoretical molecular weight of peptides. *Stand for that the molecular weight was measured by ESI-MASS; C: circular dichroism spectra of PMI, SP0, SP1, SP2 and SP3. The experiment was repeated independently for 3 times with similar results
Dimethylbromobenzene-cysteine stapled peptide specifically targets intracellular complexes of p53/MDM2 and p53/MDMX
Dubbed the “guardian of the genome”[33], p53 is critical for maintaining genetic stability and preventing tumor development[4]. MDM2 binds the N-terminal transactivation domain of p53 with high affinity to block p53 regulating responsive gene expression, resulting in the p53 inactivation[34]. Moreover, MDM2 controls p53 stability by targeting the tumor suppressor protein for ubiquitin-mediated constitutive degradation[35]. Although MDMX lacks E3 ubiquitin ligase activity, the MDM2 homologue acts as an effective transcriptional antagonist of p53, and impedes p53-induced growth inhibitory and apoptotic responses[36]. Thus, the ideal p53 activators are dual specific inhibitors to target both MDM2 and MDMX, and SPx (SP1, SP2 or SP3) may well be one of them.
For verification, the inhibitory effects of stapled PMI SPx on the interaction between p53 and MDM2/MDMX were measured by fluorescence polarization-based competition assays, in which different concentrations of stapled PMI were applied to pre-incubated MDM2/18-26p53-FITC or MDMX/18-26p53-FITC complexes, respectively, [Figure 3B and C] and the IC50 and Kd values are tabulated in Figure 3D. Compared with the N-acetylated and C-amidated wild-type peptide PMI, SP1 and SP2 were bound more strongly to MDM2 and MDMX. Meanwhile, SP3 showed a moderately higher ability to block MDMX/p53 interaction than PMI, but was slightly inferior than PMI to MDM2/p53. Notably, the half-maximal inhibitory concentrations (IC50 values) of SP1, SP2 and SP3 were as the same order of magnitude as PMI, demonstrating that all of the three stapled PMI were capable of blocking p53-MDM2/MDMX interaction, thereby reactivating p53 to suppress tumor growth.
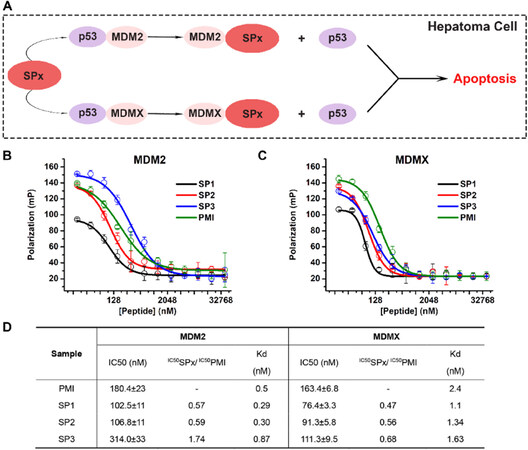
Figure 3. The stapled peptides specifically targets intracellular complexes of p53/MDM2 and p53/MDMX. A: Schematic diagram for that stapled peptides can compete with p53 for MDM2 or MDMX binding; B and C: fluorescence polarization-based competitive binding assay of p53-MDM2/MDMX inhibitor (PMI) or stapled peptides to MDM2/p53 complex (B) and MDMX/p53 complex. For fluorescence polarization measurements at room temperature on a Tecan Infinite M2000 plate reader, FITC was covalently conjugated to the N-terminal of 15-29p53. Non-linear regression analyses were performed to give rise to IC50 values (mean ± SEM, n = 3); D: table for the results from B and C. Kd of PMI was cited from our previous reports, and Kd of SP1, SP2 and SP3 were calculated by the IC50 ratio between SPx to PMI
Functional characterization of dimethylbromobenzene-cysteine PMI
Previous reports have shown that structurally permissible stapling of peptide, while enhancing α-helicity and improving targets binding, is not sufficient to endow the peptide with an ability to kill tumor cells[15]. In fact, the amino acid composition and topological structure of a stapled peptide are critical for its ability to traverse the cell membrane to exert biological activity[15]. To address it, we firstly measured the cellular uptakes of FITC-labled SP0, SP1, SP2 and SP3 in HCT116 cells after 6 h incubation in 37 °C at a concentration of 100 μmol/L. As shown in Supplementary Figure 1, SP3 showed the strongest ability of cellular internalization (> 75%), whereas neither SP1 nor SP2 showed exceed 15% cellular internalization. Moreover, there exists no cellular uptakes for the three stapled peptides at 4 °C incubation [Supplementary Figure 1], suggesting that the cellular uptakes of the stapled peptide most likely result from the ATP-dependent endocytosis. Furthermore, it is necessary to verify the biological activity of SPx to induce the cancer cells apoptosis in a p53-dependent manner. To functionally validate SPx, we subjected them and their unstapled control to a cell viability assay in a FBS-free medium using a pair of cell lines with the same genetic background carried wild-type p53 (HCT116 p53+/+) or deleted p53 (HCT116 p53-/-). While the control peptide exhibited no anti-proliferative activity against both cell lines at concentrations of up to 100 μmol/L, SP3 displayed dose- and p53-dependent growth inhibitory activity against HCT116 p53+/+, but not HCT116 p53-/-, with an IC50 value of ~18.6 μmol/L at 72 h [Figure 4A and B]. Besides, SP3 also showed an obvious suppression for a hepatocellular carcinoma cell line carried wild-type p53, named Sk-Hep-1, with an IC50 value of ~24.8 μmol/L in the absence of serum [Figure 4C and Supplementary Figure 2]. Consistent with this result, the induction of apoptosis of HCT116 p53+/+ and SK-Hep-1 cells by SP3 was verified by fluorescence-activated cell sorting [Figure 4D-I]. Taken together, these findings support that SP3 actively traversed the cell membrane and killed tumor cells by reactivating the p53 pathway. It is worth pointing out that as is often the case with other stapled peptide activators, although its efficient blocking the interaction between p53-MDM2/MDMX, are rather weak in killing HCT116 p53+/+ and SK-Hep-1 cells. The weak in vitro activity implies that stapling alone is insufficient to achieve optimal therapeutic efficacy of helical peptides, dictated by cell internalization, endosomal escape, proteolytic stability, spatio-temporal distribution, etc.
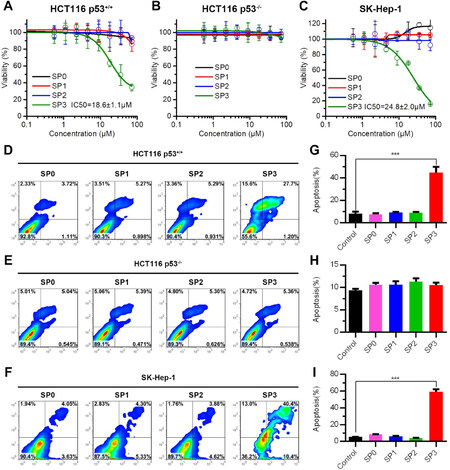
Figure 4. Functional characterization of dimethylbromobenzene-cysteine p53-MDM2/MDMX inhibitor. A-C: dose-dependent growth inhibition of HCT116 p53+/+ (A), HCT116 p53+/+ (B) and SK-hep-1 (C) cells upon various treatments as determined by the MTT assay to monitor the pesticide effects. Three cell lines were plated in 96-well plates at a density of 2,500 cells/well (100 μL). After 24 h, cells were treated with drug sample at the indicated concentrations and times in FBS-free mediums, respectively. The in vitro cytotoxicity was then measured by using a standard MTT (Thermo Fisher scientific) assay after 72 h drug treatment. (mean ± SD, n = 4); D-F: apoptosis levels measured by FACS in three cell lines treated with SP0, SP1 SP2 and SP3 for 48 h incubation at concentration of 50 μmol/L; G-I: the average means of the apoptosis calculated three independent experiments like D-F. P values were calculated by t-test (*P < 0.05; **P < 0.01; ***P < 0.001)
Discussion
The tumor suppressor protein p53 induces powerful cancer cell antiproliferation and apoptotic responses to cellular stress, plays a pivotal role in preventing damaged cells from cancerous[4]. Not surprisingly, the impairment of p53 signaling pathway is a hallmark of almost all human cancers, where either the TP53 gene is mutated or wild-type p53 is functionally inactivated by the E3 ubiquitin ligase MDM2 and its homolog MDMX[37,38]. In many tumor cells harboring wild-type p53, the up-regulated MDM2 and/or MDMX often cooperate to inhibit p53 transactivation activity and urge p53 for degradation, conferring tumor development and progression[2]. A great number of studies have validated that MDM2 and/or MDMX antagonism as a viable therapeutic regimen for cancer therapy, and several small-molecule antagonists specific for MDM2 are in various phases of clinical trials[39,40]. As for HCC, our results in Figure 1 further illustrates that p53-MDM2/MDM2 is an important target for therapy, thus, the development of potent antagonists specific for MDM2 and MDMX is meaningful for HCC therapy.
Growing evidence suggests that the interplay between MDM2 and MDMX confers robust p53 inactivation in tumorigenesis and that antagonizing both MDM2 and MDMX affords a powerful, synergistic and sustained inhibition of tumor growth[41,42]. However, traditional small-molecule drugs are always limited by the comparatively small interaction area, resulting in the failure as dual specific inhibitors to target both MDM2 and MDMX simultaneously. To this end, a peptide therapeutics termed PMI was developed to competes with p53 for MDM2 and MDMX binding at high affinity[14,43]. However, major pharmacological hurdles still impede the development of anticancer peptide therapeutics with optimal therapeutic efficacy, including: short circulation half-life due to proteolytic degradation and poor cellular uptake. To overcome these technical obstacles, we developed a novel peptide stapling method to link the side chains of Cys and Cys at (i, i+4) positions by two bromine methyl group in benzene para-, ortho- or meta- positions to form the dimethylbromobenzene-cysteine structure. Of note, this stapling method is appropriate for all α-helix after mutating two nonfunctional residues into Cys at (i, i+4) positions. After a series of characterization and functional verification, SP3, a stapled PMI crosslinked the side chains by 1,4-dimethylbromobenzene, can potently inhibit the growth of cancer cell in a p53 dependent manner. Of note, the remaining position of the benzene in the dimethylbromobenzene can be further modification for more hydrophilic and more appropriate charge characteristics.
In this work, we found that the expression of p53, MDM2 and MDMX were closely related to the survival of ~70% HCC patients carrying wild-type p53, and provided strong evidence that reactivating p53 from MDM2 and MDMX was a potentially feasible program for HCC therapy. After that, we have developed a novel stapling strategy for peptide drug design using the reaction between mercapto group and bromine to crosslink the side chains of the two Cys at (i, i+4) positions. By this way, we successfully induced the formation of and stabilized a productive α-helical conformation of PMI - a dual-specificity peptide antagonist of MDM2 and MDMX, enabling it to traverse the cell membrane and kill tumor cells by reactivating the p53 pathway. This stapling functionally rescued PMI that, on its own, failed to activate p53 because of its poor membrane permeability and susceptibility to proteolytic degradation. Taken together, this work not only illustrates that the restoration of p53 is a potentially feasible program for HCC therapy, but promises an important new tool for peptide drug discovery and development for a variety of human diseases.
Declarations
Authors’ contributionsDid experiments: Jiang W, Jin L
Designed this work: Liu M, Hou P, He WX
Wrote this paper: Hou P, He WX
Revised the manuscripts: All authors
Availability of data and materialsThe patient mRNA data was from TCGA. All experimental data did by the authors listed in this paper.
Financial support and sponsorshipThis work was supported by the Clinical Research Award of the First Affiliated Hospital of Xi’an Jiaotong University (XJTU1AF-CRF-2017-003) to Hou P and He WX.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
1. Meng X, Franklin DA, Dong J, Zhang Y. MDM2-p53 pathway in hepatocellular carcinoma. Cancer Res 2014;74:7161-7.
2. Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer 2013;13:83-96.
3. Bian Z, Yan J, Wang S, Li Y, Guo Y, et al. Awakening p53 in vivo by D-peptides-functionalized ultra-small nanoparticles: overcoming biological barriers to D-peptide drug delivery. Theranostics 2018;8:5320-35.
4. Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer 2009;9:862-73.
5. Jemal A, Bray F, Center MM, Ferlay J, Ward E, et al. Global cancer statistics. CA Cancer J Clin 2011;61:69-90.
6. Staib F, Perwez Hussain S, Hofseth LJ, Wang XW, et al. TP53 and liver carcinogenesis. Hum Mutat 2003;21:201-16.
7. Pekow JR, Bhan AK, Zheng H, Chung RT. Hepatic steatosis is associated with increased frequency of hepatocellular carcinoma in patients with hepatitis C-related cirrhosis. Cancer 2007;109:2490-6.
8. Siegel AB, Zhu AX. Metabolic syndrome and hepatocellular carcinoma: two growing epidemics with a potential link. Cancer 2009;115:5651-61.
11. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004;303:844-8.
12. Shangary S, Qin D, McEachern D, Liu M, Miller RS, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci USA 2008;105:3933-8.
13. Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996;274:948-53.
14. Pazgier M, Liu M, Zou G, Yuan W, Li C, et al. Structural basis for high-affinity peptide inhibition of p53 interactions with MDM2 and MDMX. Proc Natl Acad Sci USA 2009;106:4665-70.
15. Bernal F, Tyler AF, Korsmeyer SJ, Walensky LD, Verdine GL. Reactivation of the p53 tumor suppressor pathway by a stapled p53 peptide. J Am Chem Soc 2007;129:2456-7.
16. Madden MM, Vera CIR, Song W, Lin Q. Facile synthesis of stapled, structurally reinforced peptide helices via a photoinduced intramolecular 1, 3-dipolar cycloaddition reaction. Chem Commun 2009:5588-90.
17. Boal AK, Guryanov I, Moretto A, Crisma M, Lanni EL, et al. Facile and e-selective intramolecular ring-closing metathesis reactions in 310-helical peptides: a 3D structural study. J Am Chem Soc 2007;129:6986-7.
18. Schievano E, Bisello A, Chorev M, Bisol A, Mammi S, et al. Aib-rich peptides containing lactam-bridged side chains as models of the 310-helix. J Am Chem Soc 2001;123:2743-51.
19. Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, et al. A stapled BID BH3 helix directly binds and activates BAX. Mol Cell 2006;24:199-210.
20. Zhang F, Sadovski O, Xin SJ, Woolley GA. Stabilization of folded peptide and protein structures via distance matching with a long, rigid cross-linker. J Am Chem Soc 2007;129:14154-5.
21. Ousaka N, Inai Y, Kuroda R. Chain-terminus triggered chiral memory in an optically inactive 310-helical peptide. J Am Chem Soc 2008;130:12266-7.
22. Cantel S, Le Chevalier Isaad A, Scrima M, Levy JJ, DiMarchi RD, et al. Synthesis and conformational analysis of a cyclic peptide obtained via i to i+ 4 intramolecular side-chain to side-chain azide-alkyne 1, 3-dipolar cycloaddition. J Org Chem 2008;73:5663-74.
23. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6:pl1.
24. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401-4.
25. He W, Mazzuca P, Yuan W, Varney K, Bugatti A, et al. Identification of amino acid residues critical for the B cell growth-promoting activity of HIV-1 matrix protein p17 variants. Biochim Biophys Acta Gen Subj 2019;1863:13-24.
26. He W, Yan J, Sui F, Wang S, Su X, et al. Turning a luffa protein into a self-assembled biodegradable nanoplatform for multitargeted cancer therapy. ACS Nano 2018;12:11664-77.
27. Dolcetti R, Giagulli C, He W, Selleri M, Caccuri F, et al. Role of HIV-1 matrix protein p17 variants in lymphoma pathogenesis. Proc Natl Acad Sci U S A 2015;112:14331-6.
28. Liu M, Li C, Pazgier M, Li C, Mao Y, et al. D-peptide inhibitors of the p53-MDM2 interaction for targeted molecular therapy of malignant neoplasms. Proc Natl Acad Sci USA 2010;107:14321-6.
29. Yan J, He W, Yan S, Niu F, Liu T, et al. Self-assembled peptide-lanthanide nanoclusters for safe tumor therapy: overcoming and utilizing biological barriers to peptide drug delivery. ACS Nano 2018;12:2017-26.
30. Niu F, Yan J, Ma B, Li S, Shao Y, et al. Lanthanide-doped nanoparticles conjugated with an anti-CD33 antibody and a p53-activating peptide for acute myeloid leukemia therapy. Biomaterials 2018;167:132-42.
31. Yu M, Yan J, He W, Li C, Ma PX, et al. Synthetic θ-defensin antibacterial peptide as a highly efficient nonviral vector for redox-responsive miRNA delivery. Adv Biosys 2017;1:1700001.
32. Bu B, Tong X, Li D, Hu Y, He W, et al. N-terminal acetylation preserves α-synuclein from oligomerization by blocking intermolecular hydrogen bonds. ACS Chem Neurosci 2017;8:2145-51.
34. Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. cell 1992;69:1237-45.
35. Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature 1997;387:299-303.
36. Linares LK, Hengstermann A, Ciechanover A, Müller S, Scheffner M. HdmX stimulates Hdm2-mediated ubiquitination and degradation of p53. Proc Natl Acad Sci USA 2003;100:12009-14.
38. Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov 2014;13:217-36.
39. Burgess A, Chia KM, Haupt S, Thomas D, Haupt Y, et al. Clinical overview of MDM2/X-targeted therapies. Front Oncol 2016;6:7.
40. Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu Rev Pharmacol Toxicol 2009;49:223-41.
41. Tan BX, Liew HP, Chua JS, Ghadessy FJ, Tan YS, et al. Anatomy of Mdm2 and Mdm4 in evolution. J Mol Cell Biol 2017;9:3-15.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Jiang W, Jin L, Liu M, Hou P, He WX. A dimethylbromobenzene-cysteine stapled peptide dual inhibitor of the p53-MDM2/MDMX interactions. Hepatoma Res 2019;5:5. http://dx.doi.org/10.20517/2394-5079.2018.97
AMA Style
Jiang W, Jin L, Liu M, Hou P, He WX. A dimethylbromobenzene-cysteine stapled peptide dual inhibitor of the p53-MDM2/MDMX interactions. Hepatoma Research. 2019; 5: 5. http://dx.doi.org/10.20517/2394-5079.2018.97
Chicago/Turabian Style
Jiang, Wei, Liang Jin, Min Liu, Peng Hou, Wang-Xiao He. 2019. "A dimethylbromobenzene-cysteine stapled peptide dual inhibitor of the p53-MDM2/MDMX interactions" Hepatoma Research. 5: 5. http://dx.doi.org/10.20517/2394-5079.2018.97
ACS Style
Jiang, W.; Jin L.; Liu M.; Hou P.; He W.X. A dimethylbromobenzene-cysteine stapled peptide dual inhibitor of the p53-MDM2/MDMX interactions. Hepatoma. Res. 2019, 5, 5. http://dx.doi.org/10.20517/2394-5079.2018.97
About This Article
Copyright

Data & Comments
Data


 Cite This Article 6 clicks
Cite This Article 6 clicks

 Like This Article 17
likes
Like This Article 17
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.