
Oncogenicity of viral hepatitis B and C in the initiation of hepatic cancer stem cells


Abstract
Chronic infection of hepatitis B virus (HBV) or/and hepatitis C virus (HCV) is one of major risk factors in the development of the hepatocellular carcinoma. Recent studies had shown the capacity of viral proteins in inducing the presence of the population of so-called the cancer stem cells (CSC). The integration of HBV S and X gene in the host genome indicates its direct oncogenicity. In addition, the presence HBV and HCV proteins were shown to modulate intracellular molecular pathways and epigenetic modification. This review summarizes current literature regarding direct oncogenic properties of HBV and HCV in the initiation of CSC both in in vitro and in vivo studies.
Keywords
Introduction
Chronic infection of viral hepatitis B or C is a major risk factor for the development of hepatocellular carcinoma (HCC). In fact, global distribution of HCC is associated with the prevalence of hepatitis viruses: hepatitis B virus (HBV) or hepatitis C virus (HCV). The infection of endemic HBV is the major cause of HCC in eastern Asia and sub-Saharan Africa for around 70%, while in Europe and North American countries, the infection of HCV ranges from 50% to 70% of all cases[1-3]. In addition, due to different oncogenic mechanisms of viruses, as well as various genetic host background and long-term development of the disease, viral-related HCCs show high heterogeneity.
Hepatocarcinogenesis is multifactorial, consisting of various steps in a long-term course. At its initiation, disturbance in the molecular and cellular pathways might result in the malignant transformation from normal to malignant cells. Natural pathogenesis of hepatitis viruses usually involve a sequentially damaging process. It starts with cellular immunological response, triggering DNA damage, mitochondrial dysfunction, and endoplasmic reticulum stress, thus resulting in liver fibrosis, cirrhosis and finally HCC. On the other hand, in the initial step, infection of viral hepatitis can play a significant role in the switch of the fate of the cells by directly triggering the appearance of the cancer stem cells (CSC) [Figure 1].
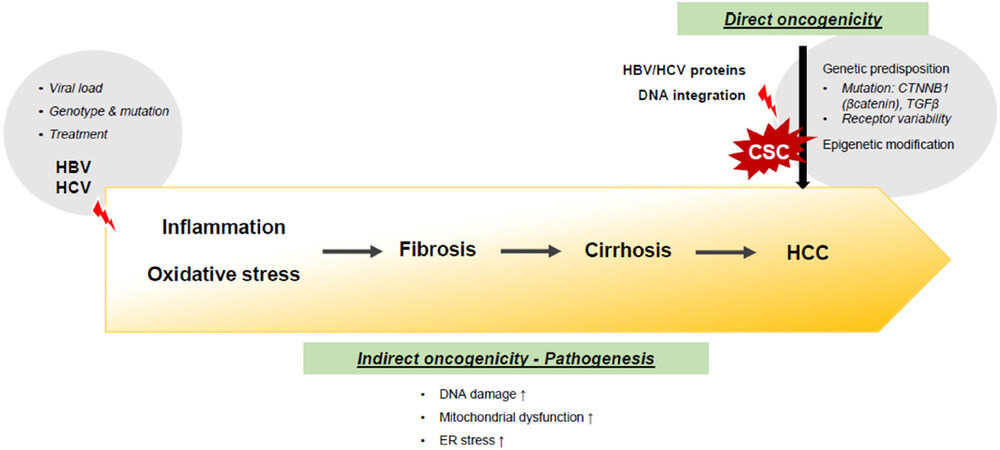
Figure 1. The oncogenicity of viral hepatitis in the development of hepatocellular carcinoma. HBV: hepatitis B virus; HCV: hepatitis C virus; TGF: transforming growth factor; HCC: hepatocellular carcinoma; ER: endoplasmic reticulum
CSC is the highest-ranking cell population in cancer with the tumorigenic capacity to initiate cancer. It has the capability to divide and differentiate to partially or fully-differentiated cancer cells that comprise the majority of cancer mass. This hierarchy model shows that CSC population is unique, with protective mechanism to be responsible for the maintenance and propagation of the tumor[4]. These cells act as the main players in the highest level of the cancer hierarchy and may still have stem cells properties such as self-renewal and ability to multiple cell types. Non-tumorigenic cells are thought to compose the bulk of tumors but have little capacity to contribute to cancer progression[5,6].
The first evidence of CSC in HCC was demonstrated by the isolation of the side population in vitro[7,8] showing the involvement of CSC in drug resistance. The search and identification method of hepatic CSC progressed by performing sphere colony formation and more commonly, by using CSC markers.
Various markers of CSC from established HCC cell lines and primary tumors had been identified and validated by in vivo xenograft assay. Cell protein markers CD133 (PROM1)[9-11], CD90 (THY-1)[12,13], epithelial cell adhesion molecule (EpCAM)[14,15], CD24[16], CD13 (ANPEP)[7,17] are the most common method to define a hepatic CSC population. Until now, at least 12 different phenotypical CSC markers had been proposed. The combination of these CSC markers was further used to characterize several subpopulations in a CSC population, resulting in a wide variety of CSC phenotypes.
To understand the mechanism of early initiation of HCC, the oncogenic role of HBV and HCV proteins in hepatic CSC has been started to be explored. They were analyzed by determining the extent of up-regulation and the presence of various hepatic CSC markers, after the exposure of viral proteins into hepatic cells. In addition, these findings were also supported by functional analysis such as cell aggressiveness, migration, and more importantly, by xenograft in vivo model, several published data was shown in Table 1.
Several studies on the effect of viral hepatitis in the acquisition of cancer stem cells traits
| Gene | Experimental model | CSC marker | Functional analysis | Ref. | |
|---|---|---|---|---|---|
| HBV | Pre-S1 | Cell lines L02, HepG2, Huh7 | CD133, CD117, CD90 | Facilitation of growth and migration; induction of tumorigenesis | [22] |
| Pre-S1/Pre-S2/S | Transgenic mouse Tg(Alb1-HBV)Bri44 | CD133, EpCAM, CK19, CD34 | Follow-up of hepatocarcinogenesis | [21] | |
| X | Cell line HepG2 | OCT4, Nanog, Klf4, EpCAM | Stimulation of cell growth and migration | [25] | |
| Cell lines 4pX-1 (from AML12), HepAD38 | EpCAM | Active DNA demethylation | [29] | ||
| HCV | SGR | Cell lines FCA4 (from Huh7), GS5 (from Huh7.5) | CD133, AFP, CK19 | Tumorigenicity | [36] |
| Core | Cell lines PHH, THH (from IHH) | c-Kit | Sphere formation, tumorigenicity | [35] | |
| NS5A | Transgenic mice NS5A TG (FVB strain), Tlr4-/- | Nanog, CD133 | Liver damage and tumor formation | [38,42] |
Oncogenicity of viral hepatitis in hepatic CSC acquisition traits
HBV
HBV, a member of Hepadnaviridae family, is a partially double-stranded DNA virus with 3.2 kb genome size. HBV genome encodes four overlapping open reading frames (ORFs: S, C, P, and X)[18]. Up to now, most of the studies in literature focused on the involvement of HBV S and X proteins in the initiation oncogenesis.
ORF S with three translational start sites Pre-S1, Pre-S2, and S, encodes for large, middle and small surface protein (HBs), respectively, which acts as the main factor in the natural pathogenesis of the virus. The accumulation of HBs antigen (HBsAg) in hepatocytes triggers cellular inflammation and oxidative stress driving a sustained prolonged liver injury until the development of HCC.
On the other hand, direct oncogenic effect of HBV in the development of HCC is closely related with the integration of the HBV DNA sequence into the host genome. HBV DNA integration was considered as a strong oncogenic effect in hepatocarcinogenesis. A recent study reported that in HCC patients with occult hepatitis B, HBV DNA integration was found in around 75% of cases, in which the inserted viral genes were mainly X and PreS/S, followed by C and P sequences[19].
A HBV-transgenic mouse model with the insertion of whole S gene region expressed high level of HBsAg, showing inflammation and appearance of glass ground hepatocytes. Interestingly, the damage induced pre-neoplastic lesion and finally HCC in major number of animals[20], indicating a direct oncogenic contribution of this gene. Our time-course study in this HBV-transgenic mouse showed a progressive increase of the expression of CSC and hepatic progenitor marker during the course of hepatocarcinogenesis, up to 18 months. The expression of several markers such as CD133, EpCAM, and CK19 were significantly increased along liver injury. Further, there was a significant correlation between CSC markers and diagnosis[21]. Furthermore, it was recently demonstrated that PreS1 of the S gene activated the expressions of CSC markers CD133, CD117, and CD90 in normal hepatocytes and HCC cells. It indicated the new role of PreS1 as a new oncoprotein to play a key role in the appearance and self-renewal of CSC during HCC development[22].
ORF X encodes for HBx, which has pleiotropic functions as an important regulator in viral life cycle, a transcriptional activator, and a stimulator in the cytoplasmic signal transduction pathway. In HCC clinical specimens, high HBx expression was correlated with the expansion of EpCAM or OV6 progenitor cells, aggressive clinicopathological features[23,24] and activated β-catenin signalling[25]. In depth, a direct in vitro model had shown that the insertion of HBx induced the pluripotent stem cell transcription factors Oct4, Nanog, Klf4, as well as CSC markers EpCAM and β-catenin. The presence of HBx proteins stimulated cell growth and migration[25]. This in vitro data were then confirmed by using HBx transgenic mice where a high number of EpCAM cells with characteristics of human progenitor cells was observed[23]. Transformation of rat oval cells with HBx and the subsequent injection in nude mice treated with aflatoxin B1 in vivo, gave rise to tumor that expressed markers of adult hepatocytes as albumin and CK18, undifferentiated marker alpha-fetoprotein (AFP), and oncoprotein c-Myc[26].
The truncation of HBx protein in the C-terminal region (HBx-ΔC) is a common event because of HBV X sequence integration in the genome. A recent study had shown that HBx-ΔC promoted the appearance of a CD133 hepatic CSC subset and confer cancer and stem cell-like features in HCC[27]. It is associated with cancer cell invasiveness and reduction of apoptotic response, tumorigenicity, chemoresistance, and migration[27,28].
Regardless of the data provided, the exact mechanism by which HBV proteins altered the early fate of the cells was still unclear. Several studies had shown that DNA demethylation could be a major mechanism in the increase of the expression of CSC markers in normal hepatocytes[29], also correlated with HBV DNA integration[30] and the axis of HBx-DLL3 (Delta-like 3) of Notch receptor[31]. In the last study, the treatment of HBV-transformed cells with a histone deacetylase inhibitor induced DLL3 expression[31]. In a recent 2018 study, it was shown that in the very early stage of HCC, the global DNA methylation 5hmC and 5fC contents were decreased significantly. It was found to be correlated with HBV infection, decreased ten-eleven translocation enzyme activity and uncoordinated expression of DNA methylation-related enzymes[32].
HCV
HCV, a member of flaviviridae family, is a single stranded RNA virus with 9.6 kb genome size. HCV genome is processed into structural proteins core, E1, and E2, and non-structural (NS) proteins p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B[33]. Since HCV being an RNA virus cannot integrate into human genome, at the beginning, the mechanism in HCV-related HCC pathogenesis is supposed exclusively to indirect via chronic inflammation and oxidative stress. Subsequently, it leads to fibrosis and eventually cirrhosis as observed in the other HCC etiologies[34]. However, current literature in experimental models also showed direct oncogenic effect of the HCV proteins, including on the involvement of the CSC.
A previous study showed that HCV-infected hepatocytes transformed into sphere formation with a number of epithelial-mesenchymal transition (EMT) and CSC markers, including high level of the stem cell factor receptor c-Kit. These spheres were potent in promoting tumor growth in immunodeficient mice. However, these spheres were highly sensitive to cell death from the treatment of sorafenib, a multikinase inhibitor, and stattic, an inhibitor of the Stat3 molecule[35]. Furthermore, by inserting HCV sub-genomic replicon in cultured cells, the acquisition of CSC traits, including an enhanced expression of doublecortin and CaM kinase-like-1, Lgr5, CD133, AFP, CK19, Lin28, and c-Myc, was demonstrated. Conversely, curing of the replicon from these cells diminished the expression of these factors. In vivo analysis of liver tissues from HCV-positive patients and liver tissue microarrays supported these observations[36].
It had been shown that HCV core and NS proteins, can induce cell transformation in vitro and in vivo mice transgenic model[37]. By using intercross breeding of transgenic mouse models, HCV NS5A protein induced the toll-like receptor 4 (TLR4). This induction mediated liver damage and tumor formation in synergy with alcohol-induced endotoxemia. Consequently, the expression of stem cell marker Nanog and the presence of CD133/Nanog-positive cells were observed[38].
This study was then continued by in vivo animal study of NS5A mice fed with high in cholesterol and saturated fat diet (HCFD). Liver tissues of these HCFD mice had increased levels of TLR4, Nanog, phosphorylated signal transducer and activator of transcription (pStat3), and Twist1. Further analysis of isolated tumor-initiating stem-like cells (TISCs) with the phenotype of CD133+ CD49f+ showed that TISCs expressed higher levels of stemness genes and Twist1[39].
It was known that HCV core protein induces the upregulation of transforming growth factor beta 1 (TGFβ1), showing a direct role in fibrogenesis[40]. A recent study on HCV core protein demonstrated that the TISCs obtained from the model had the capacity to recruit and activate fibroblasts in a xenograft, exhibited by high expression of fibrogenesis and EMT markers. It showed that in HCV infection, preneoplastic or tumorigenic state of the hepatocytes influenced the network for the tumor environment[41], presumably with the involvement of the hepatic cells stemness. As seen in NS5A transgenic mouse, a study in HCV core transgenic mouse with HCV core insertion showed a corresponding result. The TISCs isolated from this mouse were tumorigenic both in vitro and in vivo and the TLR4-Nanog pathway was necessary for the maintenance of tumorigenic properties[42].
The Wnt/β-catenin pathway might be the major or one of the major molecular mechanisms involved in the oncogenicity of HCV. The activation of this pathway was noted in transgenic expressions of both HCV core and NS5A proteins. Pharmacological inhibition or loss of the Wnt/β-catenin signal represses TISCs growth in vitro, and decreases the accumulation of TISCs in vivo[43].
Regarding the core protein, since HCV core is closely related with TGF-β pathway, it is expected that TGF-β is involved in the induced CSC population. A previous study showed that CSC generation by HCV core protein was dependent on the endoglin (CD105), a TGF-β receptor complex. Besides the increase of CSC proteins anti-apoptosis and proliferation are enhanced during infection or ectopic expression of HCV core[44].
As in HBV, epigenetic mechanisms such as DNA methylation could give a hint on the molecular mechanism of the oncogenicity HCV. It was shown that demethylation of CpGs induced Sal-like protein 4, an embryonic stem cell transcriptional regulator. This re-expression was noticed in subgroups of HCC associated with HBV or HCV infection[45].
Relevance of CSC markers in clinicopathological feature of HCC
The complexity of HCC showed that the heterogeneity is not limited among patients (intertumoral heterogeneity) but also within the same person (intratumoral heterogeneity). Cell morphology, molecular profile, and expression of specific markers can be used to stratify and classify discrete tumor subtypes[46]. Consequently, HBV and/or HCV infection contributes to phenotypic and molecular characteristics in hepatic cell populations, including the CSC.
Multiple clinical studies had shown that the high expression of CSC marker CD133 in HCC tissues, in particular in the cytoplasm, is correlated with a poor prognosis[47-51]. In addition to prognosis, CD90 is high-expressed in HCC nodule[52] and is correlated with HCC differentiation grades[53,54].
Protein analysis showed that the levels of hepatic CD133 were higher in HBV+ than those in HBV- HCC tissues[22], pointing to the oncogenicity of the PreS1. Recent data showed that in HBV-related HCC cases, CD133 in combination with the level of serum AFP, HCC could be subclassified into four subtypes, with different clinicopathological features and various prognosis. A high expressions of both CD133 and serum AFP was associated with a relatively poor prognosis[55].
However, a previous study in endemic HBV area showed that CD133 expression in HCC was negatively associated with the presence of HBsAg[56]. A histological analysis of human tissue found a positive correlation between HBV and CD90[57] but since co-staining of the CSC markers and the HBV proteins was not performed, it remains unclear if and how HBV alters the physiology of CD90+ and CD133+ CSC.
Recent data also showed a correlation between HBV and CSC EpCAM. High expression of HBx in human HBV-related HCC was also correlated with the expansion of EpCAM HCC cells; EpCAM expression was detected more frequently with HBV than with other etiologies. Further, in chemotherapy treated patients, EpCAM was strongly expressed, indicating its association with treatment resistance[24].
In contrast to the clear direct oncogenicity of HBV, the association between clinical and pathological characteristics and CSC markers in HCV-related HCC is still very limited. Recently it was demonstrated that CSC spheres induced by HCV were highly sensitive to cell death from sorafenib. It can be a basis for the development of new targeted therapies against hepatic CSC[35,58].
Perspectives
While the pathogenesis of HBV and HCV proteins in the development of HCC has been intensely investigated, information on their significance in the initiation of hepatic CSC is still very limited. It is because the theory of CSC in HCC is still relatively new and further evidence must be demonstrated. Moreover, even though this hypothesis is exciting, CSC theory in HCC is debatable and controversial. Recent studies had indicated that in gastrointestinal cancers, the so-called CSC should be defined as tumor-initiating cells/TISCs. These cells were not pluripotent, but bi- or multipotential to give rise to diverse tumor types and tumor initiation potential in mouse models[59]. The complexity of the liver, as well as the limitation in the experimental models, still limit the proof of the CSC concept. Further, genomic diversity and genetic characteristic of the virus (genotypes, subgenotypes, and quasispecies) significantly contribute to different clinical outcome and viral susceptibilities[33].
In addition to the type of the virus, another point to be considered is the state of the hepatic cells during viral exposure. A recent study had shown that the susceptibility of the hepatic cells to HCV was different during cellular maturation course. In this study, an epigenetic transduction by pluripotency factors reprogrammed mature cells into hepatic oval (progenitor) cells. In this progeny stage, cells lost their susceptibility to HCV infection and viral RNA replication. Upon hepatic differentiation, however, a permissiveness to HCV RNA replication was re-obtained. In contrast to HCV, in HBV infection, viral susceptibility was maintained along the course. It indicated that during hepatic maturation process, cells receptor susceptibility are specific to particular virus[60].
Even though basic in vitro studies and studies in transgenic animals, as well as clinical data from HCC patients, had shown expanded evidence on “stemness” oncogenicity of HBV and HCV, the mechanism of how viral particle induces hepatocarcinogenesis is still unclear and open for discussion. We presume that there would not be a single answer because of the complexity and heterogeneity of both virus and host factors. Finding strong evidence on this field will keep us busy for some time but the application of potentiality of this trip is intriguing.
Declarations
Authors’ contributionsDesigned the manuscript: Sukowati CHC, Tiribelli C
Wrote, read and approved the manuscript: Sukowati CHC, Reyes PAC, Tell G, Tiribelli C
Availability of data and materialsNot applicable.
Financial support and sponsorshipSukowati CHC was supported by a Research Grant of Associazione Italiana per la Ricerca sul Cancro (AIRC - #IG19862 to Tell G) and Reyes PAC by Philippine Council for Health Research and Development (PCHRD) and the Hepatology Society of the Philippines (HSP).
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
2. Bosetti C, Levi F, Boffetta P, Lucchini F, Negri E, et al. Trends in mortality from hepatocellular carcinoma in Europe, 1980-2004. Hepatology 2008;48:137-45.
5. Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell 2012;21:283-96.
7. Haraguchi N, Utsunomiya T, Inoue H, Tanaka F, Mimori K, et al. Characterization of a side population of cancer cells from human gastrointestinal system. Stem Cells Dayt Ohio 2006;24:506-13.
8. Chiba T, Kita K, Zheng YW, Yokosuka O, Saisho H, et al. Side population purified from hepatocellular carcinoma cells harbors cancer stem cell-like properties. Hepatology 2006;44:240-51.
9. Suetsugu A, Nagaki M, Aoki H, Motohashi T, Kunisada T, et al. Characterization of CD133+ hepatocellular carcinoma cells as cancer stem/progenitor cells. Biochem Biophys Res Commun 2006;351:820-4.
10. Ma S, Chan KW, Hu L, Lee TK, Wo JY, et al. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology 2007;132:2542-56.
11. Yin S, Li J, Hu C, Chen X, Yao M, et al. CD133 positive hepatocellular carcinoma cells possess high capacity for tumorigenicity. Int J Cancer 2007;120:1444-50.
12. Yang ZF, Ngai P, Ho DW, Yu WC, Ng MN, et al. Identification of local and circulating cancer stem cells in human liver cancer. Hepatology 2008;47:919-28.
13. Yang ZF, Ho DW, Ng MN, Lau CK, Yu WC, et al. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell 2008;13:153-66.
14. Yamashita T, Ji J, Budhu A, Forgues M, Yang W, et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology 2009;136:1012-24.
15. Kimura O, Takahashi T, Ishii N, Inoue Y, Ueno Y, et al. Characterization of the epithelial cell adhesion molecule (EpCAM)+ cell population in hepatocellular carcinoma cell lines. Cancer Sci 2010;101:2145-55.
16. Lee TK, Castilho A, Cheung VC, Tang KH, Ma S, et al. CD24(+) liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell 2011;9:50-63.
17. Haraguchi N, Ishii H, Mimori K, Tanaka F, Ohkuma M, et al. CD13 is a therapeutic target in human liver cancer stem cells. J Clin Invest 2010;120:3326-39.
19. Saitta C, Tripodi G, Barbera A, Bertuccio A, Smedile A, et al. Hepatitis B virus (HBV) DNA integration in patients with occult HBV infection and hepatocellular carcinoma. Liver Int 2015;35:2311-7.
20. Dunsford HA, Sell S, Chisari FV. Hepatocarcinogenesis due to chronic liver cell injury in hepatitis B virus transgenic mice. Cancer Res 1990;50:3400-7.
21. Anfuso B, El-Khobar KE, Ie SI, Avellini C, Radillo O, et al. Activation of hepatic stem cells compartment during hepatocarcinogenesis in a HBsAg HBV-transgenic mouse model. Sci Rep 2018;8:13168.
22. Liu Z, Dai X, Wang T, Zhang C, Zhang W, et al. Hepatitis B virus PreS1 facilitates hepatocellular carcinoma development by promoting appearance and self-renewal of liver cancer stem cells. Cancer Lett 2017;400:149-60.
23. Wang C, Yang W, Yan HX, Luo T, Zhang J, et al. Hepatitis B virus X (HBx) induces tumorigenicity of hepatic progenitor cells in 3,5-diethoxycarbonyl-1,4-dihydrocollidine-treated HBx transgenic mice. Hepatology 2012;55:108-20.
24. Kimura O, Kondo Y, Kogure T, Kakazu E, Ninomiya M, et al. Expression of EpCAM increases in the hepatitis B related and the treatment-resistant hepatocellular carcinoma. Biomed Res Int 2014;2014:172913.
25. Arzumanyan A, Friedman T, Ng IO, Clayton MM, Lian Z, et al. Does the hepatitis B antigen HBx promote the appearance of liver cancer stem cells? Cancer Res 2011;71:3701-8.
26. Li CH, Wang YJ, Dong W, Xiang S, Liang HF, et al. Hepatic oval cell lines generate hepatocellular carcinoma following transfection with HBx gene and treatment with aflatoxin B1 in vivo. Cancer Lett 2011;311:1-10.
27. Ng KY, Chai S, Tong M, Guan XY, Lin CH, et al. C-terminal truncated hepatitis B virus X protein promotes hepatocellular carcinogenesis through induction of cancer and stem cell-like properties. Oncotarget 2016;7:24005-17.
28. Ching RHH, Sze KMF, Lau EYT, Chiu YT, Lee JMF, et al. C-terminal truncated hepatitis B virus X protein regulates tumorigenicity, self-renewal and drug resistance via STAT3/Nanog signaling pathway. Oncotarget 2017;8:23507-16.
29. Fan H, Zhang H, Pascuzzi PE, Andrisani O. Hepatitis B virus X protein induces EpCAM expression via active DNA demethylation directed by RelA in complex with EZH2 and TET2. Oncogene 2015;35:715-26.
30. Zhang X, Liu S, Shen C, Wu Y, Zhang L, et al. DNA methylation consistency implicates the primary tumor cell origin of recurrent hepatocellular carcinoma. Epigenomics 2015;7:581-92.
31. Hamamoto H, Maemura K, Matsuo K, Taniguchi K, Tanaka Y, et al. Delta-like 3 is silenced by HBx via histone acetylation in HBV-associated HCCs. Sci Rep 2018;8:4842.
32. Liu J, Jiang J, Mo J, Liu D, Cao D, et al. Global DNA 5-hydroxymethylcytosine and 5-formylcytosine contents are decreased in the early stage of hepatocellular carcinoma. Hepatology 2018; doi: 10.1002/hep.30146.
33. Shlomai A, de Jong YP, Rice CM. Virus associated malignancies: the role of viral hepatitis in hepatocellular carcinoma. Semin Cancer Biol 2014;26:78-88.
34. Sukowati CH, El-Khobar KE, Ie SI, Anfuso B, Muljono DH, et al. Significance of hepatitis virus infection in the oncogenic initiation of hepatocellular carcinoma. World J Gastroenterol 2016;22:1497-512.
35. Kwon YC, Bose SK, Steele R, Meyer K, Di Bisceglie AM, et al. Promotion of cancer stem-like cell properties in hepatitis C virus-infected hepatocytes. J Virol 2015;89:11549-56.
36. Ali N, Allam H, May R, Sureban SM, Bronze MS, et al. Hepatitis C virus-induced cancer stem cell-like signatures in cell culture and murine tumor xenografts. J Virol 2011;85:12292-303.
37. Bartosch B, Thimme R, Blum HE, Zoulim F. Hepatitis C virus-induced hepatocarcinogenesis. J Hepatol 2009;51:810-20.
38. Machida K, Tsukamoto H, Mkrtchyan H, Duan L, Dynnyk A, et al. Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog. Proc Natl Acad Sci U S A 2009;106:1548-53.
39. Uthaya Kumar DB, Chen CL, Liu JC, Feldman DE, Sher LS, et al. TLR4 signaling via NANOG cooperates with STAT3 to activate Twist1 and promote formation of tumor-initiating stem-like cells in livers of mice. Gastroenterology 2016;150:707-19.
40. Taniguchi H, Kato N, Otsuka M, Goto T, Yoshida H, et al. Hepatitis C virus core protein upregulates transforming growth factor-beta 1 transcription. J Med Virol 2004;72:52-9.
41. Sasaki R, Devhare P, Ray RB, Ray R. Hepatitis C virus-induced tumor-initiating cancer stem-like cells activate stromal fibroblasts in a xenograft tumor model. Hepatology 2017;66:1766-78.
42. Machida K, Chen CL, Liu JC, Kashiwabara C, Feldman D, et al. Cancer stem cells generated by alcohol, diabetes, and hepatitis C virus. J Gastroenterol Hepatol 2012;27 Suppl 2:19-22.
43. Debebe A, Medina V, Chen CY, Mahajan IM, Jia C, et al. Wnt/β-catenin activation and macrophage induction during liver cancer development following steatosis. Oncogene 2017;36:6020-9.
44. Kwon YC, Sasaki R, Meyer K, Ray R. Hepatitis C virus core protein modulates endoglin (CD105) signaling pathway for liver pathogenesis. J Virol 2017; doi: 10.1128/JVI.01235-17.
45. Fan H, Cui Z, Zhang H, Mani SK, Diab A, et al. DNA demethylation induces SALL4 gene re-expression in subgroups of hepatocellular carcinoma associated with hepatitis B or C virus infection. Oncogene 2017;36:2435-45.
46. Anfuso B, El-Khobar KE, Sukowati CH, Tiribelli C. The multiple origin of cancer stem cells in hepatocellular carcinoma. Clin Res Hepatol Gastroenterol 2015;39 Suppl 1:S92-7.
47. Chen YL, Lin PY, Ming YZ, Huang WC, Chen RF, et al. The effects of the location of cancer stem cell marker CD133 on the prognosis of hepatocellular carcinoma patients. BMC Cancer 2017;17:474.
48. Chan AW, Tong JH, Chan SL, Lai PB, To KF. Expression of stemness markers (CD133 and EpCAM) in prognostication of hepatocellular carcinoma. Histopathology 2014;64:935-50.
49. Sasaki A, Kamiyama T, Yokoo H, Nakanishi K, Kubota K, et al. Cytoplasmic expression of CD133 is an important risk factor for overall survival in hepatocellular carcinoma. Oncol Rep 2010;24:537-46.
50. Ma YC, Yang JY, Yan LN. Relevant markers of cancer stem cells indicate a poor prognosis in hepatocellular carcinoma patients: a meta-analysis. Eur J Gastroenterol Hepatol 2013;25:1007-16.
51. Song W, Li H, Tao K, Li R, Song Z, et al. Expression and clinical significance of the stem cell marker CD133 in hepatocellular carcinoma. Int J Clin Pract 2008;62:1212-8.
52. Sukowati CHC, Anfuso B, Torre G, Francalanci P, Crocè LS, et al. The expression of CD90/Thy-1 in hepatocellular carcinoma: an in vivo and in vitro study. PLoS One 2013;8:e76830.
53. Liu R, Shen Y, Nan K, Mi B, Wu T, et al. Association between expression of cancer stem cell markers and poor differentiation of hepatocellular carcinoma: a meta-analysis (PRISMA). Medicine (Baltimore) 2015;94:e1306.
54. Lingala S, Cui YY, Chen X, Ruebner BH, Qian XF, et al. Immunohistochemical staining of cancer stem cell markers in hepatocellular carcinoma. Exp Mol Pathol 2010;89:27-35.
55. Dai XM, Yang SL, Zheng XM, Chen GG, Chen J, et al. CD133 expression and α-fetoprotein levels define novel prognostic subtypes of HBV-associated hepatocellular carcinoma: a long-term follow-up analysis. Oncol Lett 2018;15:2985-91.
56. Yeh CT, Kuo CJ, Lai MW, Chen TC, Lin CY, et al. CD133-positive hepatocellular carcinoma in an area endemic for hepatitis B virus infection. BMC Cancer 2009;9:324.
57. Lu JW, Chang JG, Yeh KT, Chen RM, Tsai JJ, et al. Overexpression of Thy1/CD90 in human hepatocellular carcinoma is associated with HBV infection and poor prognosis. Acta Histochem 2011;113:833-8.
58. Bose SK, Meyer K, Bisceglie AMD, Ray RB, Ray R. Hepatitis C virus induces epithelial-mesenchymal transition in primary human hepatocytes. J Virol 2012;86:13621-8.
59. Machida K. Existence of cancer stem cells in hepatocellular carcinoma: myth or reality? Hepatol Int 2017;11:143-7.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Sukowati CHC, Reyes PAC, Tell G, Tiribelli C. Oncogenicity of viral hepatitis B and C in the initiation of hepatic cancer stem cells. Hepatoma Res 2019;5:2. http://dx.doi.org/10.20517/2394-5079.2018.106
AMA Style
Sukowati CHC, Reyes PAC, Tell G, Tiribelli C. Oncogenicity of viral hepatitis B and C in the initiation of hepatic cancer stem cells. Hepatoma Research. 2019; 5: 2. http://dx.doi.org/10.20517/2394-5079.2018.106
Chicago/Turabian Style
Sukowati, Caecilia H. C., Peter A. C. Reyes, Gianluca Tell, Claudio Tiribelli. 2019. "Oncogenicity of viral hepatitis B and C in the initiation of hepatic cancer stem cells" Hepatoma Research. 5: 2. http://dx.doi.org/10.20517/2394-5079.2018.106
ACS Style
Sukowati, CHC.; Reyes PAC.; Tell G.; Tiribelli C. Oncogenicity of viral hepatitis B and C in the initiation of hepatic cancer stem cells. Hepatoma. Res. 2019, 5, 2. http://dx.doi.org/10.20517/2394-5079.2018.106
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 11 clicks
Cite This Article 11 clicks

 Like This Article 38
likes
Like This Article 38
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.