
The fibroblast growth factor receptor pathway in hepatocellular carcinoma


Abstract
Hepatocellular carcinoma is the third most common cause of cancer-related death globally and portends a poor prognosis. The fibroblast growth factor receptor (FGFR) pathway is increasingly acknowledged to play a role in the pathogenesis of hepatocellular carcinoma (HCC) and is postulated to be upregulated as a mechanism of resistance to anti-VEGF treatment. We attempt to review the importance of the FGFR pathway in HCC oncogenesis, as well as the current clinical evidence on the efficacy and safety of FGFR pathway inhibitors in HCC.
Keywords
Introduction
Hepatocellular carcinoma (HCC) is the third most common cause of cancer-related death globally[1]. Most patients have advanced disease on diagnosis. In unresectable advanced disease, sorafenib used to be the only available systemic therapy option available and prognosis was poor with a one-year survival rate of less than 50%[2].
HCC tumours harbour an average of 30-40 mutations, of which 20% may be driver mutations[3]. The molecular complexity and heterogeneity of HCC likely underlies the reason for failure of multiple phase III trials of targeted agents over the years. With improving technologies, we have been able to learn more about the molecular mechanisms underlying the oncogenesis of HCC, and in recent past have seen breakthroughs with several new drugs being added to our armamentarium both in the front-line and second-line setting[4], and many more compounds showing great promise on the horizon[5].
One signaling pathway that is increasingly recognized to play a role in the carcinogenesis of HCC is the fibroblast growth factor (FGF)/fibroblast growth factor receptor (FGFR) pathway, which has roles in oncogenesis, mediating cell proliferation and neo-angiogenesis[6,7]. Preclinical models suggest that inhibition of the FGFR pathway is a feasible therapeutic strategy[7] and many clinical trials using FGF/FGFR pathway inhibitors have since been conducted or are ongoing in hepatocellular carcinoma.
We attempt to review the importance of the FGF/FGFR pathway and current clinical evidence to date for use of the pathway inhibitors in HCC.
FGF/FGFR pathway and its aberrations in cancer
The human FGF family consists of 22 structurally related molecules that interact with four FGFRs. Each FGFR comprises three components, an extracellular domain which interacts with the FGF ligand, a transmembrane domain, and an intracellular domain. FGFs act as ligands which can bind to more than one kind of FGFR, causing downstream activation of several pathways including the mitogen-activated protein kinase pathway regulating cellular proliferation, and the phosphoinositide-3 kinase-Akt pathway controlling cellular survival[8]. FGF/FGFR signaling is involved in normal embryonic development of the liver and lungs[9] as well as adult wound healing and angiogenesis[10].
FGFRs are widely expressed in adult tissue, although their relative levels differ in the various organ systems. Under normal conditions, hepatocytes express high levels of FGFR3 and FGFR4 and have lower levels of FGFR1 and FGFR2[11].
FGFR signaling has significant effects on tumour neo-angiogenesis, both via the direct promotion of endothelial cell proliferation through effects on the tumour microenvironment[12], as well as indirectly via interactions and synergism with the vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF) pathways[13].
FGFR pathway activation has also been shown to be an important resistance mechanism in response to therapeutic pressure with use of anti-VEGF therapy[6,14]. In both the preclinical[15] and clinical[16] settings, tumours progressing on anti-VEGF treatment have been shown to have a higher level of expression of FGF2. As such, upfront dual inhibition of VEGFR and FGFR, or introduction of FGFR inhibition after progression on a VEGF pathway inhibitor[17] can potentially result in greater clinical benefit compared to inhibition of the VEGF pathway alone.
FGFR aberrations occur in approximately 7% of all solid tumours and in almost every tumour type, though the frequency and type of aberration differ[18]. Pathway aberrations identified include[19]: (1) gene amplification, or post-transcriptional changes giving rise to receptor overexpression; (2) gene mutations, resulting in constitutionally activated receptors or receptors that have a reduced dependence of ligand binding for activation; (3) translocations, resulting in expression of FGFR-fusion proteins with constitutive FGFR kinase activity; (4) alternative splicing of FGFR and isoform switching, changing ligand specificity and increasing the range of FGFs that can activate the FGFR; (5) upregulation of FGF ligand expression.
Overall the most common aberration seen in solid tumours is FGFR gene amplification, most commonly in FGFR1. FGFR mutations in cancer differ from those seen in hereditary disorders in that they are not limited to the kinase domains, but may occur in any part of the gene[19].
Relevance of the FGF pathway in HCC
The importance of the FGF/FGFR pathway in HCC can be seen in the fact that more than 80% of HCCs overexpress at least one FGF and/or FGFR[20]. The main FGFRs expressed in liver tissue are FGFR3[21] and FGFR4[22].
Whilst healthy hepatocytes express minimal levels of FGF1 or FGF2, these levels increase when there is cirrhosis and increasing levels correlate with the progression of cirrhosis into HCC. Higher levels of FGF1 and FGF2 are also seen in more advanced tumour stages[23]. There is hence interest in using FGF1 and FGF2 expression levels as a prognostic marker[24], though its utility as a diagnostic marker or for follow-up of HCC patients is limited by its non-specificity[25].
In preclinical models, FGF1 and FGF2 were shown to stimulate proliferation of HCC cell lines[26] through the activation of tumour invasion and angiogenesis resulting in an increase in capillarised sinusoids[27]. There is however substantial redundancy in FGF1- and FGF2-mediated signaling, suggesting that direct targeting of these ligands may have limited therapeutic efficacy[28].
The FGF8 subfamily, comprising FGFs 8, 17 and 18, also promotes oncogenesis through stimulating hepatocyte proliferation. At least one member of the FGF8 subfamily or its corresponding receptors FGFR2, FGFR3 and FGFR4 is upregulated in more than 50% of HCCs[20]. The use of small interfering RNA (siRNA) targeting FGF18 has been shown to reduce the viability and proliferation of HCC cells[20].
The FGF19 subfamily, comprising FGFs 19, 21 and 23, act as endocrine factors mediating metabolic effects through FGFR signaling. FGF19, which comes mainly from the ileum, plays a role in the physiological regulation of bile acid and cholesterol metabolism as well as insulin sensitivity. FGF19 binds exclusively to FGFR4 with the co-receptor β-Klotho (KLB) stabilising the interaction. FGF19/FGFR4 signaling is thought to be of particular importance in the carcinogenesis of HCC[29], with FGF19 expression increased, through focal amplification of 11q, in approximately 6%-12% of HCC cases[30]. FGFR4 expression is also upregulated in almost half of HCCs[31]. In addition, FGF19 levels may be prognostic, with higher expression in resected HCC specimens being associated with larger tumour size and stage and higher risk of recurrence after hepatectomy[32].
In vitro studies show that FGF19 induces HCC cell proliferation[29] and inhibits apoptosis[33]. Mice models also confirm that the ectopic expression of FGF19 promotes hepatocyte proliferation, dysplastic change and precipitates the formation of HCC[34]. Similarly, FGFR4 knockout mice showed increased hepatocyte injury when challenged with the hepato-toxin carbon tetrachloride[35]. Targeting the FGF19/FGFR4 interaction through various approaches appears to be effective in inhibiting hepatocarcinogenesis and HCC growth in preclinical models, be it through the use of a neutralizing antibody against FGF19[36], through genetic knockdown[30], or though siRNA[33]. Using siRNA to knockdown FGFR4 also showed similar results in mice models, which had impaired regeneration and increased liver injury after partial hepatectomy[37].
As previously mentioned, the FGF/FGFR pathway has been shown to be upregulated after initial blockade of the anti-VEGF pathway[38], and may be an important resistance mechanism to anti-VEGF therapy including that of sorafenib. For a long time, sorafenib was the only systemic treatment option for advanced HCC, having demonstrated an improvement in overall survival of 2-3 months in two large phase III trials[39,40]. Whilst having inhibitory effects on multiple targets including VEGFR, PDGFR and Raf kinases, sorafenib has no anti-FGFR activity[41]. Concomitant dual blockade of FGF/FGFR and VEGF pathways are hence a potentially attractive approach in the efforts to overcome this resistance[38].
Overview of FGF/FGFR pathway inhibitors and their toxicities
Current available inhibitors against the FGF/FGFR pathway can be classified into Figure 1: (1) monoclonal antibodies which competitively inhibit FGF binding to the FGFR extracellular domain; (2) FGF-ligand traps; and (3) small molecule tyrosine kinase inhibitors (FGFR TKIs).
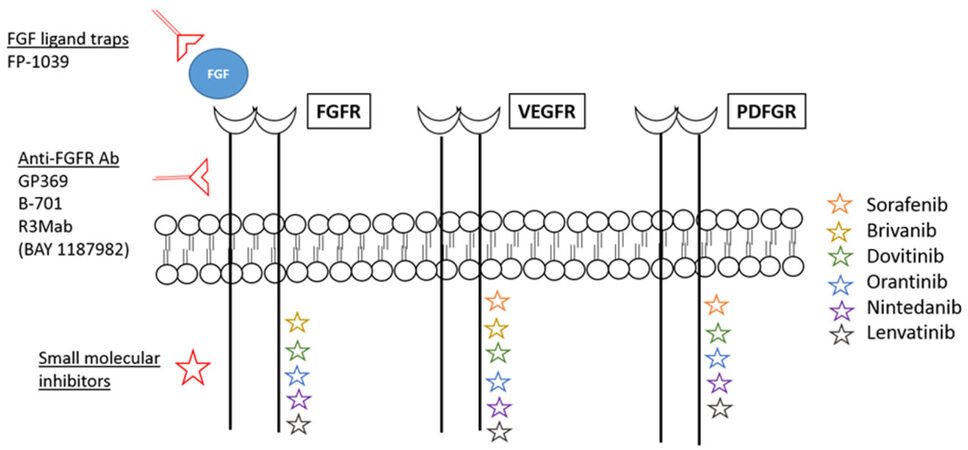
Most of the FGF/FGFR pathway inhibitors currently in development belong to the last category. These TKIs can be further divided into multi-kinase inhibitors and the selective FGFR TKIs.
Most of the multi-kinase inhibitors have inhibitory effects on both VEGFR and FGFR because of the structural similarities in the kinase domains of both receptors, though they may vary in their relative potency for inhibition for the two groups of receptors, with the majority having a higher potency for VEGFR than FGFR. Whilst multi-kinase inhibitors may potentially increase therapeutic efficacy by simultaneously disrupting resistance pathways, toxicity and off-target effects inevitably increase, which may limit the ability to achieve doses required for effective FGFR inhibition[19,42].
Selective FGFR inhibitors on the other hand, may have unique on-target dose-limiting toxicities. Preclinical models with selective FGFR TKIS caused hyperphosphataemia-mediated tissue calcification through the inhibition of FGF23 signaling in the kidney and bone, where it plays a critical role in vitamin D and phosphate homeostasis[43,44]. This was replicated in the clinical setting with 83% of patients treated at the maximum tolerated dose in the BGJ398 phase I trial developing hyperphosphataemia[45]. This resulted in repeated dose interruptions and reductions, and ultimately prompted trial sponsors to explore an alternative intermittent dosing schedule[45]. An increase in serum FGF23, phosphate and vitamin D levels is being studied as potential on-target biomarkers for effective FGFR inhibition[46]. Other mechanism-based toxicities observed in preclinical models and clinical studies include cutaneous toxicities such as nail toxicities, xerostomia, stomatitis, as well as dose-dependent keratopathy and retinal pigment epithelial detachment. Although multikinase VEGFR/FGFR inhibitors may cause hypertension and proteinuria, these problems seem to occur with a lesser frequency with selective FGFR inhibitors.
Completed clinical studies of FGF/FGFR pathway inhibitors in HCC
An overview of the completed clinical studies of FGF/FGFR pathway inhibitors in HCC is given below [Table 1].
Summary of completed clinical trials of FGFR multikinase inhibitors in hepatocellular carcinoma (adapted and updated from[76]
| Trial | Endpoints | |
|---|---|---|
| Brivanib | PII: 1L systemic therapy in advanced HCC[49] n = 55 NCT00355238 | 6m PFS 18.2% mPFS 2.7m mOS 10 m |
| PII: 2L systemic therapy in advanced HCC n = 41 NCT00355238 | mTTP 2 m | |
| PIII: 1L systemic therapy in advanced HCC (non-inferiority trial)[50] n = 1155 NCT00858871 | mOS 9.5 m (brivanib) vs. 9.9 m (sorafenib) | |
| PIII: 2L systemic therapy in advanced HCC n = 295 NCT00825955 | mOS 9.4 m (brivanib) vs. 8.2 m (placebo) (NS) mTTP 4.2 m (brivanib) vs. 2.8 m (placebo) (SS) ORR 10% (brivanib) vs. 2% (placebo) (SS) | |
| PIII: in combination with TACE as adjuvant[52] NCT00908752 | mOS 26.4 m (TACE/brivanib) vs. 26.1 m (TACE/placebo) | |
| Dovitinib | RPII: 1L systemic therapy in advanced HCC in Asia-Pacific population n = 165 NCT01232296 | mOS 8.0 m (dovitinib) vs. 8.4 m (sorafenib) mTTP 4.1 m (dovitinib) vs. 4.1 m (sorafenib) |
| Orantinib (TSU-68) | PI/II: any line systemic therapy advanced HCC[56] n = 12 (PI) n = 35 (PII) NCT00784290 | ORR: 2.9% CR, 5.7% PR, 42.8% SD mTTP 2.1 m, mOS 13.1 m |
| PIII: in combination with TACE as adjuvant[58] n = 889 NCT01465464 | mOS 31.1 m (TACE/orantinib) vs. 32.3 m (TACE/placebo) | |
| Nintedanib (BIBF 1120) | PI/RPII: 1L systemic therapy in advanced HCC in Western population[60] n = 93 (PII) NCT01004003 | mTTP 5.5 m (nintedanib) vs. 4.6 m (sorafenib) mOS 11.9 m (nintedanib) vs. 11.4 m (sorafenib) mPFS 5.3m (nintedanib) vs. 3.9m (sorafenib) G3 or higher AE 68% (nintedanib) vs. 90% (sorafenib) |
| PI/RPII: 1L systemic therapy in advanced HCC in Asian patients n = 95 (RPII)[61] NCT00987935 | mTTP 2.8 m (nintedanib) vs. 3.0 m (sorafenib) mOS 10.2 m (nintedanib) vs. 10.7 m (sorafenib) G3 or higher AE 56% (nintedanib) vs. 84% (sorafenib) | |
| Lenvatinib (E7080) | PII: 1L systemic therapy in advanced HCC in Asian patients[63] n = 46 NCT00946153 | mTTP 7.4 m mOS 18.7 m ORR 37% DCR 78% |
| RPIII: 1L systemic therapy in advanced HCC (non-inferiority trial)[64] n = 954 NCT01761266 | mOS 13.6 m (lenvatinib) vs. 12.3 m (sorafenib) |
Brivanib
Brivanib is a selective inhibitor of VEGFR2 and FGFR1. In preclinical studies, it attenuated hepatic fibrosis in vivo[47] and hence was postulated to be useful in slowing the progression of cirrhosis to HCC[48]. In a single-arm phase II study in advanced HCC, brivanib was shown to have anti-tumour activity in both the frontline and second-line setting, reporting a 6-month progression free survival rate of 18% when used as first line treatment[49]. The registration phase III trial (BRISK-FL) however was a negative trial, with brivanib failing to demonstrate non-inferiority to sorafenib in the first-line setting, though it had similar anti-tumour activity albeit a less well-tolerated safety profile with higher rates of drug discontinuation[50]. A second-line phase III study of brivanib against placebo after sorafenib failure or intolerance (BRISK-PS) also failed to show an overall survival advantage though it had a better improved time to progression and overall response rate[51]. Following the results of these two trials, the phase III trial of brivanib as adjuvant therapy to transarterial chemoembolization (TACE) was prematurely terminated though analysis similarly suggested no improvement in survival with brivanib use[52].
Dovitinib
Dovitinib is a non-selective FGFR inhibitor which also has effects on VEGFR, PDGFR, FGFR, c-KIT and other targets. In HCC xenograft models, dovitinib inhibited tumour growth and angiogenesis, and reduced the development of metastases and prolonged mouse survival[53]. In other preclinical work, it also induced apoptosis in sorafenib-resistant cell lines[54]. When translated to the clinical setting however, the randomized phase II study comparing dovitinib versus sorafenib as first-line treatment in advanced HCC in Asian-Pacific patients failed to show improved overall survival and anti-tumour activity with dovitinib. Of note though, subgroup analysis showed that the subset of patients with higher baseline plasma soluble VEGFR1 (sVEGFR1) levels had longer median overall survival[55], and although inconclusive, it suggests that the enrichment of a patient population through biomarker selection may be a feasible approach for future studies. No phase III trials were or are being conducted using dovitinib for the indication of HCC.
Orantinib (TSU-68)
Orantinib, a multi-kinase inhibitor of FGFR, VEGFR and PDGFR, showed promising efficacy in pretreated patients with advanced HCC, with 51% of patients achieving disease control, and a good safety profile in phase I/II HCC studies[56]. Following a similarly designed phase II study suggesting prolonged progression free survival[57], a randomized phase III trial was conducted in Asia in patients with unresectable HCC studying either orantinib or placebo after TACE. This study was however terminated early for futility after interim analysis showed no improvement in overall survival with the use of orantinib[58].
Nintedanib (BIBF 1120)
Nintedanib, a multikinase VEGFR/PDGFR/FGFR inhibitor, showed inhibition of HCC cell line growth in vitro and decreased tumour growth and angiogenesis in a xenograft mouse model of HCC[59]. Two phase I/randomized phase II trials comparing nintedanib and sorafenib in patients with unresectable HCC were performed in the Western population[60] and the Asian population[61] with similar results. Both trials reported similar overall survival and time to progression results with both drugs, with fewer serious drug-related adverse events but higher drug discontinuation rates. We await further studies of this compound in patients with advanced HCC.
Lenvantinb (E7080)
Lenvantinib is a multi-kinase inhibitor with inhibitory effects against VEGFR, FGFR1 - 4, KIT and RET. Although higher doses have been tested in other solid tumour types, a lower dose of 12 mg was tested in a phase I trial of lenvantinib in HCC patients[62], and used subsequently in a Phase II trial conducted in Japan and South Korea[63]. This led to the phase III study comparing lenvatinib and sorafenib in patients with unresectable HCC (REFLECT), showing non-inferiority of lenvatinib in terms of overall survival, and improvements in secondary endpoints of progressive free survival and objective response rate with lenvatinib[64]. Following this study, further studies of lenvatinib in advanced HCC are being conducted or planned, such as a trial studying the combination of lenvatinib and anti-programmed death 1 (anti-PD1) inhibitors in the first line setting (NCT03418922, NCT03006926), as well as a trial studying the safety and efficacy of subsequently second-line treatment after initial lenvatinib use (NCT03433703).
Ongoing clinical studies of other FGF/FGFR inhibitors in HCC
Although most of the completed clinical studies in HCC used multi-kinase inhibitors, several ongoing clinical studies are being conducted with promising selective FGFR inhibitors.
Erdafinitib (JNJ-4276493)
Erdafinitib is an oral selective pan-FGFR inhibitor which has shown a manageable safety profile in a phase I study in advanced or refractory solid tumours. Common drug-related adverse events encountered in the phase I study included hyperphosphataemia, nausea, stomatitis and dysguesia, with one dose-limiting toxicity of bilateral retinal pigment epithelium detachment necessitating treatment discontinuation[65]. An ongoing phase I/IIa study is currently recruiting targeting Asian patients with advanced HCC with FGF19 amplification (NCT02421185). Phase II and III trials are also being conducted with the drug in other tumour types, and notably, the drug received FDA breakthrough therapy designation in the treatment of FGFR-alteration positive urothelial cancer recently, following promising results in a phase II clinical trial[66].
BLU-554
BLU-554, a selective and potent inhibitor of FGFR4, was derived from an earlier compound BLU9931 which suppressed proliferation in HCC tumour xenograft models with an activated FGFR4 signaling pathway[67]. A phase I first-in-human study of BLU-554 in patients with HCC (NCT02508467) is ongoing, and preliminary results reported suggest promising clinical activity in FGF19 immunohistochemistry positive (IHC+) patients who have failed prior systemic therapy[68].
Other promising FGF/FGFR inhibitors in clinical studies
BGJ398
BGJ398 is a selective and potent pan-FGFR inhibitor which has shown to have preliminary clinical activity in a variety of solid tumours including FGFR3-mutant bladder and urothelial cancers, FGFR1-dependent squamous lung and head and neck cancers[45] as well as FGFR-altered cholangiocarcinoma[69]. Ongoing clinical trials are being conducted and/or planned in the above tumour types.
AZD4547
AZD4547 is a selective FGFR1 - 3 inhibitor with activity in FGFR2-amplied gastric cancer models[70] as well as FGFR1-amplified NSCLC models[71]. The randomised phase II trial in FGFR2-amplified gastric cancer did not show an improved progression free survival for AZD4547 compared to paclitaxel though exploratory biomarker analyses suggests that marked intratumoural heterogeneity of FGF2 amplification could have contributed to the negative results[72]. The phase II/III study of AZD4547 as second-line therapy in treating FGFR-positive patients with stage IV squamous cell lung cancer is ongoing (NCT02965378).
Anti-FGFR antibodies
GP369, a monoclonal antibody against the extracellular domain of the FGFR2-IIIB receptor has shown potent anti-tumour activity in breast and gastric cancer cell lines with FGFR2 amplification[73]. MFGR1877S (R3Mab) (NCT01363024) and B-701, both monoclonal antibodies targeting FGFR3, show promise in urothelial cancers, with the latter compound being tested in combination with pembrolizumab in the second line-setting (NCT03123055).
On the other hand, the auristatin-based antibody drug conjugate BAY 1187982 also shows significant tumour growth inhibition in models of FGF2 amplified human gastric and breast cancers[74], which led to a phase I dose-escalation trial in FGFR2-expressing solid tumours (NCT02368951) though the trial had to be terminated early due to concerns over toxicity.
FGF-ligand traps
FP-1039 comprises of a soluble fusion protein consisting of extracellular FGFR1-IIIc fused to the Fc domain of IgG1 hence acting as a ligand trap of FGF1, FGF2 and FGF4. A phase II trial is currently recruiting to study FP-1039 alone and in combination with chemotherapy (docetaxel or paclitaxel and carboplatin) in solid tumours (NCT01868022).
Conclusion
Although the majority of clinical studies with FGF/FGFR pathway inhibitors have been negative in hepatocellular carcinoma aside from REFLECT, the results suggest that these compounds do have anti-tumoural activity and better biomarker-based enrichment of a target population is likely the key in planning more successful future trials[75]. Several ongoing clinical trials of FGF/FGFR pathway inhibitors in a biomarker-enriched population are ongoing and we await the results of these promising studies.
Declarations
Authors’ contributionsThe two authors are responsible for all the work of this article.
Availability of data and materialsThe data were strictly obtained from medical records according to the privacy policy and ethics code of our institute.
Financial support and sponsorshipNone.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011;61:69-90.
2. Altekruse SF, McGlynn KA, Reichman ME. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol 2009;27:1485-91.
3. Llovet JM, Villanueva A, Lachenmayer A, Finn RS. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat Rev Clin Oncol 2015;12:408-24.
4. Bruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G, Pracht M, Yokosuka O, Rosmorduc O, Breder V, Gerolami R, Masi G, Ross PJ, Song T, Bronowicki JP, Ollivier-Hourmand I, Kudo M, Cheng AL, Llovet JM, Finn RS, LeBerre MA, Baumhauer A, Meinhardt G, Han G, Investigators R. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017;389:56-66.
5. Kelley RK, Verslype C, Cohn AL, Yang TS, Su WC, Burris H, Braiteh F, Vogelzang N, Spira A, Foster P, Lee Y, Van Cutsem E. Cabozantinib in hepatocellular carcinoma: results of a phase 2 placebo-controlled randomized discontinuation study. Ann Oncol 2017;28:528-34.
6. Lieu C, Heymach J, Overman M, Tran H, Kopetz S. Beyond VEGF: inhibition of the fibroblast growth factor pathway and antiangiogenesis. Clin Cancer Res 2011;17:6130-9.
7. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 2010;10:116-29.
8. Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov 2009;8:235-53.
9. Serls AE, Doherty S, Parvatiyar P, Wells JM, Deutsch GH. Different thresholds of fibroblast growth factors pattern the ventral foregut into liver and lung. Development 2005;132:35-47.
10. Werner S, Peters KG, Longaker MT, Fuller-Pace F, Banda MJ, Williams LT. Large induction of keratinocyte growth factor expression in the dermis during wound healing. Proc Natl Acad Sci U S A 1992;89:6896-900.
11. Hughes SE. Differential expression of the fibroblast growth factor receptor (FGFR) multigene family in normal human adult tissues. J Histochem Cytochem 1997;45:1005-19.
12. Streuli CH, Akhtar N. Signal co-operation between integrins and other receptor systems. Biochem J 2009;418:491-506.
13. Nissen LJ, Cao R, Hedlund EM, Wang Z, Zhao X, Wetterskog D, Funa K, Brakenhielm E, Cao Y. Angiogenic factors FGF2 and PDGF-BB synergistically promote murine tumor neovascularization and metastasis. J Clin Invest 2007;117:2766-77.
14. Tille JC, Wood J, Mandriota SJ, Schnell C, Ferrari S, Mestan J, Zhu Z, Witte L, Pepper MS. Vascular endothelial growth factor (VEGF) receptor-2 antagonists inhibit VEGF- and basic fibroblast growth factor-induced angiogenesis in vivo and in vitro. J Pharmacol Exp Ther 2001;299:1073-85.
15. Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005;8:299-309.
16. Kopetz S, Hoff PM, Morris JS, Wolff RA, Eng C, Glover KY, Adinin R, Overman MJ, Valero V, Wen S, Lieu C, Yan S, Tran HT, Ellis LM, Abbruzzese JL, Heymach JV. Phase II trial of infusional fluorouracil, irinotecan, and bevacizumab for metastatic colorectal cancer: efficacy and circulating angiogenic biomarkers associated with therapeutic resistance. J Clin Oncol 2010;28:453-9.
17. Allen E, Walters IB, Hanahan D. Brivanib, a dual FGF/VEGF inhibitor, is active both first and second line against mouse pancreatic neuroendocrine tumors developing adaptive/evasive resistance to VEGF inhibition. Clin Cancer Res 2011;17:5299-310.
18. Helsten T, Elkin S, Arthur E, Tomson BN, Carter J, Kurzrock R. The FGFR landscape in cancer: analysis of 4,853 tumors by next-generation sequencing. Clin Cancer Res 2016;22:259-67.
19. Dienstmann R, Rodon J, Prat A, Perez-Garcia J, Adamo B, Felip E, Cortes J, Iafrate AJ, Nuciforo P, Tabernero J. Genomic aberrations in the FGFR pathway: opportunities for targeted therapies in solid tumors. Ann Oncol 2014;25:552-63.
20. Gauglhofer C, Sagmeister S, Schrottmaier W, Fischer C, Rodgarkia-Dara C, Mohr T, Stattner S, Bichler C, Kandioler D, Wrba F, Schulte-Hermann R, Holzmann K, Grusch M, Marian B, Berger W, Grasl-Kraupp B. Up-regulation of the fibroblast growth factor 8 subfamily in human hepatocellular carcinoma for cell survival and neoangiogenesis. Hepatology 2011;53:854-64.
21. Qiu WH, Zhou BS, Chu PG, Chen WG, Chung C, Shih J, Hwu P, Yeh C, Lopez R, Yen Y. Over-expression of fibroblast growth factor receptor 3 in human hepatocellular carcinoma. World J Gastroenterol 2005;11:5266-72.
22. French DM, Lin BC, Wang M, Adams C, Shek T, Hotzel K, Bolon B, Ferrando R, Blackmore C, Schroeder K, Rodriguez LA, Hristopoulos M, Venook R, Ashkenazi A, Desnoyers LR. Targeting FGFR4 inhibits hepatocellular carcinoma in preclinical mouse models. PLoS One 2012;7:e36713.
23. Tsunematsu H, Tatsumi T, Kohga K, Yamamoto M, Aketa H, Miyagi T, Hosui A, Hiramatsu N, Kanto T, Hayashi N, Takehara T. Fibroblast growth factor-2 enhances NK sensitivity of hepatocellular carcinoma cells. Int J Cancer 2012;130:356-64.
24. Poon RT, Ng IO, Lau C, Yu WC, Fan ST, Wong J. Correlation of serum basic fibroblast growth factor levels with clinicopathologic features and postoperative recurrence in hepatocellular carcinoma. Am J Surg 2001;182:298-304.
25. Hsu PI, Chow NH, Lai KH, Yang HB, Chan SH, Lin XZ, Cheng JS, Huang JS, Ger LP, Huang SM, Yen MY, Yang YF. Implications of serum basic fibroblast growth factor levels in chronic liver diseases and hepatocellular carcinoma. Anticancer Res 1997;17:2803-9.
26. Asada N, Tanaka Y, Hayashido Y, Toratani S, Kan M, Kitamoto M, Nakanishi T, Kajiyama G, Chayama K, Okamoto T. Expression of fibroblast growth factor receptor genes in human hepatoma-derived cell lines. In Vitro Cell Dev Biol Anim 2003;39:321-8.
27. Motoo Y, Sawabu N, Yamaguchi Y, Terada T, Nakanuma Y. Sinusoidal capillarization of human hepatocellular carcinoma: possible promotion by fibroblast growth factor. Oncology 1993;50:270-4.
28. Sandhu DS, Baichoo E, Roberts LR. Fibroblast growth factor signaling in liver carcinogenesis. Hepatology 2014;59:1166-73.
29. Wu X, Ge H, Lemon B, Vonderfecht S, Weiszmann J, Hecht R, Gupte J, Hager T, Wang Z, Lindberg R, Li Y. FGF19-induced hepatocyte proliferation is mediated through FGFR4 activation. J Biol Chem 2010;285:5165-70.
30. Sawey ET, Chanrion M, Cai C, Wu G, Zhang J, Zender L, Zhao A, Busuttil RW, Yee H, Stein L, French DM, Finn RS, Lowe SW, Powers S. Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by oncogenomic screening. Cancer Cell 2011;19:347-58.
31. Alvarez-Sola G, Uriarte I, Latasa MU, Urtasun R, Barcena-Varela M, Elizalde M, Jimenez M, Rodriguez-Ortigosa CM, Corrales FJ, Fernandez-Barrena MG, Berasain C, Avila MA. Fibroblast Growth Factor 15/19 in Hepatocarcinogenesis. Dig Dis 2017;35:158-65.
32. Hyeon J, Ahn S, Lee JJ, Song DH, Park CK. Expression of fibroblast growth factor 19 is associated with recurrence and poor prognosis of hepatocellular carcinoma. Dig Dis Sci 2013;58:1916-22.
33. Miura S, Mitsuhashi N, Shimizu H, Kimura F, Yoshidome H, Otsuka M, Kato A, Shida T, Okamura D, Miyazaki M. Fibroblast growth factor 19 expression correlates with tumor progression and poorer prognosis of hepatocellular carcinoma. BMC Cancer 2012;12:56.
34. Nicholes K, Guillet S, Tomlinson E, Hillan K, Wright B, Frantz GD, Pham TA, Dillard-Telm L, Tsai SP, Stephan JP, Stinson J, Stewart T, French DM. A mouse model of hepatocellular carcinoma: ectopic expression of fibroblast growth factor 19 in skeletal muscle of transgenic mice. Am J Pathol 2002;160:2295-307.
35. Yu C, Wang F, Jin C, Wu X, Chan WK, McKeehan WL. Increased carbon tetrachloride-induced liver injury and fibrosis in FGFR4-deficient mice. Am J Pathol 2002;161:2003-10.
36. Desnoyers LR, Pai R, Ferrando RE, Hotzel K, Le T, Ross J, Carano R, D'Souza A, Qing J, Mohtashemi I, Ashkenazi A, French DM. Targeting FGF19 inhibits tumor growth in colon cancer xenograft and FGF19 transgenic hepatocellular carcinoma models. Oncogene 2008;27:85-97.
37. Padrissa-Altes S, Bachofner M, Bogorad RL, Pohlmeier L, Rossolini T, Bohm F, Liebisch G, Hellerbrand C, Koteliansky V, Speicher T, Werner S. Control of hepatocyte proliferation and survival by FGF receptors is essential for liver regeneration in mice. Gut 2015;64:1444-53.
38. Gao L, Wang X, Tang Y, Huang S, Hu CA, Teng Y. FGF19/FGFR4 signaling contributes to the resistance of hepatocellular carcinoma to sorafenib. J Exp Clin Cancer Res 2017;36:8.
39. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, Bolondi L, Greten TF, Galle PR, Seitz JF, Borbath I, Haussinger D, Giannaris T, Shan M, Moscovici M, Voliotis D, Bruix J, Group SIS. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359:378-90.
40. Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, Luo R, Feng J, Ye S, Yang TS, Xu J, Sun Y, Liang H, Liu J, Wang J, Tak WY, Pan H, Burock K, Zou J, Voliotis D, Guan Z. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol 2009;10:25-34.
41. Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther 2008;7:3129-40.
42. Gavine PR, Mooney L, Kilgour E, Thomas AP, Al-Kadhimi K, Beck S, Rooney C, Coleman T, Baker D, Mellor MJ, Brooks AN, Klinowska T. AZD4547: an orally bioavailable, potent, and selective inhibitor of the fibroblast growth factor receptor tyrosine kinase family. Cancer Res 2012;72:2045-56.
43. Wohrle S, Bonny O, Beluch N, Gaulis S, Stamm C, Scheibler M, Muller M, Kinzel B, Thuery A, Brueggen J, Hynes NE, Sellers WR, Hofmann F, Graus-Porta D. FGF receptors control vitamin D and phosphate homeostasis by mediating renal FGF-23 signaling and regulating FGF-23 expression in bone. J Bone Miner Res 2011;26:2486-97.
44. Gattineni J, Alphonse P, Zhang Q, Mathews N, Bates CM, Baum M. Regulation of renal phosphate transport by FGF23 is mediated by FGFR1 and FGFR4. Am J Physiol Renal Physiol 2014;306:F351-8.
45. Nogova L, Sequist LV, Perez Garcia JM, Andre F, Delord JP, Hidalgo M, Schellens JH, Cassier PA, Camidge DR, Schuler M, Vaishampayan U, Burris H, Tian GG, Campone M, Wainberg ZA, Lim WT, LoRusso P, Shapiro GI, Parker K, Chen X, Choudhury S, Ringeisen F, Graus-Porta D, Porter D, Isaacs R, Buettner R, Wolf J. Evaluation of BGJ398, a fibroblast growth factor receptor 1-3 kinase inhibitor, in patients with advanced solid tumors harboring genetic alterations in fibroblast growth factor receptors: results of a global phase I, dose-escalation and dose-expansion study. J Clin Oncol 2017;35:157-65.
46. Dienstmann R, Brana I, Rodon J, Tabernero J. Toxicity as a biomarker of efficacy of molecular targeted therapies: focus on EGFR and VEGF inhibiting anticancer drugs. Oncologist 2011;16:1729-40.
47. Nakamura I, Zakharia K, Banini BA, Mikhail DS, Kim TH, Yang JD, Moser CD, Shaleh HM, Thornburgh SR, Walters I, Roberts LR. Brivanib attenuates hepatic fibrosis in vivo and stellate cell activation in vitro by inhibition of FGF, VEGF and PDGF signaling. PLoS One 2014;9:e92273.
48. Huynh H, Ngo VC, Fargnoli J, Ayers M, Soo KC, Koong HN, Thng CH, Ong HS, Chung A, Chow P, Pollock P, Byron S, Tran E. Brivanib alaninate, a dual inhibitor of vascular endothelial growth factor receptor and fibroblast growth factor receptor tyrosine kinases, induces growth inhibition in mouse models of human hepatocellular carcinoma. Clin Cancer Res 2008;14:6146-53.
49. Park JW, Finn RS, Kim JS, Karwal M, Li RK, Ismail F, Thomas M, Harris R, Baudelet C, Walters I, Raoul JL. Phase II, open-label study of brivanib as first-line therapy in patients with advanced hepatocellular carcinoma. Clin Cancer Res 2011;17:1973-83.
50. Johnson PJ, Qin S, Park JW, Poon RT, Raoul JL, Philip PA, Hsu CH, Hu TH, Heo J, Xu J, Lu L, Chao Y, Boucher E, Han KH, Paik SW, Robles-Avina J, Kudo M, Yan L, Sobhonslidsuk A, Komov D, Decaens T, Tak WY, Jeng LB, Liu D, Ezzeddine R, Walters I, Cheng AL. Brivanib versus sorafenib as first-line therapy in patients with unresectable, advanced hepatocellular carcinoma: results from the randomized phase III BRISK-FL study. J Clin Oncol 2013;31:3517-24.
51. Llovet JM, Decaens T, Raoul JL, Boucher E, Kudo M, Chang C, Kang YK, Assenat E, Lim HY, Boige V, Mathurin P, Fartoux L, Lin DY, Bruix J, Poon RT, Sherman M, Blanc JF, Finn RS, Tak WY, Chao Y, Ezzeddine R, Liu D, Walters I, Park JW. Brivanib in patients with advanced hepatocellular carcinoma who were intolerant to sorafenib or for whom sorafenib failed: results from the randomized phase III BRISK-PS study. J Clin Oncol 2013;31:3509-16.
52. Kudo M, Han G, Finn RS, Poon RT, Blanc JF, Yan L, Yang J, Lu L, Tak WY, Yu X, Lee JH, Lin SM, Wu C, Tanwandee T, Shao G, Walters IB, Dela Cruz C, Poulart V, Wang JH. Brivanib as adjuvant therapy to transarterial chemoembolization in patients with hepatocellular carcinoma: a randomized phase III trial. Hepatology 2014;60:1697-707.
53. Huynh H, Chow PK, Tai WM, Choo SP, Chung AY, Ong HS, Soo KC, Ong R, Linnartz R, Shi MM. Dovitinib demonstrates antitumor and antimetastatic activities in xenograft models of hepatocellular carcinoma. J Hepatol 2012;56:595-601.
54. Tai WT, Cheng AL, Shiau CW, Liu CY, Ko CH, Lin MW, Chen PJ, Chen KF. Dovitinib induces apoptosis and overcomes sorafenib resistance in hepatocellular carcinoma through SHP-1-mediated inhibition of STAT3. Mol Cancer Ther 2012;11:452-63.
55. Cheng AL, Thongprasert S, Lim HY, Sukeepaisarnjaroen W, Yang TS, Wu CC, Chao Y, Chan SL, Kudo M, Ikeda M, Kang YK, Pan H, Numata K, Han G, Balsara B, Zhang Y, Rodriguez AM, Zhang Y, Wang Y, Poon RT. Randomized, open-label phase 2 study comparing frontline dovitinib versus sorafenib in patients with advanced hepatocellular carcinoma. Hepatology 2016;64:774-84.
56. Kanai F, Yoshida H, Tateishi R, Sato S, Kawabe T, Obi S, Kondo Y, Taniguchi M, Tagawa K, Ikeda M, Morizane C, Okusaka T, Arioka H, Shiina S, Omata M. A phase I/II trial of the oral antiangiogenic agent TSU-68 in patients with advanced hepatocellular carcinoma. Cancer Chemother Pharmacol 2011;67:315-24.
57. Inaba Y, Kanai F, Aramaki T, Yamamoto T, Tanaka T, Yamakado K, Kaneko S, Kudo M, Imanaka K, Kora S, Nishida N, Kawai N, Seki H, Matsui O, Arioka H, Arai Y. A randomised phase II study of TSU-68 in patients with hepatocellular carcinoma treated by transarterial chemoembolisation. Eur J Cancer 2013;49:2832-40.
58. Kudo M, Cheng AL, Park JW, Park JH, Liang PC, Hidaka H, Izumi N, Heo J, Lee YJ, Sheen IS, Chiu CF, Arioka H, Morita S, Arai Y. Orantinib versus placebo combined with transcatheter arterial chemoembolisation in patients with unresectable hepatocellular carcinoma (ORIENTAL): a randomised, double-blind, placebo-controlled, multicentre, phase 3 study. Lancet Gastroenterol Hepatol 2018;3:37-46.
59. Kudo K, Arao T, Tanaka K, Nagai T, Furuta K, Sakai K, Kaneda H, Matsumoto K, Tamura D, Aomatsu K, De Velasco MA, Fujita Y, Saijo N, Kudo M, Nishio K. Antitumor activity of BIBF 1120, a triple angiokinase inhibitor, and use of VEGFR2+pTyr+ peripheral blood leukocytes as a pharmacodynamic biomarker in vivo. Clin Cancer Res 2011;17:1373-81.
60. Palmer DH, Ma YT, Peck-Radosavljevic M, Ross P, Graham J, Fartoux L, Deptala A, Studeny M, Schnell D, Hocke J, Loembe AB, Meyer T. A multicentre, open-label, phase-I/randomised phase-II study to evaluate safety, pharmacokinetics, and efficacy of nintedanib vs. sorafenib in European patients with advanced hepatocellular carcinoma. Br J Cancer 2018;118:1162-8.
61. Cheng AL, Yen CJ, Kim TY, Feng YH, Chao Y, Lin DY. Efficacy and safety of nintedanib versus sorafenib in Asian patients with advanced hepatocellular carcinoma (HCC): a randomised phase II trial. J Clin Oncol 2013;33:339.
62. Ikeda M, Okusaka T, Mitsunaga S, Ueno H, Tamai T, Suzuki T, Hayato S, Kadowaki T, Okita K, Kumada H. Safety and pharmacokinetics of lenvatinib in patients with advanced hepatocellular carcinoma. Clin Cancer Res 2016;22:1385-94.
63. Ikeda K, Kudo M, Kawazoe S, Osaki Y, Ikeda M, Okusaka T, Tamai T, Suzuki T, Hisai T, Hayato S, Okita K, Kumada H. Phase 2 study of lenvatinib in patients with advanced hepatocellular carcinoma. J Gastroenterol 2017;52:512-9.
64. Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, Baron A, Park JW, Han G, Jassem J, Blanc JF, Vogel A, Komov D, Evans TRJ, Lopez C, Dutcus C, Guo M, Saito K, Kraljevic S, Tamai T, Ren M, Cheng AL. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet 2018;391:1163-73.
65. Tabernero J, Bahleda R, Dienstmann R, Infante JR, Mita A, Italiano A, Calvo E, Moreno V, Adamo B, Gazzah A, Zhong B, Platero SJ, Smit JW, Stuyckens K, Chatterjee-Kishore M, Rodon J, Peddareddigari V, Luo FR, Soria JC. Phase I dose-escalation study of JNJ-42756493, an oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J Clin Oncol 2015;33:3401-8.
66. Loriot Y, Necchi A, Park SH, García-Donas J, Huddart RA, Burgess EF, Fleming MT, Rezazadeh A, Mellado B, Varlamov S, Joshi M, Duran I, OHagan A OH, Avadhani AN, Zhong B, Stuyckens K, Dosne AG, Siefker-Radtke AO, Group OBotBS OB. Erdafitinib (ERDA; JNJ-42756493), a pan-fibroblast growth factor receptor (FGFR) inhibitor, in patients (pts) with metastatic or unresectable urothelial carcinoma (mUC) and FGFR alterations (FGFRa): phase 2 continuous versus intermittent dosing. J Clin Oncol 2018;36:411.
67. Hagel M, Miduturu C, Sheets M, Rubin N, Weng W, Stransky N, Bifulco N, Kim JL, Hodous B, Brooijmans N, Shutes A, Winter C, Lengauer C, Kohl NE, Guzi T. First selective small molecule inhibitor of FGFR4 for the treatment of hepatocellular carcinomas with an activated FGFR4 signaling pathway. Cancer Discov 2015;5:424-37.
68. Kim R, Sarker D, Macarulla T, Yau T, Choo SP, Meyer T, Hollebecque A, Whisenant J, Sung M, Yoon JH, Lim HY, Zhu A, Park JW, Faivre S, Mazzaferro V, Shi H, Schmidt-Kittler O, Clifford C, Wolf B, Kang YK. 365OPhase 1 safety and clinical activity of BLU-554 in advanced hepatocellular carcinoma (HCC). Ann Oncol 2017;28.
69. Javle M, Lowery M, Shroff RT, Weiss KH, Springfeld C, Borad MJ, Ramanathan RK, Goyal L, Sadeghi S, Macarulla T, El-Khoueiry A, Kelley RK, Borbath I, Choo SP, Oh DY, Philip PA, Chen LT, Reungwetwattana T, Van Cutsem E, Yeh KH, Ciombor K, Finn RS, Patel A, Sen S, Porter D, Isaacs R, Zhu AX, Abou-Alfa GK, Bekaii-Saab T. Phase II Study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J Clin Oncol 2018;36:276-82.
70. Xie L, Su X, Zhang L, Yin X, Tang L, Zhang X, Xu Y, Gao Z, Liu K, Zhou M, Gao B, Shen D, Zhang L, Ji J, Gavine PR, Zhang J, Kilgour E, Zhang X, Ji Q. FGFR2 gene amplification in gastric cancer predicts sensitivity to the selective FGFR inhibitor AZD4547. Clin Cancer Res 2013;19:2572-83.
71. Zhang J, Zhang L, Su X, Li M, Xie L, Malchers F, Fan S, Yin X, Xu Y, Liu K, Dong Z, Zhu G, Qian Z, Tang L, Schottle J, Zhan P, Ji Q, Kilgour E, Smith PD, Brooks AN, Thomas RK, Gavine PR. Translating the therapeutic potential of AZD4547 in FGFR1-amplified non-small cell lung cancer through the use of patient-derived tumor xenograft models. Clin Cancer Res 2012;18:6658-67.
72. Van Cutsem E, Bang YJ, Mansoor W, Petty RD, Chao Y, Cunningham D, Ferry DR, Smith NR, Frewer P, Ratnayake J, Stockman PK, Kilgour E, Landers D. A randomized, open-label study of the efficacy and safety of AZD4547 monotherapy versus paclitaxel for the treatment of advanced gastric adenocarcinoma with FGFR2 polysomy or gene amplification. Ann Oncol 2017;28:1316-24.
73. Bai A, Meetze K, Vo NY, Kollipara S, Mazsa EK, Winston WM, Weiler S, Poling LL, Chen T, Ismail NS, Jiang J, Lerner L, Gyuris J, Weng Z. GP369, an FGFR2-IIIb-specific antibody, exhibits potent antitumor activity against human cancers driven by activated FGFR2 signaling. Cancer Res 2010;70:7630-9.
74. Sommer A, Kopitz C, Schatz CA, Nising CF, Mahlert C, Lerchen HG, Stelte-Ludwig B, Hammer S, Greven S, Schuhmacher J, Braun M, Zierz R, Wittemer-Rump S, Harrenga A, Dittmer F, Reetz F, Apeler H, Jautelat R, Huynh H, Ziegelbauer K, Kreft B. Preclinical efficacy of the auristatin-based antibody-drug conjugate BAY 1187982 for the treatment of FGFR2-positive solid tumors. Cancer Res 2016;76:6331-9.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Lee JJX, Choo SP. The fibroblast growth factor receptor pathway in hepatocellular carcinoma. Hepatoma Res 2018;4:52. http://dx.doi.org/10.20517/2394-5079.2018.42
AMA Style
Lee JJX, Choo SP. The fibroblast growth factor receptor pathway in hepatocellular carcinoma. Hepatoma Research. 2018; 4: 52. http://dx.doi.org/10.20517/2394-5079.2018.42
Chicago/Turabian Style
Lee, Joycelyn Jie Xin, Su Pin Choo. 2018. "The fibroblast growth factor receptor pathway in hepatocellular carcinoma" Hepatoma Research. 4: 52. http://dx.doi.org/10.20517/2394-5079.2018.42
ACS Style
Lee, JJX.; Choo SP. The fibroblast growth factor receptor pathway in hepatocellular carcinoma. Hepatoma. Res. 2018, 4, 52. http://dx.doi.org/10.20517/2394-5079.2018.42
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 7 clicks
Cite This Article 7 clicks

 Like This Article 6
likes
Like This Article 6
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.