
Aberrant pre-mRNA splicing regulation in the development of hepatocellular carcinoma


Abstract
Alternative splicing is a highly regulated process that plays a critical role in diversification of the transcriptome and proteome in the cell. Several diseases, including different types of cancers, have been associated with aberrant regulation of alternative splicing. Thus, correcting alternative splicing is an attractive strategy to restore normal cell physiology in patients with cancer including hepatocellular carcinoma (HCC). This review summarizes the role of alternative splicing events related to HCC and potential therapeutic applications for it.
Keywords
Hepatocellular carcinoma: the cause of disease and mortality
Liver cancer is the fifth most leading cause of cancer death worldwide[1]. More than 700,000 people are diagnosed with this cancer and 600,000 people die each year throughout the world. Hepatocellular carcinoma (HCC) is the most common form of primary liver cancer, accounting 70%-85% of all liver cancer in adults, primarily caused by chronic liver injury and inflammation, e.g., viral hepatitis or alcoholic and non-alcoholic cirrhosis and nonalcoholic fatty liver disease (NAFLD)[2,3].
From the molecular point of view, HCCs are complex tumors[4]. The prognosis of HCC is unsatisfactory due to lack of reliable early diagnostic and screening tests and effective treatment options. Seventy percent of HCCs have been detected in an advanced stage at diagnosis. The molecular pathogenesis of the disease has also remained poorly understood. Therefore, a better understanding of HCC biology and identification of the prognostic molecular markers with benefits for HCC risk assessment and development of novel therapeutic approaches is urgently required.
Regulation of alternative pre-mRNA splicing
Alternative splicing (AS) is a process by which multiple messenger RNAs (mRNAs) are generated from a single pre-mRNA, resulting in functionally distinct protein products that may have different or even opposing roles[5]. Genome-wide studies showed that nearly all multi-exon genes in human undergo alternative splicing and produce multiple mRNA isoforms from a single pre-mRNA in a tissue or developmental stage-specific manner[6,7]. Thus, AS is an important mechanism to vastly expand transcriptomic and proteomic diversity from a finite genome[8]. This is accomplished by the differential recognition of splice sites by RNA binding splicing factors in the pre-mRNA[9]. The different types of AS are shown schematically in Figure 1. The most common type of AS consists of a single cassette exon that is either included or skipped in the mRNA. Other forms of AS include alternative selection of 5’ and 3’ splice sites, selection of mutually exclusive exon, and intron retention. Different cis-regulatory elements in the pre-mRNA play a critical role in alternative selection of splice sites by binding to splicing regulatory proteins. Based on the location of binding in the pre-mRNA and function, there are four cis-regulatory elements: exonic splicing enhancers, exonic splicing silencers, intronic splicing enhancers and intronic splicing silencers. These cis-regulatory elements which are present within the alternative exon itself or upstream/downstream intron sequences bind trans-regulatory splicing factors and either promote or inhibit the usage of the alternative exon(s). Though there are a number of RNA-binding proteins that regulate alternative pre-mRNA splicing, two of the well-studied families are serine/arginine-rich (SR) proteins and heterogeneous nuclear ribonucleoproteins (hnRNPs)[10]. Other less common families include the CELF/BRUNOL family, and the RBM family[10-12]. Both SR proteins and hnRNPs can promote or inhibit exon recognition depending on location of the binding and sequence context[10].
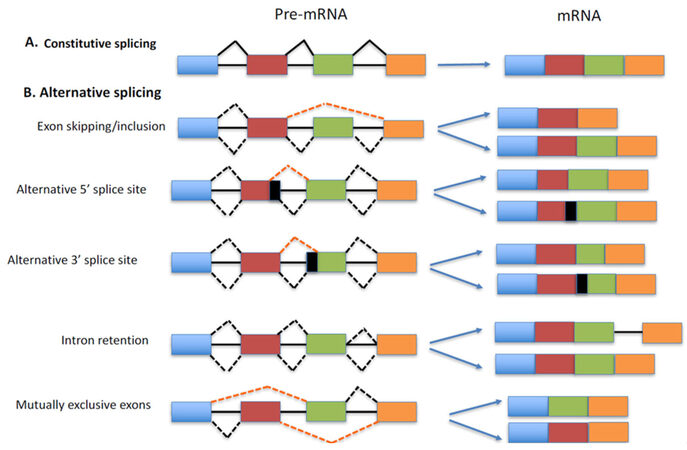
Figure 1. Different types of splicing events are shown schematically. (A) In constitutive splicing, all introns are spliced out and all exons are joined together to produce mRNA. (B) By alternative splicing, pre-mRNA can encode more than one mRNA isoform. Different isoforms can be generated by exon skipping/inclusion of alternative exons, the selection of alternative 5' or 3' splice sites, the retention of intron(s) or selection of the mutually exclusive exon(s). Exons and mRNAs are illustrated as boxes, while introns are represented by solid lines
Alternative splicing and HCC
Alternative splicing is a major post-transcriptional regulatory event that can modulate key aspects of cancer cell biology including cell proliferation, metabolism, apoptosis, survival, invasiveness, angiogenesis, drug-resistance, and metastasis[13-15], thus playing a very critical role in the development and progression of cancers. In case of HCC, splicing alterations of genes such as DNA methyltransferase 3b (DNMT3b), Aurora kinase B (AURKB), E3 ubiquitin ligase (MDM2), TENSIN2, MAD1, SVH, TP53, and Fibronectin1 (FN1)[16] have long been reported. Recent studies have shown that the list of tumor-specific aberrantly spliced mRNAs is increasing and implicated in HCC[17].
Alternative splicing facilitates the development of HCC either by generating oncogenic variants or by inactivating the tumor suppressors. For example, an alternative POLDIP3 transcript promotes HCC progression[18]. POLDIP3 is a target of ribosomal protein S6 kinase 1, and regulates DNA replication and mRNA translation. The alternative POLDIP3 transcript (POLDIP3-β), which lacks exon 3, was found to be significantly up-regulated in clinical HCC tissue compared to paired adjacent noncancerous hepatic tissue. This POLDIP3-β isoform has been shown to increase HCC cell proliferation, inhibit HCC cell apoptosis, enhance HCC cell migration, and promote xenograft growth. Another example is the cell fate determinant protein, Numb, which is aberrantly spliced in HCC and produces an isoform that contains a long proline-rich region (PRRL)[19]. In HCC cell lines, PRRL generally promotes and PRRS (short proline-rich region) suppresses proliferation, migration, invasion, and colony formation. PRRL-Numb expression has been shown to increase in HCC and be associated with early recurrence and thus reduces overall survival after surgery[19].
It was observed that, in HCC cell lines and tumors, insulin receptor (IR) is aberrantly spliced and promotes expression of the mitogenic isoform of insulin receptor (IR-A) that is generally expressed in the embryonic tissues but not in the adult liver. In contrary to the isoform IR-B that is normally expressed in the adult liver and promotes metabolic effects of insulin, IR-A signals proliferative effects via binding to insulin-like growth factor II (IGF-II). An IR-B to IR-A switch has been frequently observed in HCC tumors regardless of tumor etiology[20].
Another study demonstrated the oncogenic role of the truncated isoform of estrogen receptor α (ER-α36) in primary HCC[21]. In contrast to wild-type ER (WT-ERα), the major variant in normal liver tissue, the ERα36 splice variant has the opposite function in primary HCC, and that ERα36 increases in primary HCC tissue. Also, the high levels of WT-ERα mRNA appear to predict better survival of patients with HCC though the mechanism is yet to be explored.
Similarly, variants 5 and 6 of trans-membrane protein CD44[22], the variants a and b of extracellular matrix protein osteopontin (OPN)[23], and variant J of the transcription factor7-like 2 (TCF7L2), also known as T-cell factor 4 (TCF-4)[24], are some oncogenic isoforms that contribute to the development of HCC. Thus, selectively targeting these oncogenic isoforms would be a promising therapeutic strategy for HCC.
Regarding tumor suppressors, aberrant splicing of Hugl-1 transcripts has been identified in HCC specimens. The majority of these aberrant Hugl-1 transcripts encode truncated proteins lacking one or more conserved WD-40 repeat motifs that resulted from skipping part of and/or entire exon or insertion of intron sequences. Two truncated Hugl-1 proteins were found exclusively in HCC tissues. Aberrant Hugl-1 transcripts (78.3%, 20 of 23) had a short “direct repeat” sequence flanking their deleted regions. Over-expression of two representative HCC-derived aberrant Hugl-1 variants was shown to promote HCC cell migration, invasion, and tumorigenicity in nude mice. Moreover, the abnormal Hugl-1 was significantly correlated with poor differentiation and large tumor size of HCC. This suggests that Hugl-1 mRNA is frequently mutated by aberrant splicing in HCC, which may be involved in HCC[25,26].
Some tumor suppressors are also self-inactivated in HCC by alternative splicing. The tumor suppressor isoform of TP73 gene is TAp73, which promotes apoptosis and limits the anchorage-independent growth of tumor cells. Truncated isoforms (ΔEx2p73, ΔEx2/3p73, and ΔN’p73) of TP73 are generated by aberrant splicing and serve as dominant negative inhibitors of TAp73 and inhibit its tumor suppressor activity[27]. Several studies have shown that these isoforms are over-expressed in HCC compared to normal liver and correlated with poor patient prognosis[28]. In HCC, ΔEx2p73 expression is correlated with activation of the epidermal growth factor receptor (EGFR) and the down-regulation of the mRNA splicing factor Slu7. From a mechanistic perspective, activation of EGFR by its ligand amphiregulin (AR), whose expression is up-regulated in HCC, and c-Jun N-terminal kinase-1 activity facilitates TAp73 alternative splicing and ΔEx2p73 production[27].
Tumor suppressor KLF6 that regulates many genes involved in cell cycle, apoptosis and differentiation also inactivated by its dominant-negative SV1 isoform in HCC[29]. Studies found that SV1 isoform of KLF6 is over-expressed in HCC that promotes cellular proliferation and KLF6 full form is decreased in HCC tissue. The oncogenic activation of the Ras/PI3K/Akt pathway and subsequent down regulation of splice regulatory protein ASF/SF2 or SRSF1 leads to this aberrant splicing of KLF6 in HCC. Also, upstream of Ras, the EGFR tyrosine kinase activity could potentially trigger KLF6 SV1 generation[29]. These findings suggest potential antagonistic functions of the two isoforms in HCC and relative abundance of the isoforms might dictate the cellular fate. Thus, unraveling the regulatory mechanisms that promote these aberrant splicing might provide effective molecular targets for HCC therapy.
Aberrant regulation of splicing factors and oncofetal transformation in HCC
Aberrant expression or activity of splicing factors is a major cause of splicing deregulation; thus, it is quite expected that, increased or decreased expression of crucial splicing factors leads to disease. Indeed, deregulation of splicing regulators such as SRSF1, SRSF10, RBFOX2, MBNL1/2, and QKI proteins has been observed and accounts for hundreds of altered alternative splicing events present in multiple cancer types[30-32]. In HCC, the splicing dysregulation may be influenced by down-regulation of splicing factors ESRP2, CELF2 and SRSF5 and up-regulation of splicing factors SRSF1 or SF2/ASF, SRSF2, hnRNPA1, hnRNPA2B1, hnRNPH and CUGBP1. In multiple HCC samples, decreased expression of ESRP2, CELF2 and SRSF5 were observed[17], whereas, a significant correlation was found between the increased expression of IR-A and up-regulation of splicing factors SRSF1, hnRNPA1, hnRNPA2B1, hnRNPH, and CUGBP1[20]. This observation is in agreement with the previous in vitro studies that showed, SRSF1, CUGBP1 and hnRNPA1 promote IR-A expression in hepatoma cell-lines[33,34]. Studies showed that overexpression of the SR proteins SRSF1 and SRSF3 promote tumor growth in nude mice and these proteins are elevated in certain cancers[35]. Interestingly, in the mouse model, hepatocyte-specific deletion of SRSF3 caused spontaneous HCC with aging[36], suggesting that the function of individual splicing factor depends on the cellular context. It was observed that the splicing factors that play important roles in the maturation of liver, down-regulation of those factors promoting HCC. Studies have shown that embryonic liver development and HCC share similar alterations in many genetic programs, and HCC patients with gene expression profiles similar to embryonic stem cells had a worse prognosis[37,38]. Also, in case of HCC, it has been observed that different mRNA isoforms that are developmentally regulated and not generally expressed in the adult liver, are often expressed in cancer tissue. However, little is known about the mechanisms driving hepatocellular dedifferentiation during chronic liver diseases and tumor development.
Expression of splicing factor Esrp2 is increased in the adult liver as this splicing factor plays an important role in mesenchymal to epithelial transformation (MET) that is the opposite of epithelial to mesenchymal transformation (EMT), observed in cancer tissues. Studies showed that, homozygous knockout of Esrp2[39] led to impaired adult splicing patterns in the liver in the mouse model, suggesting the role of this splicing factor in the fetal to adult transition in hepatocytes. Consistent with these findings, knockdown of pre-mRNA splicing regulator SLU7 in human liver cells and mouse liver resulted in profound changes in pre-mRNA splicing of genes essential for hepatocellular differentiation and reversion to a fetal-like gene expression pattern[40]. Moreover, Slu7 expression has been found to be significantly compromised in chronic liver diseases and in HCC[27] suggesting a role of SLU7 down-regulation in the progression of liver pathogenesis. Interestingly, SLU7 also preserves survival of HCC cells and other solid tumors via oncogenic miR-17-92 cluster expression[41] indicating a complex regulatory role of this splicing factor in pathogenesis of liver diseases.
Hepatocyte-specific deletion of SRSF3 caused impaired hepatocyte maturation and also glucose and lipid metabolism in early adult life[42]. Loss of SRSF3 facilitates expression of the mitogenic isoform of insulin receptor (IR-A) that is generally not expressed in adult liver allowing aberrant activation of mitogenic signaling. Loss of SRSF3 in hepatocytes also promotes aberrant splicing and expression of EMT genes and activates Wnt/beta-catenin signaling leading to c-Myc induction. Additionally, loss of SRSF3 promotes inclusion of the profibrogenic EDA exon in fibronectin gene (FN1) and expression of the short isoform of XBP1 (XBP1s) in hepatocytes and SRSF3 knock-out mice developed spontaneous HCC with aging[36]. In support of this, SRSF3 has also been found to be reduced or mislocalized in human HCC[40], suggesting a potential preventive role of SRSF3 in HCC. Interestingly, a recent report suggests XBP1s as a newly discovered molecule involved in the HCC progression by promoting EMT[43] by enhancing the expression of Twist and Snail. Pathological analysis showed that the expression of XBP1s was closely correlated with distant metastasis.
Recently, Yuan et al.[44] identified an important oncofetal protein, MBNL3, and an oncofetal splicing event, inclusion or skipping of lncRNA-PXN-AS1 exon 4, both of which play vital roles in hepatocarcinogenesis and serve as prognostic biomarkers and therapeutic targets for HCC. This suggests that identifying the common molecular events between embryonic liver development and HCC would promote the understanding of molecular pathogenesis of HCC and the development of more effective targeted therapies.
Summary and perspectives
The findings reviewed here, though handful, are sufficient to show that the AS plays a very critical role in regulating HCC progression and diagnostic. Thus, understanding the mechanisms of alternative pre-mRNA splicing for HCC related genes are important for the development of new therapeutic strategies such as targeting HCC specific isoform as biomarkers and targeting oncogenic isoform.
With the fast development of technologies, next generation sequencing provides a powerful way to study the transcriptome to uncover the aberrant splicing events in different cancers including HCC. For example, analyzing the ultra-deep transcriptome landscape of human liver cancer, Lin et al.[45] identified potential biomarkers for HCC, including ALG1L, SERPINA11, TMEM82 and DUNQU1 and the AS event of FGFR2. Using antisense oligonucleotides or splicing switch oligonucleotides that can complimentarily bind to a target site in pre-mRNAs and regulate the splicing could be used to selectively target specific isoforms of RNA with oncogenic potential[46,47]. Targeting specific isoforms of RNA and protein has the potential to improve drug efficacy and reduce side effects. In summary, we are hoping that the integration of pre-mRNA alternative splicing in the pathogenesis of HCC will contribute to the better understanding of the disease and development of new therapies.
Declarations
Authors’ contributionsThe author contributed solely to the review.
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestThe author declares that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2018.
REFERENCES
3. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011;61:69-90.
4. Zucman-Rossi J, Laurent-Puig P. Genetic diversity of hepatocellular carcinomas and its potential impact on targeted therapies. Pharmacogenomics 2007;8:997-1003.
5. Singh RK, Cooper TA. Pre-mRNA splicing in disease and therapeutics. Trends Mol Med 2012;18:472-82.
6. Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature 2008;456:470-6.
7. Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 2008;40:1413-5.
8. Yang X, Coulombe-Huntington J, Kang S, Sheynkman GM, Hao T, Richardson A, Sun S, Yang F, Shen YA, Murray RR, Spirohn K, Begg BE, Duran-Frigola M, MacWilliams A, Pevzner SJ, Zhong Q, Trigg SA, Tam S, Ghamsari L, Sahni N, Yi S, Rodriguez MD, Balcha D, Tan G, Costanzo M, Andrews B, Boone C, Zhou XJ, Salehi-Ashtiani K, Charloteaux B, Chen AA, Calderwood MA, Aloy P, Roth FP, Hill DE, Iakoucheva LM, Xia Y, Vidal M. Widespread expansion of protein interaction capabilities by alternative splicing. Cell 2016;164:805-17.
10. Lee Y, Rio DC. Mechanisms and regulation of alternative pre-mRNA splicing. Annu Rev Biochem 2015;84:291-323.
11. Barreau C, Paillard L, Mereau A, Osborne HB. Mammalian CELF/Bruno-like RNA-binding proteins: molecular characteristics and biological functions. Biochimie 2006;88:515-25.
12. Sutherland LC, Rintala-Maki ND, White RD, Morin CD. RNA binding motif (RBM) proteins: a novel family of apoptosis modulators? J Cell Biochem 2005;94:5-24.
13. David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev 2010;24:2343-64.
14. Dominguez D, Tsai YH, Weatheritt R, Wang Y, Blencowe BJ, Wang Z. An extensive program of periodic alternative splicing linked to cell cycle progression. Elife 2016;5.
16. Berasain C, Goni S, Castillo J, Latasa MU, Prieto J, Avila MA. Impairment of pre-mRNA splicing in liver disease: mechanisms and consequences. World J Gastroenterol 2010;16:3091-102.
17. Zhang L, Liu X, Zhang X, Chen R. Identification of important long non-coding RNAs and highly recurrent aberrant alternative splicing events in hepatocellular carcinoma through integrative analysis of multiple RNA-Seq datasets. Mol Genet Genomics 2016;291:1035-51.
18. Liu XN, Yuan JH, Wang TT, Pan W, Sun SH. An alternative POLDIP3 transcript promotes hepatocellular carcinoma progression. Biomed Pharmacother 2017;89:276-83.
19. Lu Y, Xu W, Ji J, Feng D, Sourbier C, Yang Y, Qu J, Zeng Z, Wang C, Chang X, Chen Y, Mishra A, Xu M, Lee MJ, Lee S, Trepel J, Linehan WM, Wang X, Yang Y, Neckers L. Alternative splicing of the cell fate determinant Numb in hepatocellular carcinoma. Hepatology 2015;62:1122-31.
20. Chettouh H, Fartoux L, Aoudjehane L, Wendum D, Claperon A, Chretien Y, Rey C, Scatton O, Soubrane O, Conti F, Praz F, Housset C, Rosmorduc O, Desbois-Mouthon C. Mitogenic insulin receptor-A is overexpressed in human hepatocellular carcinoma due to EGFR-mediated dysregulation of RNA splicing factors. Cancer Res 2013;73:3974-86.
21. Zhang J, Ren J, Wei J, Chong CC, Yang D, He Y, Chen GG, Lai PB. Alternative splicing of estrogen receptor alpha in hepatocellular carcinoma. BMC Cancer 2016;16:926.
22. Endo K, Terada T. Protein expression of CD44 (standard and variant isoforms) in hepatocellular carcinoma: relationships with tumor grade, clinicopathologic parameters, p53 expression, and patient survival. J Hepatol 2000;32:78-84.
23. Chae S, Jun HO, Lee EG, Yang SJ, Lee DC, Jung JK, Park KC, Yeom YI, Kim KW. Osteopontin splice variants differentially modulate the migratory activity of hepatocellular carcinoma cell lines. Int J Oncol 2009;35:1409-16.
24. Koga H, Tsedensodnom O, Tomimaru Y, Walker EJ, Lee HC, Kim KM, Yano H, Wands JR, Kim M. Loss of the SxxSS motif in a human T-cell factor-4 isoform confers hypoxia resistance to liver cancer: an oncogenic switch in Wnt signaling. PLoS One 2012;7:e39981.
25. Liu L, Xie S, Zhang C, Zhu F. Aberrant regulation of alternative pre-mRNA splicing in hepatocellular carcinoma. Crit Rev Eukaryot Gene Expr 2014;24:133-49.
26. Lu X, Feng X, Man X, Yang G, Tang L, Du D, Zhang F, Yuan H, Huang Q, Zhang Z, Liu Y, Strand D, Chen Z. Aberrant splicing of Hugl-1 is associated with hepatocellular carcinoma progression. Clin Cancer Res 2009;15:3287-96.
27. Castillo J, Goni S, Latasa MU, Perugorria MJ, Calvo A, Muntane J, Bioulac-Sage P, Balabaud C, Prieto J, Avila MA, Berasain C. Amphiregulin induces the alternative splicing of p73 into its oncogenic isoform DeltaEx2p73 in human hepatocellular tumors. Gastroenterology 2009;137:1805-15.
28. Stiewe T, Zimmermann S, Frilling A, Esche H, Putzer BM. Transactivation-deficient DeltaTA-p73 acts as an oncogene. Cancer Res 2002;62:3598-602.
29. Yea S, Narla G, Zhao X, Garg R, Tal-Kremer S, Hod E, Villanueva A, Loke J, Tarocchi M, Akita K, Shirasawa S, Sasazuki T, Martignetti JA, Llovet JM, Friedman SL. Ras promotes growth by alternative splicing-mediated inactivation of the KLF6 tumor suppressor in hepatocellular carcinoma. Gastroenterology 2008;134:1521-31.
30. Danan-Gotthold M, Golan-Gerstl R, Eisenberg E, Meir K, Karni R, Levanon EY. Identification of recurrent regulated alternative splicing events across human solid tumors. Nucleic Acids Res 2015;43:5130-44.
31. Zhou X, Li X, Cheng Y, Wu W, Xie Z, Xi Q, Han J, Wu G, Fang J, Feng Y. BCLAF1 and its splicing regulator SRSF10 regulate the tumorigenic potential of colon cancer cells. Nat Commun 2014;5:4581.
32. Anczukow O, Akerman M, Clery A, Wu J, Shen C, Shirole NH, Raimer A, Sun S, Jensen MA, Hua Y, Allain FH, Krainer AR. SRSF1-regulated alternative splicing in breast cancer. Mol Cell 2015;60:105-17.
33. Sen S, Talukdar I, Webster NJ. SRp20 and CUG-BP1 modulate insulin receptor exon 11 alternative splicing. Mol Cell Biol 2009;29:871-80.
34. Talukdar I, Sen S, Urbano R, Thompson J, Yates JR 3rd, Webster NJ. hnRNP A1 and hnRNP F modulate the alternative splicing of exon 11 of the insulin receptor gene. PLoS One 2011;6:e27869.
35. Webster NJG. Alternative RNA splicing in the pathogenesis of liver disease. Front Endocrinol (Lausanne) 2017;8:133.
36. Sen S, Langiewicz M, Jumaa H, Webster NJ. Deletion of serine/arginine-rich splicing factor 3 in hepatocytes predisposes to hepatocellular carcinoma in mice. Hepatology 2015;61:171-83.
37. Lee JS, Heo J, Libbrecht L, Chu IS, Kaposi-Novak P, Calvisi DF, Mikaelyan A, Roberts LR, Demetris AJ, Sun Z, Nevens F, Roskams T, Thorgeirsson SS. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med 2006;12:410-6.
38. Becker D, Sfakianakis I, Krupp M, Staib F, Gerhold-Ay A, Victor A, Binder H, Blettner M, Maass T, Thorgeirsson S, Galle PR, Teufel A. Genetic signatures shared in embryonic liver development and liver cancer define prognostically relevant subgroups in HCC. Mol Cancer 2012;11:55.
39. Bhate A, Parker DJ, Bebee TW, Ahn J, Arif W, Rashan EH, Chorghade S, Chau A, Lee JH, Anakk S, Carstens RP, Xiao X, Kalsotra A. ESRP2 controls an adult splicing programme in hepatocytes to support postnatal liver maturation. Nat Commun 2015;6:8768.
40. Elizalde M, Urtasun R, Azkona M, Latasa MU, Goni S, Garcia-Irigoyen O, Uriarte I, Segura V, Collantes M, Di Scala M, Lujambio A, Prieto J, Avila MA, Berasain C. Splicing regulator SLU7 is essential for maintaining liver homeostasis. J Clin Invest 2014;124:2909-20.
41. Urtasun R, Elizalde M, Azkona M, Latasa MU, Garcia-Irigoyen O, Uriarte I, Fernandez-Barrena MG, Vicent S, Alonso MM, Muntane J, Prieto J, Avila MA, Berasain C. Splicing regulator SLU7 preserves survival of hepatocellular carcinoma cells and other solid tumors via oncogenic miR-17-92 cluster expression. Oncogene 2016;35:4719-29.
42. Sen S, Jumaa H, Webster NJ. Splicing factor SRSF3 is crucial for hepatocyte differentiation and metabolic function. Nat Commun 2013;4:1336.
43. Wu S, Du R, Gao C, Kang J, Wen J, Sun T. The role of XBP1s in the metastasis and prognosis of hepatocellular carcinoma. Biochem Biophys Res Commun 2018;500:530-7.
44. Yuan JH, Liu XN, Wang TT, Pan W, Tao QF, Zhou WP, Wang F, Sun SH. The MBNL3 splicing factor promotes hepatocellular carcinoma by increasing PXN expression through the alternative splicing of lncRNA-PXN-AS1. Nat Cell Biol 2017;19:820-32.
45. Lin KT, Shann YJ, Chau GY, Hsu CN, Huang CY. Identification of latent biomarkers in hepatocellular carcinoma by ultra-deep whole-transcriptome sequencing. Oncogene 2014;33:4786-94.
46. Wood M, Yin H, McClorey G. Modulating the expression of disease genes with RNA-based therapy. PLoS Genet 2007;3:e109.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Sen S. Aberrant pre-mRNA splicing regulation in the development of hepatocellular carcinoma. Hepatoma Res 2018;4:37. http://dx.doi.org/10.20517/2394-5079.2018.39
AMA Style
Sen S. Aberrant pre-mRNA splicing regulation in the development of hepatocellular carcinoma. Hepatoma Research. 2018; 4: 37. http://dx.doi.org/10.20517/2394-5079.2018.39
Chicago/Turabian Style
Sen, Supriya. 2018. "Aberrant pre-mRNA splicing regulation in the development of hepatocellular carcinoma" Hepatoma Research. 4: 37. http://dx.doi.org/10.20517/2394-5079.2018.39
ACS Style
Sen, S. Aberrant pre-mRNA splicing regulation in the development of hepatocellular carcinoma. Hepatoma. Res. 2018, 4, 37. http://dx.doi.org/10.20517/2394-5079.2018.39
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 43 clicks
Cite This Article 43 clicks

 Like This Article 34
likes
Like This Article 34
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.