
Congenital absence of the portal vein complicated by hepatocellular carcinoma in the liver of an adult woman: review of imaging, literature and management

Abstract
We present a case of absence of the portal vein and Laennec’s cirrhosis in a 51-year-old female who was diagnosed with hepatocellular carcinoma (HCC). Only 101 cases of this malformation of the splanchnic vasculature have been reported of which 4 were reported to have HCC. Patient had disease progression while waiting for a liver transplant. Patient was treated with 3 separate conventional transarterial chemoembolization procedures at an outside hospital. At our institution, radioembolization of the right hepatic lobe was performed. She succumbed to liver insufficiency 8 years after being diagnosed with HCC. The features of this patient’s clinical course are reviewed.
Keywords
Introduction
The adult liver has a complex vascular architecture composed of two distinct circulatory systems. The liver is supplied by blood mostly from the portal vein (PV) and its intrahepatic branches, as well as the hepatic artery and its intrahepatic branches. The PV is responsible for carrying blood from the organs of the abdominal cavity such as the gastrointestinal tract, the spleen, pancreas, and biliary apparatus. In conventional anatomy, the splenic vein (SV) and the superior mesenteric vein (SMV) join to form the PV. The PV is then subdivided into right and left branches, which form small vessels throughout the liver that eventually drain into the sinus venosus[1].
During embryological development, the PV originates from the right and left vitelline veins between gestational weeks 4 to 10. There is selective involution and persistence of the peri-intestinal vitelline venous loops. The vitelline veins originally emerge from the yolk sac, cross the septum transversum, and drain into the sinus venosus. During the 3rd to 8th gestational week, abnormal patterns of involution and persistence may result in pre-duodenal, pre-biliary, or duplicated PV. Excessive involution can result in the absence of the PV as seen in type 1 portocaval shunts. Type 2 portocaval shunts may develop due to persistence of the right vitelline vein, where the shunt drains into the retrohepatic inferior vena cava (IVC), or the left vitelline vein, where the shunt drains into the suprahepatic IVC or right atrium[2].
A London surgeon by the name of John Abernethy[3] first described congenital absence of the PV in 1793 during a postmortem examination of a 10-month-old girl. Since, there have been 101 reported cases with 66% in women and most cases being in children. Most patients presented with encephalopathy, hepatopulmonary syndrome, or hepatorenal syndrome. Almost half of cases have liver masses at presentation such as focal nodular hyperplasia (FNH), adenomas, hepatoblastoma, or hepatocellular carcinoma (HCC)[4].
In 1994, Morgan and Superina[5] proposed a classification of portosystemic anomalies. Type 1 shunts are characterized by the absence of intrahepatic PV. Liver is not perfused with portal blood because of a complete shunt. A type 2 shunt is characterized as a partial shunt. The liver is perfused with portal blood in the presence of a partial shunt to systemic circulation. The type 1 shunts are subdivided into two further types, depending on the anatomy of the PV. The SV and the SMV drain separately into the IVC in a type 1a shunt. The SMV either drains into the IVC or the left renal vein. A confluence of SMV and SV is usually present in a type 1b shunt, but it does not supply the liver. While type 1 shunts are managed with liver transplant, type 2 shunts may be surgically ligated[5].
In this report we will review a case of congenital absence of the portal vein (CAPV) in a 51-year-old woman who was diagnosed with HCC and had a history of Laennec’s cirrhosis and a type Ib Abernethy malformation.
Case Report
A 51-year-old female who was diagnosed in 2008 with HCC was referred to the interventional radiology clinic from the liver transplant service. She had been managed with conventional transarterial chemoembolization (c-TACE) on three separate occasions and she had signs of disease progression around the prior treated areas as marked by lipiodol. Imaging revealed PV agenesis (type 1b). Her clinical course was marked by Laennec’s cirrhosis related to alcohol abuse complicated by occasional hepatic encephalopathy resulting in hospitalization. Limited pediatric history included only an episode of meningitis of unclear etiology and struggles with psychiatric illness. Histologic evaluation of liver parenchyma from a biopsy at presentation to transplant team revealed ballooning hepatocytes, mixed with collapsed hepatocytes, Mallory-Denk bodies, and glycogenated nuclei, which can be seen in the setting of alcoholic hepatitis. These were accompanied by bridging and pericellular fibrosis as seen after trichome staining to the extent of stage 3 or severe fibrosis. A trial of sorafenib failed due to development of a rash, fatigue and weight loss. Social history was positive for prior alcohol abuse but patient stopped drinking 3 years after being diagnosed with HCC.
Interventional radiology was consulted for another TACE procedure to downstage her disease to allow for a transplant 6 years after HCC initial diagnosis. At that point her liver profile was: alkaline phosphatase 720 U/L, aspartate aminotransferase 69 U/L, total bilirubin 2.6 mg/dL, ammonia 30 mcmol/L, albumin 3.0 g/dL. Her coagulation profile was normal (international normalized ratio was 1.02). Her alpha fetal protein (AFP) level was 357.1 ng/mL. Her Eastern Cooperative Oncology Group (ECOG) performance status was 0. Child Pugh score was B (8) therefore she had a expected 2-year overall survival of ~57%[6].
Her physical examination was unremarkable notably without asterixis. She had no significant cardiac history (ejection fraction of 55% on stress test). Surgical history was non contributive. When she was presented in transplant tumor board, abdominal ultrasound showed several lesions in the liver. Computed tomography (CT) with contrast showed a mass identified in segments 6 and 7 measuring 4.6 cm × 4.0 cm × 4.5 cm with surrounding hypoattenuation of the liver parenchyma [Figure 1]. There was an additional hypodense lesion in segment 2 measuring 3.1 cm × 4.9 cm × 3.5 cm with some areas of hyperdensity. Both lesions were deemed to be changes secondary to prior TACE. Follow-up CT showed arterial enhancing lesions in the right liver lobe the dominant lesion had increased in size from 4.6 cm × 4.0 cm × 4.5 cm to 9.2 cm × 8.9 cm × 11.0 cm with washout, characteristics HCC findings [Figure 2]. Absence of the right and left PV and confluence of the SV and SMV into the IVC was also noted [Figure 3]. No collateral vessels to suggest cavernous transformation nor extrahepatic portal vein remnant can be seen. Based on the presence of multinodular disease without vascular invasion (although difficult to qualify given lack of PV), good performance status (ECOG 0), and liver function (Child Pugh B), her disease was classified as intermediate stage disease by the Barcelona Clinic Liver Classification (BCLC) system or BCLC B[7].
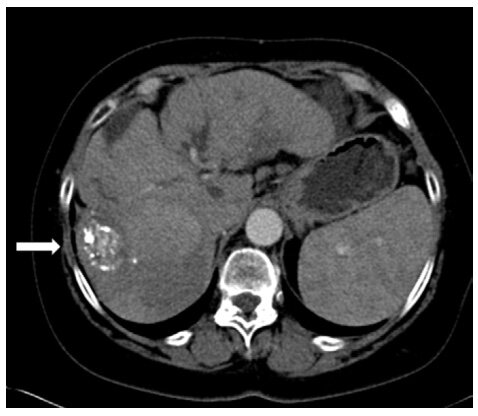
Figure 1. Hyper attenuating lesion from prior conventional transarterial chemoembolization containing lipiodol (white arrow)
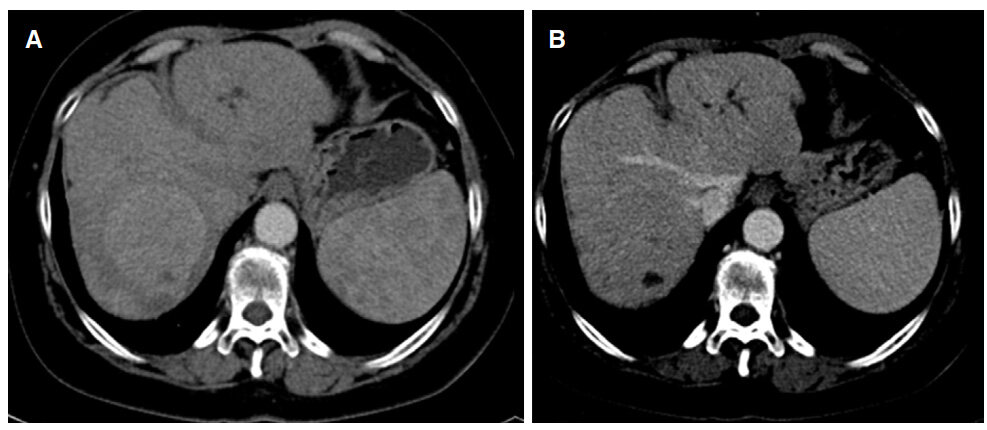
Figure 2. Contrast enhanced computed tomography revealing a 9.2 cm × 8.9 cm × 11.0 cm mass in the right lobe of the liver. (A) Arterial phase shows diffuse enhancement; (B) venous phase shows washout
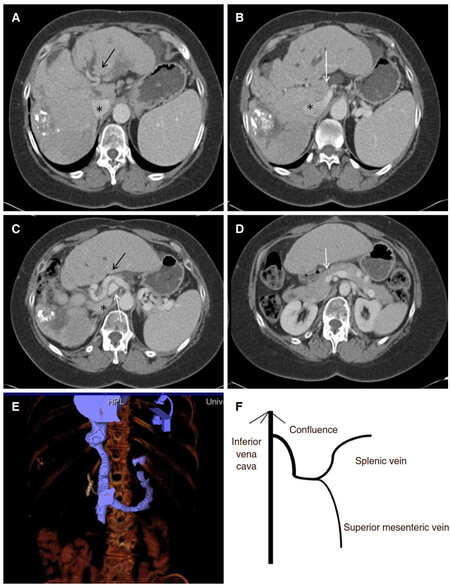
Figure 3. Type 1B Abernathy malformation. (A-D) Axial computed tomography images showing course of hepatic artery (black arrow), confluence of superior mesenteric vein and splenic vein (white arrow), inferior vena cava (asterisk), and the superior mesenteric artery (white arrow); (E) coronal reformats of findings; (F) diagram of malformation
At that point recommendation from the liver multidisciplinary tumor board was to repeat TACE. Yet, at the time of her evaluation in the interventional radiology clinic, TACE was not offered due to increased risk for abscess formation and progressive liver dysfunction. After referral to the oncology team, she received two intra-arterial chemoinfusions of cisplatin into the proper hepatic artery. Patient’s disease continued to progress as markedly elevated AFP of 8779. Despite risk of hepatoxicity and elevated lung shunt of 21%, she subsequently underwent radioembolization to the right lobe of the liver. She received a dose of 1.06 gBq (29.1 mCi) of Yttrium-90 (Y-90) embolic resin spheres delivered to the right lobe of the liver.
Three months follow-up CT scan showed dramatic partial response with no further enhancement in the dominant mass [Figure 4]. Incongruent to imaging findings AFP increased dramatically normalizing approximately 9 months after treatment [Table 1]. Unfortunately liver dysfunction was exacerbated due to treatment [Table 1] consistent with radiation embolization induced liver disease (REILD). Only further treatment received was octreotide and supportive care. She passed away after struggling with depression 8 years after initial diagnosis of HCC and 22 months after radioembolization therapy.
Changes of total bilirubin and AFP in reference to radioembolization
| Component | Bilirubin (mg/dL) | AFP (ng/mL) |
|---|---|---|
| Latest reference | 0.0-1.2 | 0.0-8.3 |
| 21 months after radioembolization | 12.7 | 6.7 |
| 12 months after radioembolization | 13.9 | 20.7 (H) |
| 11 months after radioembolization | 13.8 | 55.3 (H) |
| 10 months after radioembolization | 8.4 | 178.2 (H) |
| 9 months after radioembolization | 6.9 | 805.4 (H) |
| 7 months after radioembolization | 6.9 | 5450.0 (H) |
| 6 months after radioembolization | 7.4 | 19,394.0 (H) |
| 4 months after radioembolization | 7.4 | > 60,500.0 (H) |
| 3 months after radioembolization | 4.8 | 55,658.0 (H) |
| 2 weeks after radioembolization | 2.9 | 18,662.0 (H) |
| 1 month prior to radioembolization | 2.1 | 8779.0 (H) |
| 2 months prior to radioembolization | 1.8 | 3678.0 (H) |
| 4 months prior to radioembolization | 1.6 | 1032.0 (H) |
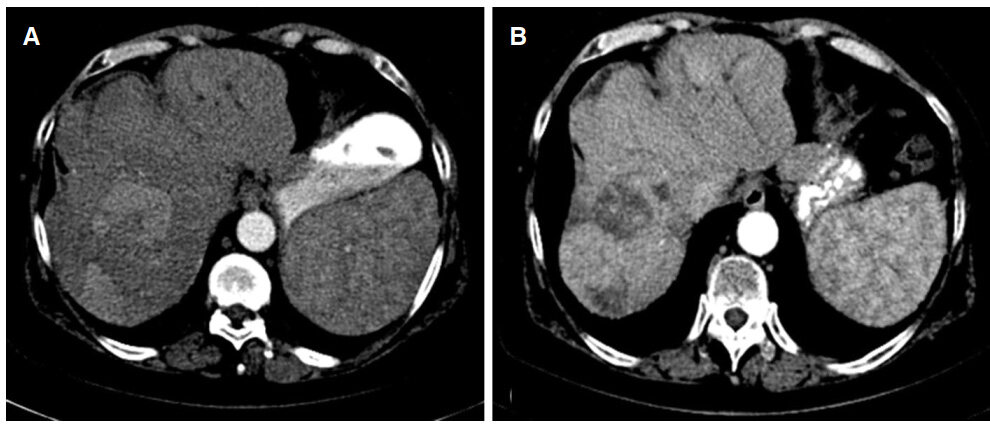
Figure 4. Follow-up images. (A) Computed tomography (CT) scan in 3 months demonstrates partial response with decreased enhancement in the central aspect of the dominant mass; (B) CT scan in 18 months shows dramatic partial response with atrophy of the treated right lobe
The determination of PV agenesis in this case was by imaging features only. No surgical or histopathological confirmation is available despite patient’s ultimate demise. To the knowledge of the authors’ no autopsy was performed.
Discussion
In the case presented the mesenteric venous system and the SV joined to form a confluence of vessels yet this confluence drained directly into the suprarenal IVC. This malformation can be attributed to the embryological development of the portal venous system. This would be classified as type 1b shunt.
There are currently 101 reported cases of CAPV. Of the reported cases, 66% of patients are females and about 70% had been diagnosed by age of 18 years; < 10% were associated with a type 2 malformation[4]. This patient presented to our institution to undergo liver transplant evaluation. Additional associated anomalies such as congenital heart disease were absent in this case.
CAPV is associated with hepatic tumors. Hepatic changes such as FNH, HCC and hepatoblastoma were seen in 40% of cases[1]. In this case, the patient presented with HCC. Research has shown that insulin, glucagon, and epidermal growth factor are delivered to the liver through the splanchnic venous system. These substances are vital for the hepatic regeneration. Therefore, it is suggested that absence of PV flow may result into abnormal hepatic development, function, and regenerative capacity as seen in this patient. Increased arterial hepatic flow may subsequently play a role in the development of hepatic neoplasms[8].
To date, 4 cases of patients with CAPV have been reported to have HCC[1]. One case was reported in a 14-year-old female, however nature of the review focused on intestinal flora compensating to result in normal ammonia levels rather than tumor description and presentation[9]. In 2001, Lundstedt et al.[10] eported a case of asymptomatic CAPV (type 1b shunt) found at time of resection of a 12-cm HCC thought to have arisen secondary to hepatitis B virus in a 51-year-old male. The patient remained disease free over 2-year follow-up period[10] Unlike our patient, there was no history of encephalopathy. Only the aspartate transaminase and alanine transaminase were mildly elevated[10]. Morotti et al.[11] reported a case of an 8-year-old female with Turner syndrome who was found to have CAPV at time of transplant. Liver transplantation was performed due to liver dysfunction (bilirubin 13.9 mg/dL) and concern for enlarging right and left liver lesions originally shown on biopsy to be FNH[11]. Explant specimen revealed well-differentiated HCC[11]. It was suggested that the combination of the hormonal therapy for Turner syndrome, and vascular anomaly may have contributed to the development of HCC[11]. Pichon et al.[12] noted PV absence on an ultrasound (US) for a 36-year-old female undergoing evaluation for abdominal pain and follow-up of liver masses. The SMV and SV were found on indirect venogram at angiography and surgical evaluation to have direct but separate drainage into the IVC consistent with a type 1a shunt[12]. A 12-cm dominant right HCC surrounded by small peripheral nodules were noted in the right hepatectomy specimen and the patient did well for the course of 2-year follow-up period[12]. As in the case presented here, the CAPV was found incidentally while undergoing evaluation for management of HCC.
How the absence of the PV effects imaging features and resultant diagnosis of HCC is unknown. Detection of the PV abnormality can be done with US, CT or magnetic resonance imaging (MRI). The former has the benefit of no radiation, but detecting alternate shunts is difficult with US. For this reason CT or MRI is preferred to trace the course of the SMV and SV[13]. It has been shown that PV thrombosis can results in parenchyma perfusion changes readily concealing the presence of tumor on enhanced images[14]. Hyperintensity on T2 and diffusion sequences can indicate the presence of HCC, particularly infiltrative HCC with corresponding hypointensity in comparison to liver parenchyma on T1 sequences[14]. It has been shown in the setting of PV thrombosis, arterial hypervascularity is not well perceived likely due to increased arterial supply to background liver parenchyma[15]. Washout however on portal venous phases was apparent in majority of these cases[15]. Washout kinetics are poorly understood but may be related to the proportion of intravascular space to interstitial space which is greater in tumor or increased arterial pressure leading to decreased intra-tumoral portal venous blood supply[15-18]. In our case though heterogeneous appearance on arterial phase, both arterial enhancement and washout on venous phase were apparent.
Liver dysfunction has been reported in most cases of CAPV. Our patient had a background of Laennec’s cirrhosis related to alcohol abuse as well as hepatic encephalopathy. Her liver function as discussed in the case presentation fluctuated throughout her clinical course.
The patient discussed in this case also had a history of fractures and osteopenia. Osteopenia and osteoporosis are important and common complications of chronic liver disease, receiving the generic definition of hepatic osteodystrophy (HO). The development of HO may be due to both increased bone resorption and decreased bone formation. Pathogenic mechanisms are diverse and very little is known about some of them: genetic factors, alterations in calcium-vitamin D metabolism, hyperbilirubinemia, and vitamin K and insulin-like growth factor-1 deficiency[19].
The prognosis of patients affected with CAPV generally depends on associated heart and liver anomalies in infancy. Long-term prognosis depends on the control of hepatic dysfunction and metabolic irregularities. Forty-six cases have been reported to be associated with a congenital anomaly, of which 16 were congenital cardiac disease[4]. Congenital cardiac disease typically seen with CAPV includes: patent foramen ovale, patent ductus arteriosus, ventral septal defects, and atrial septal defects[1].
Liver transplantation has been performed to effectively treat symptomatic patients with congenital agenesis of the PV. CAPV should not be considered a contraindication to hepatic transplantation[1]. During an orthotopic liver transplant, the congenital portocaval shunt can be divided while repairing the caval defect and performing a PV anastomosis[20]. Patients with hyperammonemia, portosystemic encephalopathy, hepatopulmonary syndrome, or hepatic tumors may benefit dramatically from liver transplant. Cases have been reported where transplantation has successfully reversed the hepatopulmonary syndrome caused by the Abernathy malformation as well[20]. Other treatment modalities include balloon-occluded retrograde transvenous obliteration, embolization of shunt with coiling, and surgical modification of shunts[4].
Unfortunately at time of consultation in our institution the patient presented here was not eligible for transplant given the extent of HCC. This prompted multidisciplinary care focused downstaging her cancer. Radioembolization in the setting of PV thrombosis has been shown to be as effective and better tolerated than TACE as PV thrombosis increases risk of necrosis[21,22]. Given progression after c-TACE, and lack of portal supply, radioembolization was favored as a treatment. It is suggested in some cases that a background of cirrhosis can protect from such injury[22]. In this case, c-TACE had already been performed at an outside facility. TACE has been shown to be safe and effective in patients with advanced or BCLC C disease which includes patients with varying degrees of PV thrombosis[23,24]. However, PV thrombosis does portend a poorer prognosis[24]. As anticipated post embolization syndrome is the most common side effect reported post TACE, while encephalopathy was found in approximately 5% of patients, and elevated liver function tests as high as 20% of cases[23]. In a comparison, c-TACE and drug eluting bead TACE had similar safety profiles and survival rates comparable to treatment with sorafenib[23].
The use of AFP as an oncologic marker of response to loco-regional therapy for HCC has been proven to be effective[25]. The median time to response has been reported to be between 2 and 4 months therefore it has been suggested that AFP used to identify patients who do not respond to treatment and prompt earlier consideration of implementation of alternative strategies[25]. Cases such as the one discussed here where there is a dramatic increase in AFP despite imaging response with delayed response in the marker (~7 months) have not widely been reported. Elevated levels of AFP have been seen in the setting of hepatic necro-inflammatory activity, which could lead to over production of AFP[26]. This may explain the incongruent increase in AFP initially with delayed response in this case.
Despite dramatic imaging response and eventual decline of AFP, the patient developed REILD, which results from normal hepatic parenchyma exposure to radiation. The clinical course is driven by a form of sinusoidal obstruction syndrome marked by jaundice, ascites and mild increase in liver function tests. After 3 months bilirubin can rise to 3 or higher[27]. The incidence of REILD is reported to be between 0%-4% overall[28]. The patient was known to be at higher risk given decompensated liver function in the past therefore lobar approach was selected. However in patient with cirrhosis REILD has been noted in 0%-33% of patients who underwent whole liver treatment and 8%-15% in patients who underwent partial liver treatment[27]. Management as in this case is supportive.
In this case the patient did derive a survival benefit from radioembolization. Patients with intermediate stage HCC are expected to have a median survival of 16 months from time of diagnosis[29]. After radioembolization the patient survived another 22 months. The biology of her disease suggests that initial disease was less aggressive given that she survived 8 years beyond diagnosis.
In conclusion, congenital agenesis of the PV is a rare congenital anomaly due to abnormal embryologic progression. The prognosis of patients affected with CAPV can vary depending on associated heart and liver anomalies in infancy or the progression of hepatic dysfunction. Those patients are at risk of developing HCC. Liver transplantation has been effective in patients with hepatic dysfunction. When transplantation cannot be offered loco-regional therapy can offer palliative disease control and improved overall survival. However liver directed therapy in this population could be associated with increased risk of liver failure.
Declarations
Authors’ contributionsRead and wrote the initial report: Mehta A
Corrected and amended areas related to imaging, intervention, and follow up: Venkat SR
Focused on the radiation aspects in terms of response and editing of clinical presentation: Portelance L
Helped review and highlighted oncology considerations: Feun LG
Financial support and sponsorshipNone.
Conflicts of interestThere are no conflicts of interest.
Patient consentPatients consent could not be obtained.
Ethics approvalWhile IRB approval at our institution is not required for a solitary case presentation, the data is collect as part of an IRB approved prospective database through Interventional Radiology.
Copyright© The Author(s) 2018.
REFERENCES
1. Mistinova J, Valacsai F, Varga I. Congenital absence of the portal vein - case report and a review of literature. Clin Anat 2010;23:750-8.
3. Abernethy J. Account of two instances of uncommon formation in the viscera of the human body. Phil Trans R Soc Lond B Biol Sci 1793;83:295-9.
4. Hao Y, Hong X, Zhao X. Congenital absence of the portal vein associated with focal nodular hyperplasia of the liver and congenital heart disease: a case report and literature review. Oncol Lett 2015;9:695-700.
5. Morgan G, Superina R. Congenital absence of the portal vein: two cases and a proposed classification system for portosystemic vascular anomalies. J Pediatr Surg 1994;29:1239-41.
6. Cholongitas E, Papatheodoridis GV, Vangeli M, Terreni N, Patch D, Burroughs AK. Systematic review: the model for end-stage liver disease--should it replace Child-Pugh's classification for assessing prognosis in cirrhosis? Aliment Pharmacol Ther 2005;22:1079-89.
8. Starzl TE, Francavilla A, Halgrimson CG, Francavilla FR, Porter KA, Brown TH, Putnam CW. The origin, hormonal nature, and action of hepatotrophic substances in portal venous blood. Surg Gynecol Obstet 1973;137:179-99.
9. Kamiya S, Taniguchi I, Yamamoto T, Sawamura S, Kai M, Ohnishi N, Tsuda M, Yamamura M, Nakasaki H, Yokoyama S. Analysis of intestinal flora of a patient with congenital absence of the portal vein. FEMS Immunol Med Microbiol 1993;7:73-80.
10. Lundstedt C, Lindell G, Tranberg KG, Svartholm E. Congenital absence of the intrahepatic portion of the portal vein in an adult male resected for hepatocellular carcinoma. Eur Radiol 2001;11:2228-31.
11. Morotti RA, Killackey M, Shneider BL, Repucci A, Emre S, Thung SN. Hepatocellular carcinoma and congenital absence of the portal vein in a child receiving growth hormone therapy for turner syndrome. Semin Liver Dis 2007;27:427-31.
12. Pichon N, Maisonnette F, Pichon-Lefievre F, Valleix D, Pillegand B. Hepatocarcinoma with congenital agenesis of the portal vein. Jpn J Clin Oncol 2003;33:314-6.
13. Hu GH, Shen LG, Yang J, Mei JH, Zhu YF. Insight into congenital absence of the portal vein: is it rare? World J Gastroenterol 2008;14:5969-79.
14. Reynolds AR, Furlan A, Fetzer DT, Sasatomi E, Borhani AA, Heller MT, Tublin ME. Infiltrative hepatocellular carcinoma: what radiologists need to know. Radiographics 2015;35:371-86.
15. Thian YL, Low AS, Chow PK, Ooi LL, Chung AY, Low SC, Xie W, Thng CH. Atypical enhancement pattern of hepatocellular carcinoma with portal vein thrombosis on multiphasic CT. Ann Acad Med Singapore 2011;40:454-9.
16. Lam A, Fernando D, Sirlin CC, Nayyar M, Goodwin SC, Imagawa DK, Lall C. Value of the portal venous phase in evaluation of treated hepatocellular carcinoma following transcatheter arterial chemoembolisation. Clin Radiol 2017;72:994.e9-16.
17. Furlan A, Marin D, Vanzulli A, Patera GP, Ronzoni A, Midiri M, Bazzocchi M, Lagalla R, Brancatelli G. Hepatocellular carcinoma in cirrhotic patients at multidetector CT: hepatic venous phase versus delayed phase for the detection of tumour washout. Br J Radiol 2011;84:403-12.
18. Monzawa S, Ichikawa T, Nakajima H, Kitanaka Y, Omata K, Araki T. Dynamic CT for detecting small hepatocellular carcinoma: usefulness of delayed phase imaging. AJR Am J Roentgenol 2007;188:147-53.
19. López-Larramona G, Lucendo AJ, González-Castillo S, Tenias JM. Hepatic osteodystrophy: an important matter for consideration in chronic liver disease. World J Hepatol 2011;3:300-7.
20. Lange PA, Stoller JK. The hepatopulmonary syndrome: effect of liver transplantation. Clin Chest Med 1996;17:115-23.
21. Lau WY, Sangro B, Chen PJ, Cheng SQ, Chow P, Lee RC, Leung T, Han KH, Poon RT. Treatment for hepatocellular carcinoma with portal vein tumor thrombosis: the emerging role for radioembolization using yttrium-90. Oncology 2013;84:311-8.
22. Joskin J, de Baere T, Auperin A, Tselikas L, Guiu B, Farouil G, Boige V, Malka D, Leboulleux S, Ducreux M, Baudin E, Deschamps F. Predisposing factors of liver necrosis after transcatheter arterial chemoembolization in liver metastases from neuroendocrine tumor. Cardiovasc Intervent Radiol 2015;38:372-80.
23. Gorodetski B, Chapiro J, Schernthaner R, Duran R, Lin M, Lee H, Lenis D, Stuart EA, Nonyane BA, Pekurovsky V, Tamrazi A, Gebauer B, Schlachter T, Pawlik TM, Geschwind JF. Advanced-stage hepatocellular carcinoma with portal vein thrombosis: conventional versus drug-eluting beads transcatheter arterial chemoembolization. Eur Radiol 2017;27:526-35.
24. Zhao Y, Duran R, Chapiro J, Sohn JH, Sahu S, Fleckenstein F, Smolka S, Pawlik TM, Schernthaner R, Zhao L, Lee H, He S, Lin M, Geschwind JF. Transarterial chemoembolization for the treatment of advanced-stage hepatocellular carcinoma. J Gastrointest Surg 2016;20:2002-9.
25. Riaz A, Ryu RK, Kulik LM, Mulcahy MF, Lewandowski RJ, Minocha J, Ibrahim SM, Sato KT, Baker T, Miller FH, Newman S, Omary R, Abecassis M, Benson AB 3rd, Salem R. Alpha-fetoprotein response after locoregional therapy for hepatocellular carcinoma: oncologic marker of radiologic response, progression, and surivival. J Clin Oncol 2009;27:5734-42.
26. Giannini EG, Sammito G, Farinati F, Ciccarese F, Pecorelli A, Rapaccini GL, Di Marco M, Caturelli E, Zoli M, Borzio F, Cabibbo G, Felder M, Gasbarrini A, Sacco R, Foschi FG, Missale G, Morisco F, Svegliati Baroni G, Virdone R, Trevisani F; Italian Liver Cancer (ITA.LI.CA) Group. Determinants of alpha-fetoprotein levels in patients with hepatocelular carcinoma: implications for its clinical use. Cancer 2014;120:2150-7.
27. Salem R, Mazzaferro V, Sangro B. Yttrium 90 radioembolization for the treatment of hepatocellular carcinoma: biological lessons, current challenges and clinical perspectives. Hepatology 2013;58:2188-97.
28. Riaz A, Lewandowski RJ, Kulik LM, Mulcahy MF, Sato KT, Ryu RK, Omary RA, Salem R. Complications following radioembolization with yttrium-90 microspheres: a comprehensive literature review. J Vasc Interv Radiol 2009;20:1121-30.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Mehta A, Venkat SR, Portelance L, Feun LG. Congenital absence of the portal vein complicated by hepatocellular carcinoma in the liver of an adult woman: review of imaging, literature and management. Hepatoma Res 2018;4:7. http://dx.doi.org/10.20517/2394-5079.2017.35
AMA Style
Mehta A, Venkat SR, Portelance L, Feun LG. Congenital absence of the portal vein complicated by hepatocellular carcinoma in the liver of an adult woman: review of imaging, literature and management. Hepatoma Research. 2018; 4: 7. http://dx.doi.org/10.20517/2394-5079.2017.35
Chicago/Turabian Style
Mehta, Ankit, Shree R. Venkat, Lorraine Portelance, Lynn G. Feun. 2018. "Congenital absence of the portal vein complicated by hepatocellular carcinoma in the liver of an adult woman: review of imaging, literature and management" Hepatoma Research. 4: 7. http://dx.doi.org/10.20517/2394-5079.2017.35
ACS Style
Mehta, A.; Venkat SR.; Portelance L.; Feun LG. Congenital absence of the portal vein complicated by hepatocellular carcinoma in the liver of an adult woman: review of imaging, literature and management. Hepatoma. Res. 2018, 4, 7. http://dx.doi.org/10.20517/2394-5079.2017.35
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 2 clicks
Cite This Article 2 clicks

 Like This Article 13
likes
Like This Article 13
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.