
Cancer Evo-Dev, a novel hypothesis derived from studies on hepatitis B virus-induced carcinogenesis

Abstract
Non-resolving inflammation, which may be maintained by infection, pollution, and metabolic stimulants and their interactions with immunogenetic predisposition, provides a fertile field for cancer development. This is strongly evident in hepatocellular carcinoma. Here, the framework of a hypothesis called Cancer Evo-Dev is presented, based on the advances in hepatitis B virus-induced hepatocarcinogenesis. Several aspects central to this theory are as follows: (1) immune imbalance caused by the interaction of immunogenetic predispositions and hepatitis B virus infection maintains non-resolving inflammation; (2) active inflammation executants promote mutations in viral and host genomes via disbalancing mutagenic forces including cytidine deaminases and mutation-repairing forces including uracil-DNA glycosylases, thus promoting cancer-related somatic mutations and viral mutations; (3) a small percentage of the cells whose somatic mutations alter the survival signalling adapt to the inflammatory microenvironment, de-differentiate via demethylating role of cytidine deaminases, and reversely develop into tumor-initiating cells (TICs); (4) under the cultivation of some factors like POSTN from tumor-infiltrating fibroblasts and M2 macrophages, TICs acquire the stemness, cancer-stem cells obtain distinct metastatic and drug-resistant potentials under the selection pressure from distinct microenvironments; (5) glycolysis persistence in the presence of oxygen provides essential energy for cell survival and the raw material for DNA synthesis. Thus, cancer development is characterized by an evolutionary process of “mutation-selection-adaptation”. The framework of Cancer Evo-Dev can be verified in other cancers. Cancer Evo-Dev lays theoretical foundation for understanding the mechanisms by which inflammation promotes cancer development, and it also plays a role in specific prophylaxis, prediction, and targeted treatment of cancers.
Keywords
Non-resolving inflammation and hepatocellular carcinoma
Inflammation, firstly characterized as “heat, redness, pain, and swelling” by a Roman physician Cornelius Celsus, is a complex biological response to harmful stimuli such as infections and tissue damage. Inflammation can be classified into acute inflammation and chronic inflammation. Acute inflammation, also termed as resolving inflammation, is an initial stage of inflammation mediated through activation of innate immunity; it lasts for short period. Chronic inflammation, also termed as non-resolving inflammation, is the second stage of inflammation that persists for a long period of time. Chronic inflammation may develop from acute inflammation if the stimuli are not eradicated or inflammation appears with a chronic process, reflecting the weak but active nature of host immunity. Although the two kinds of inflammation are closely linked to form a correlative antagonistic unity, inherent mechanisms regarding proinflammatory molecules, types of infiltrating macrophages, and inflammatory pathways are distinct, as shown in Figure 1[1]. Acute inflammation is often regarded as therapeutic inflammation to ward off infections and/or to repair the tissue damage; whereas chronic inflammation is now considered as pathogenic, being closely linked with most chronic illnesses, such as cancer, cardiovascular diseases, diabetes, obesity, pulmonary diseases, neurologic disorders, and even depression[2]. Non-resolving inflammation is the prerequisite for the development of most cancers. For examples, chronic hepatitis B or C, chronic bronchitis, chronic colitis including ulcerative colitis, chronic cervicitis, chronicatrophic gastritis, and chronic esophagitis (gastroesophageal reflux disease - caused Barrett’s esophagus) often precedes liver cancer, lung cancer, colorectal cancer, cervical cancer, gastric cancer, and esophageal cancer, respectively. Non-resolving inflammation is clearly evident in the development of hepatitis B virus (HBV)-induced hepatocellular carcinoma (HCC). It is generally believed that oral-administered antiviral therapy decreases the risk of developing HCC in patients with chronic hepatitis B (CHB)[3]. However, the risk of developing HCC is significantly higher in the oral nucleos(t)ide analogues-complete responder group compared with the inactive CHB group, regardless of the presence of baseline liver cirrhosis[4], indicating that continuous active inflammation in liver facilitates the development of HCC. Although surgical technologies for the treatment of liver cancer have been improved, postoperative prognosis remains to be precisely evaluated[5-7]. Active inflammation on chronic inflammation background, as reflected by an Ishak hepatic inflammation score (> 6), a higher neutrophil-to-lymphocyte ratio (> 5), and a higher C-reactive protein in sera (> 0.3 mg/dL), etc., also indicate a poor postoperative prognosis such as postoperative recurrence and shorter recurrence-free survival in HBV-related HCC (HBV-HCC) patients[8,9]. Nuclear factor-κB (NF-κB) and signal transducers and activators of transcription 3 (STAT3) are two most important transcription factors involved in inflammatory pathways that play predominant roles in carcinogenesis, especially in HBV-induced hepatocarcinogenesis[10,11]. Thus, inflammatory microenvironment including proinflammatory molecules, tumor-associated fibroblasts, and tumor-associated immune cellswith altered expression of the inflammatory pathways facilitates the evolution and development of cancers.
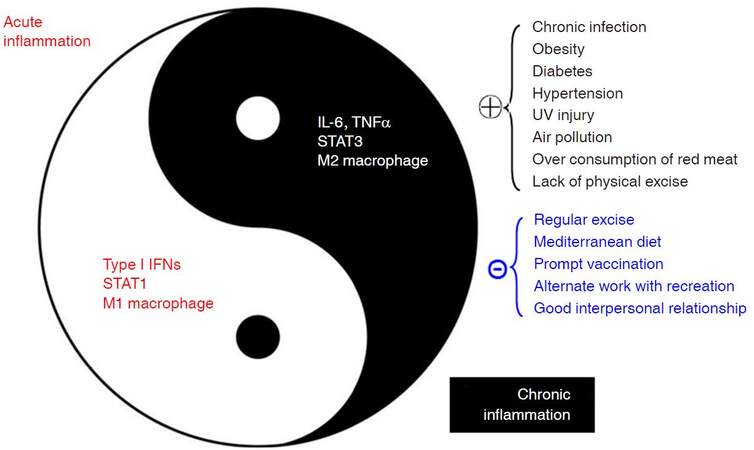
Figure 1. Factors affecting chronic inflammations and their associations with acute inflammations. IFN: interferon; STAT: signal transducers and activators of transcription; IL: interleukin; TNF: tumor necrosis factor; UV: ultraviolet
Maintenance of chronic HBV infection andhepatic inflammation
Chronic transformation of HBV infection relies on three aspects: infection occasion, the characteristics of HBV genotypes, and genetic predisposition of the key immune molecules. HBV infection in early childhood is generally believed to be one of the major causes of chronic HBV infection in adulthood. Of the infants born to hepatitis B surface antigen (HBsAg)-positive mothers globally, 42.1% who did not receive HBV passive-active immunoprophylaxis and 2.9% of infants who received the immunoprophylaxis acquired HBV infection perinatally. The perinatal infection occurred in 84.2% and 8.7% of infants born to hepatitis B e-antigen (HBeAg)-positive mothers who did not and did receive immunoprophylaxis, respectively. The infection rates were 6.7% and 0.4% for infants born to HBeAg-negative-carrier mothers, respectively. Moreover, the chronicity rates of HBV infection acquired perinatally were 28.2% in infants born to HBeAg-negative mothers and 64.5% in infants born to HBeAg-positive mothers[12]. This is possible due to the fact that the immaturity of immune system in infants make it unable to recognize HBV as an external antigen, thus establishing chronic HBV infection although the immune response can be aroused thereafter. Clearly, perinatal HBV infection is an important but not the predominant cause of chronic HBV infection in adulthood. Chronic transformation of acute hepatitis B caused by horizontal transmission among adolescents and adults contributes to the remaining proportion of chronic HBV infection. Approximately 8.5% of acute hepatitis B in adults in Shanghai, China, develops into chronic HBV infection 6 months after acute infection[13]. In mainland China where genotypes B (B2) and C (C2 and C1) are endemic, HBV subgenotype B2 is more apt to causing acute infection because of higher viral load in the virus-providing chronic carriers whereas HBV subgenotype C2 is more apt to causing chronic transformation following an acute course[13,14]. HBV C2 is more likely to develop liver cirrhosis and HCC than does HBV B2, the two major HBV subgenotypes endemic in China, possibly because of the stickiness nature of HBV subgenotype C2[15-17]. The third most important cause of chronic HBV infection and active inflammation is the genetic predispositions of some key immune and proinflammatory molecules. Genome-wide association study in the eastern Asian populations have shown that a total of 11 single nucleotide polymorphisms (SNPs) in genetic loci including HLA-DPA1 and HLA-DPB1, some SNPs in genetic loci including HLA-DQ and -DR, and a locus near HLA-Care significantly associated with CHB[18-22]. Interestingly, these SNPs in the loci encoding human leukocyte antigen-class II (HLA-II) are also significantly associated with vaccine response as well as the risks of acute-on-chronic liver failure, HBV-related liver cirrhosis, and HBV-associated HCC[23-27]. Interestingly, different human races have different allelic frequencies of SNPs that affect the expression of HLA-DP, HLA-DQ, and the inhibitory component of NF-κB complex IκBa gene NFKBIA. These genetic loci whose dominant alleles are significantly association with increased risks of chronic progression of HBV infection (or whose rare alleles are significantly associated with decreased risks of chronic HBV infection) include rs3138053 (affecting NFKBIA), rs2856718, rs7453920, and rs9275319 (affecting HLA-DQ), and rs9277378, rs2395309, rs2301220, and rs9277341 (affecting HLA-DP)[12]. The polymorphic genotypes that are significantly association with increased risks of chronic progression of HBV infection as well as immune selection of the end-stage liver diseases-associated HBV mutations are more frequent in the Han Chinese than in European populations. These data indicate that the Han Chinese are inherently more apt to progressing into chronic infection once exposed to HBV infection than European, whereas European tend to recover from HBV infection spontaneously[12]. This might be one of the reasons why chronic HBV infection and the HBV-induced end-stage liver diseases are more frequent in Chinese than in European populations. The HLA-II genetic polymorphisms may predispose immune imbalance upon HBV infection, impair immune function for HBV clearance, and maintain chronic HBV infection and hepatic inflammation, and thus facilitating the progression of CHB into liver cirrhosis and HCC.
"Dead-end" evolution of HBV
During HBV-induced hepatocarcinogenesis, both of hepatic cells and the viruses experience the process of evolution. Viral evolution serves as a valuable clue to investigate the mechanism underlying HBV-induced hepatocarcinogenesis. HBV belongs to the Hepadnaviridae family found in both mammals (orthohepadnaviruses) and birds (avihepadnaviruses) and is highly conserved in their host species during the long-term evolutionary process. Although primate hepadnaviruses are indigenous to their hosts, hepadnaviruses isolated from apes are grouped as HBV genotypes in phylogenetic analyses. With only 5% divergence from the chimpanzee viral isolates, the isolates from gorilla are categorized in the HBV genotypes. Avihepdnaviruses are the most distant relatives of HBV with a nucleic acid homology of only 40%. Compared to avihepdnaviruses, Woodchuck hepatitis virus and ground squirrel hepatitis virus as mammalian hepadnaviruses are more closely related to HBV and differ by only 17%. Genetic evolution analysis indicates that HBV and orthohepadnaviruses from non-human primates are phylogenetically clustered in the same evolutionary branch[28]. These evidences indicate that members of Hepadnaviridae family are highly conserved in their evolutionary history. However, HBV experiences a relative rapid evolution in their genome since chronic HBV infection is established in a subset of infected populations.
Previous researches by our group identified the wild-type HBV sequences (so-called the standard sequences) of HBV subgenotypes B2 and C2, based on the whole HBV genome sequenced using approximately 1,000 asymptomatic HBsAg carriers from community-based epidemiological surveys in the Yangtze river delta region of mainland China. Based on the wild-type HBV sequences, we subsequently characterized the end-stage liver diseases-related mutations and their development patterns in HBV subgenotypes B2 and C2. We found that the HBV mutations posing a significant risk of HCC or liver cirrhosis were mainly located within the enhancer II/basal core promoter/precore (EnhII/BCP/PreC) and preS regions of HBV genome[29-31]. We summarized the data concerning the association of the HBV mutations and HCC risk published up to 2009, and found that the frequencies and locations of the HBV mutations accumulate consecutively during the “trilogy” of HBV-induced carcinogenesis (CHB, liver cirrhosis, and HCC) and that these HBV mutations can be applied to predict the occurrence of liver cirrhosis and HCC[32]. In our prospective cohort study, we have identified the baseline HBV mutations (C1653T, A1762T/G1764A, and T1753V) increase the risk of HCC both independently and “dose-dependently”. The so-called “dose-dependently” is referred to that the HCC risk is significantly higher in the CHB patients carrying one of the three mutations than in those without the mutation, is significantly higher in those with two of the three mutation than in those with one of the three mutation, and is also significantly higher in those with all the three mutation than in those with two of the three mutations. Thus, the baseline HBV mutations in combination are able to predict the occurrence of HCC in CHB patients[33]. Several longitudinal studies carried in China have also demonstrated that baseline A1762T/G1764A mutation increases the risk of HCC in chronic HBV carriers or CHB patients[34-37]. Among the HCC-risk HBV mutations, the A1762T/G1764A is usually detected in the early stage in young adolescents, while other mutations including T1753V, C1653T, G1899A, and preS deletion appear only at the late stage of chronic HBV infection[12,38]. Reaction to chronic HBV infection, as characterized by immune response-induced hepatocyte damage and release of transaminase, facilitates the generation of the HBV mutations, indicating active immune selection of the HBV mutants during HBeAg seroconversion from HBeAg-positive to HBeAg-negative. One of the main features of HBV mutations is a deficiency of the CD8+ T-cell epitope, a consequence of immune selection. Reduction of CD8+ T cell epitopes of HBV is one of the common strategies to evade immune eradication. HBV that has a low density of CD8+ T cell epitope in their core and X proteins are selected during long-term evolution[39], thus CD8+ T cells play an important role in the immune selection of HCC-related HBV mutants [Figure 2].
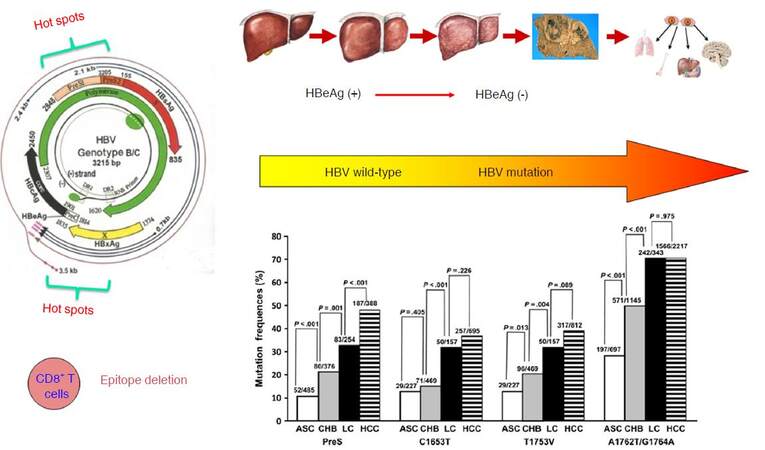
Figure 2. The HCC-related HBV mutations arehallmark molecular events during HBV-induced carcinogenesis. ASC: asymptomatic HBsAg carrier; CHB: chronic hepatitis B; LC: liver cirrhosis; HBeAg: hepatitis B e antigen; HCC: hepatocellular carcinoma
HBV acquired during infancy or early childhood, or at early infection stage in adults, is usually in the form of wild-type[12,38]. In the initial immune tolerant phase of chronic HBV infection, HBeAg is positive, viral load is high, and immune pressure is weak. With the progression of chronic infection, especially during HBeAg seroconversion, the proportion of HBV mutants gradually increases[40]. Although the HBV strains carrying the HCC-related mutations are present in the cord blood of infants, neonatal infection is usually caused by wild-type HBV strain rather than the mutant ones. In the HBV-infected children, the frequencies and locations of HCC-related mutations increase with increasing age. However, compared with their mothers who have been exposed to chronic infection for approximately 25 years, children have fewer HCC-related HBV mutations[38]. In individuals with chronic HBV infection, HBV is synthesized and packaged in hepatocytes and released into the circulation at a pace of up to 1011 viral particles daily. HBV is regularly cleared from the circulation by the host immune system, with a half-life of approximately 1.2 days. Thus, hepatocytes and their surrounding immune cells are responsible for the generation of HBV mutants[41]. HBV reverse transcriptase lacks proofreading activity, resulting in an estimated mutation rate of 4.57 × 105 nucleotide substitutions per site per year and this rate will increase after HBeAg seroconversion[42]. Inflammatory factors in the microenvironment of inflammatory liver promote the generation of HBV mutations, at least partially, via activating the human apolipoprotein B mRNA-editing enzyme catalytic polypeptide (APOBEC) family of cytidine deaminases[43,44]. Although many HBV genome fragments including the PreC/BCP/EnhII region and the S region are generally sensitive to editing by members of APOBEC3, the sequence encoding HBV X protein (HBx) is more vulnerable. APOBEC3 prefers the HBx region as its editing target and generates carboxylic acid-terminal truncated HBx (Ct-HBx)[44], the main form of HBV DNA integrated into the host genome. Insufficient immune responses elicited by HBV antigens select disease-related HBV mutations during the long-term infection process. Only the HBV variants best adapted to the host immune system will survive and thrive in liver. HBV accumulates mutations via minimizing the total number of epitopes recognized by CD8+ T cells, particularly in the HBx and the preS1/preS2/S regions, to avoid immune clearance[39]. These HBV mutations are probably selected via virus-immune interactions in the inflammatory microenvironment. Because of overlapping open reading frames, HBV mutations altering the genes necessary for viral replication are unlikely transferred into their progeny viruses. Natural selection ensures only the fittest survive to pass their genes on to the next generation. Thus, the random natural mutations are therefore constrained to special regions of the HBV genome.
The inflammatory microenvironment of liver tissue is therefore necessary for the co-evolution of HBV and the host genome[45]. Although tumor-adjacent tissues are pathologically categorized as inflammatory hepatic tissues, they are typical precancerous lesions and have already reached the middle stage of HCC evolutionary process because somatic mutations might occur in the inflammatory liver tissues. Somatic mutations in given genomic locations will become the driving mutations if they confer growth advantage of the mutated hepatocytes. The HCC that relapses more than 2 years after curative resection is believed to be mostly recurrent HCC and not from a spread of the initial HCC cell diffusion into the remnant liver tissue. The species and frequencies of certain HBV mutations in adjacent tissues are different in HBV-infected patients with HCC (HBV-HCC patients) with distinct postoperative prognosis. Together with immune markers and expression levels of inflammatory genes in adjacent hepatic tissues, the HBV mutations can be applied to predict postoperative prognosis in HCC patients[46]. The HBV mutations in the EnhII/BCP/PreC region such as A1762T/G1764A in the remnant liver after curative surgery or in the circulation before liver transplantation have been proven to be predictive markers for postoperative survival and HCC recurrence, although this result has not been repeated by some research groups[47-49]. This indicates that HBV evolution in adjacent tissues continues until the patient dies. Antiviral therapy that can attenuate viral replication and subsequent hepatic inflammation notably promotes postoperative prognosis of HBV-HCC patients[50-53], possibly because antiviral treatment can block HBV evolution in adjacent hepatic tissues and also likely in remaining HCC tissues.
Thus, Hepadnaviridae family members, including HBV, are highly conservative across species with distinct but connected evolutionary background. Wild-type HBV has the advantage of infecting hepatocytes of new hosts, facilitating viral spread from one individual to another, and contributing to the maintenance of viral species. The HCC-related HBV mutants can cause or promote malignant transformation, but might have lost the advantage of person-to-person transmission. Those mutants are therefore eliminated if their host individuals died of the end-stage liver diseases including HCC, which is termed “dead-end” evolution of HBV.
The HCC-related HBV mutated X or large S fragments promote the malignant phenotypes
As described above, the baseline HBV mutations including A1762T/G1764A, C1653T, and T1753Vin the 3’ terminal of HBx gene in sera “dose-dependently” predict the occurrence of HCC in longitudinal studies, especially cohort studies[33-37]. The frequency of HBV preS deletion also increases consecutively from ASCs to HCC and preS mutation is associated with a 3.77-fold increased risk of HCC[32]. The predictive value of HBV preS deletion on the occurrence of HCC in patients chronically infected with HBV has been confirmed in a prospective study[54]. Recent deep sequencing analysis has demonstrated that the preS deletions involving a specific fragment (nt2977-3013) in HBV genotype C are significantly associated with HCC[55]. These epidemiological evidences indicate that the HBV mutations including preS deletion, A1762T/G1764A, C1653T, and T1753V are the etiological factors of HBV-induced HCC. Experimentally, in vitro transfection with the HBx mutants with changes that correspond to A1762T/G1764A, T1753A, T1768A, or a combination of these (combo) showed that the combo mutant decreased levels of p21, increased cyclin E expression, and increased expression of S-phase kinase-associated protein 2 (SKP2) in primary human hepatocytes and HepG2 cells. The combo mutant accelerated p21 degradation and cell cycle progression in HepG2 cells. Thus, HBx mutants with changes that correspond to a combination of the core promoter mutations up-regulate SKP2, which then down-regulates p21 via ubiquitin-mediated proteasomal degradation. The core promoter mutations might increase the risk of HCC by this pathway[56]. Transfection of full-length HBV genome with the core promoter mutations in combination also upregulated SKP2 expression via activating the E2F1 transcription factor and in turn downregulate cell cycle inhibitors, thereby accelerating cellular proliferation[57]. Mutations in the preS and S regions also notably facilitate carcinogenesis. Transfection of Huh7 cells with the large S region with preS deletion has shown that HCC-associated single-nucleotide variants (SNVs) in the small surface region of HBV genome influence carcinogenesis pathways, including endoplasmic reticulum-stress and DNA repair systems[55]. The HBV large envelope protein gene fragment (preS1/preS2/S), with F141L mutation in the preS2 region, can significantly promote the proliferation of hepatocytes by downregulating the p53 and p21 pathways and upregulating the expression of cyclin-dependent kinase 4 and cyclin A. The colony-forming rates of hepatocytes expressing F141L-large envelope protein are about twice as high as those expressing the wild-type HBV large envelope protein[58]. Random integration of HBV DNA into the host genome is present in HBV-infected subjects. If the integration events endow the hepatocytes with growth advantage, the integration might facilitate the development of HCC, therefore, have the opportunity of being recorded. HBV integration is common in HBV-HCC, leading to the truncation of the HBV genome, particularly at the C terminus of HBx (Ct-HBx)[59]. Ct-HBx can enhance cell invasiveness and metastasis of HCC in a manner that is more potent than that evoked by full-length HBx and often predict the poor postoperative prognosis and ineffectiveness of antiviral prophylaxis for HCC recurrence[51,60]. These evidence indicates that some HBV X mutants and large S mutants can promote the development and progression of HCC.
Interaction of genetic predispositions of immune or inflammatory molecules with the HBV mutations in HBV-induced hepatocarcinogenesis
As described above, the HLA-II genetic polymorphisms are statistically associated with the outcomes of exposureto HBV[18-27]. This genetic background may predispose immune imbalance upon HBV infection. In our previous epidemiological studies, we found that the HLA-II genetic polymorphisms are statistically associated with the generation of the liver diseases-associated HBV mutations. The HLA-DP polymorphisms rs3077 (CT + TT vs. CC), rs3135021 (GA + AA vs. GG), rs9277535 (GA + AA vs. GG), and rs2281388 (CC vs. CT + TT) significantly decrease HBV persistence in genotype B HBV-infected subjects; HLA-DP genotypes that promote HBV clearance are associated with a lower prevalence of HBV mutations increasing HCC risk (C1653T, T1674C/G, A1846T, G1896A, preS2 mutations, and preS deletion in genotype C) and a higher prevalence of HBV mutations decreasing HCC risk (G1652A, T1673C, T1674C, G1719T, G1730C, and G1799C in genotype B and A1727T in genotype C); furthermore, significant effects of HBV mutations on cirrhosis and HCC are selectively evident in those with the HLA-DP genotypes that promote HBV persistence[61]. Thus, the HLA-DP polymorphisms affect genotype B HBV clearance, regulate immune selection of viral mutations, and influence cirrhosis and HCC risks contributed by the HBV mutations. In addition, HLA-DQ genetic polymorphisms rs2856718 variant genotypes are significantly associated with an increased frequency of HBV A1726C mutation, a cirrhosis-risk, HCC-protective mutation, in genotype C; a rs9275319 variant genotype (GG) is significantly associated with an increased frequency of preS1 start codon mutation, an HCC-risk mutation, in genotype C. Thus, the HLA-DQ polymorphisms affect the risks of cirrhosis and HCC differently in chronic HBV-infected subjects, possibly via interacting with the HBV mutations[62]. As NF-κB and STAT3 are two most important inflammatory pathways[10,11], their genetic predispositions affecting the expression of both signaling pathways may play roles in HBV-induced hepatocarcinogenesis. We have demonstrated that STAT3 SNP rs2293152 (GG vs. CC) is significantly associated with HCC risk compared with the subjects without HCC. Compared with HCC-free HBV-infected subjects, rs2293152 GG is solely associated with HCC in women. This genotype is significantly associated with high viral load (≥ 1 × 104 copies/mL) and increased frequencies of T1674C/G and A1762T/G1764A. Multiplicative interaction of STAT3 rs1053004 with T1674C/G significantly increases HCC risk, whereas rs2293152 and A1726C interaction reduces it, adjusting for covariates including HBV mutations in the EnhII/BCP/PreC region; the interaction of rs4796793 with preS2 start codon mutation significantly increases HCC risk, adjusting for covariates including HBV mutations in the preS region. Thus, STAT3 SNPs appear to predispose the host with HBV mutations to hepatocarcinogenesis[63]. We have also demonstrated that genetic polymorphisms improving NF-κB activity contribute to genotype B HBV clearance. In the genotype C HBV-infected subjects, variant genotypes of rs2233406 (NFKBIA-826C>T) are significantly associated with an increased risk of HCC compared with HCC-free HBV-infected subjects and significantly increase the frequencies of HCC-related HBV mutations including A1762T/G1764A, T1753V, preS1 start codon mutation, and preS deletion; Del allele of rs28362491 (NFKB1-94Ins>Del) significantly increase the frequency of A1762T/G1764A and reduce the frequency of preS2 start codon mutation. The variant genotypes impair NFKBIA promoter activity in hepatic cells. The interaction of rs2233406 variant genotypes (CT + TT vs. CC) with A1762T/G1764A significantly increase the risk of HCC in genotype C HBV-infected subjects[64,65]. These lines of evidence imply that immunogenetic polymorphisms may predispose chronic transformation of HBV infection, increase the frequencies of viral mutations via activating cytosine deaminases, and facilitate immune selection of HCC-causing HBV mutations via arousing active but not effective immune response against the pathogen.
Development and evolution (Dev-Evo), a novel hypothesis related to carcinogenesis
Development is referred to the process that a fertilized egg develops into an individual. In humans, the fertilized diploid cell composing of paternal haploid and maternal haploid differentiates into various functional and/or structural cells to form different organs and tissues of an infant in mother’s uterus within 40 weeks. The developmental process is a succession of functional and morphologic changes from a single cell form (fertilized egg) to a multicellular form (blastocyst), from an aquatic state (living in amniotic fluid) to a terrestrial state (pulmonary respiration). During this process, the founding diploid cell, a progenitor, divides rapidly and gives many other different types of cells via altering their gene expression profiling. The changes in gene expression profiling are achieved by epigenetic modifications such as methylation in the upstream regulatory regions of given genes, rather than altering the primary sequences of these genes. After born, lung takes over the responsibility of gas exchange, some genes only expressed in the embryonic stage are silenced and other genes solely expressed in adult cells are activated, most possibly by some epigenetic modifications. Surprisingly, the developmental process of humans resembles the process of long-term organic evolution morphologically, e.g. from single cell creatures to multicellular creatures, and from aquatic creatures and amphibian to terrestrial mammals [Figure 3]. Furthermore, some evolutionarily conserved molecular networks such as HOX, Hedgehog, and Myc play important roles in the developmental process[66-72], indicating development and evolution share some inherent mechanisms. During the past 20 years, the discovery of conserved gene networks that control embryonic development and the ability to examine genomic records has revolutionized Darwinian evolutionism that animal relationships had to be deduced by observation of external morphological characteristics. The integration between developmental biology and evolution has been named Evo-Devo[73-77]. Dr. Raff pointed out that the evolution of complex organisms such as animals and plants had involved marked changes in morphology and new features had appeared; but evolutionary change occurred not by the direct transformation of adult ancestors into adult descendants but rather when developmental processes produced the features of each generation in an evolving lineage. Thus, evolution cannot be understood if do not understand the evolution of development, and how the process of development itself biases or constrains evolution[75]. Based on these previous work, in combination with previous observations, I like to define Evo-Dev as follows: Evo-Dev is a discipline to investigate the inherent mechanisms by which the short-term developmental process resembles the long-term evolutionary process and to characterize the role of developmental process on the evolution of complex organisms.
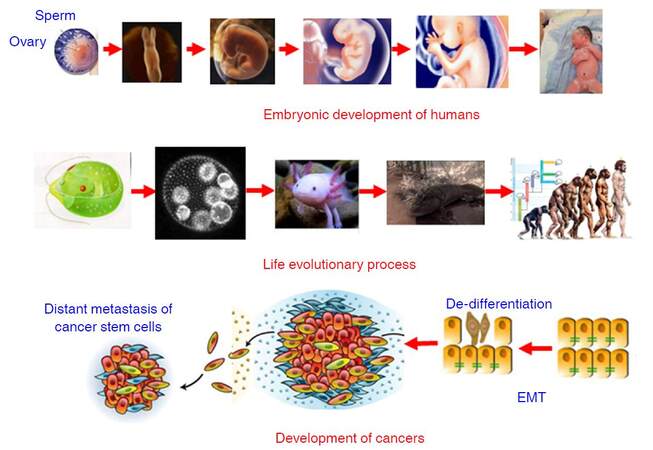
Figure 3. Synthesis of Dev-Evo and its potential link to carcinogenesis. Dev-Evo: synthesis of development and evolution; EMT: epithelial-mesenchymal transition
Carcinogenesis represents an evolutionary process. It was firstly proposed by Dr. Nowell in 1976 that most neoplasms arise from a single cell of origin, and tumor progression results from acquired genetic variability within the original clone allowing sequential selection of more aggressive sublines; tumor cell populations are apparently more genetically unstable than normal cells, perhaps from activation of specific gene loci in the neoplasm[78]. Cancer clone genetic diversification and sub-clonal selection occurs within tissue ecosystems. The implicit parallel was to Darwinian natural selection with cancer equivalent to an asexually reproducing, unicellular, quasi-species[79]. In the past decades, especially after widespread application of new generation sequencing, cancer evolution including a reiterative process of clonal expansion, genetic diversification, and clonal selection within the tissue ecosystems have been extensively investigated. Drs. Greaves and Maley[80] outlined key contents of cancer evolution as follows: (1) cancers are characterised by divergent cells of origin and mutational spectra; (2) cancers evolve over variable time frames (about 1-50 years) and tempos and the clonal structure, genotype and phenotype shifts over time; (3) the number of mutations found in any cancer can vary from 10-20 to hundreds of thousands, the great majority are “passengers” and a few are “drivers”; (4) cancers acquire, via mutational (and epigenetic) changes, a variety of critical phenotype traits that compound to empower territorial expansion, via proliferative self-renewal, migration, and invasion; (5) advanced, disseminated or very malignant cancers appear to be almost uniquely competent to evade therapy; and (6) this complexity can be explained by classical evolutionary principles. Most mutational processes have biases at the DNA sequence level and mutational spectra in cancers can reflect or implicate particular error-prone repair processes or particular genotoxic exposures, e.g. cigarette carcinogens, UV light, and chemotherapeutics[81]. We believe that somatic mutations increase with increasing age for two major reasons: (1) accumulation of exposures to various mutagens increases with increasing age; and (2) “mistake” mutations can be spontaneously generated or introduced in each cell cycle. However, the most of somatic mutations do not alter gene expression-defined key functions related to cell growth advantage or resistance to environmental insults. These mutations can be “passenger mutations”. If a somatic mutation endows the cell a growth advantage in a hostile inflammatory microenvironment, it is termed as “driver mutation”. Cells carrying “driver mutation” are positively selected ininflammatory microenvironment, facilitate the cross-talks with proinflammatory cells including tumor-associated M2 macrophages and neutrophils, promotea process termed as epithelial-mesenchymal transition (EMT) and stepwise de-differentiation into tumor-initiating cells (TICs), and adapt to new inflammatory microenvironment [Figure 3]. In our previous study, we found that expression of periostin (POSTN) from tumor-infiltrating fibroblasts significantly promoted the proliferation, anchorage independent growth, invasion, and chemo-resistance of cancer cells; whereas these effects were counteracted via targeting to PI3K/Akt or Wnt/β-catenin signaling pathway. These evidence indicates that POSTN generated in the microenvironment nurtures the cancer-stemness via activating PI3K/Akt or Wnt/β-catenin signaling pathway[82]. Thus, cancer development represents “mutation-selection-adaptation” evolutionary process in proinflammatory microenvironment, which is quite in accordance with Darwinian evolutionism.
During human embryo development, most genes expressing at this stage will be silenced after birth, but some genes including that encoding α-fetoprotein (AFP), will re-express for a short period of time when liver or testis is injured. Importantly, cancer is characterized as a reverse-developmental process, that is, develop from differentiated cells into undifferentiated cells. Oncofetal proteins are mostly referred to those that normally express at embryonic stage are silenced after birth, and then re-expressed persistently in the circulations of cancer patients. AFP serves as a diagnostic biomarker of HCC. The human homologue of the Drosophila spalt homeotic gene, SALL4, encoding an oncofetal protein Sall4, is one of the key factors for self-renewal and maintenance of embryo stem-cell pluripotency. SALL4 is expressed in the human fetal liver and silenced in the adult liver, but can be detected in a subgroup of HCC. The re-expression of SALL4 is related to the “stem function” of HCC cells and indicates invasion and unfavourable prognosis[83-86]. As a matter of fact, some cells with stem-cell-like characteristics become the main malignant subgroup in tumor tissues; embryonic or stem-like gene expression signatures expressed in cancers of distinct histotypes including HCC are robustly associated with cancer cell self-renewal, EMT, increased aggressiveness, and poor postoperative prognosis[87-92]. Furthermore, the cell aging process is accompanied by the shortening of telomeres, which does not seem to occur in cancer cells. Telomerase activation occurs through telomerase reverse transcriptase (hTERT) induction. hTERT and ZEB1 form a complex, which directly binds to the E-cadherin promoter, and then inhibits E-cadherin expression and promotes EMT[93]. Sirtuin 1 has been implicated in telomere maintenance and HCC growth. Sirtuin 2, another member of the sirtuin family, plays a role in maintaining the motility, invasiveness, and EMT phenotypes of HCC[94]. In terms of morphology, EMT is the process in which epithelial cells lose their epithelial characteristics and acquire mesenchymal characteristics, structure, and biologic function. An EMT usually occurs at a critical stage of embryonic development, and it is equally important for cancer metastasis[95]. In the process of cancer invasion through EMT, epithelial cells acquire “stemness”, including self-renewal and antiapoptotic capacities. Most tumor cells are differentiated, with limited amplification ability. However, a small proportion of tumor cells with the “stemness” feature becomes the main malignant cell subsets in tumors and is known as cancer stem cells, responsible for the disease’s malignant nature and chemo-resistance. Thus, retro-differentiation or reverse-development is the hallmark in cancer development.
Theoretical framework of cancer Evo-Dev
Cancers are caused by the accumulation of somatic mutations - a process that abides by the Darwinian evolutionism: mutation-selection-adaptation. Somatic mutation patterns related to chronic inflammation have been identified in most cancers[96]. In some cancers, the inflammation-related somatic mutations increase with time, accompanied by a decline in the mutations related to the initial exposure[97]. Those distribution characteristics and the switch in mutation domination can be analyzed from an evolutionary perspective, suggesting that inflammation sometimes induced by chronic infection might not only cause somatic mutations, but also play an important role in selecting TICs as a cancer-supportive niche. We ever proposed the scientific hypothesis of Cancer Evolution-Development ("Cancer Evo-Dev") and summarize the basic concepts and theoretical framework[98-100]. Here, some further evidence are presented to optimize the theoretical framework of Cancer Evo-Dev. This novel scientific hypothesis may help in elucidating the mechanisms by which cancer develops and optimizing the most cost-effective ways to control these life-threating diseases.
Figure 4 depicts the framework of Cancer Evo-Dev exemplified by HBV-induced hepatocarcinogenesis. Evolutionary process of HCC is a succession of important molecular events-from inflammatory precancerous lesions to carcinogenesis, postoperative recurrence, and metastasis. Those events include, but are not limited to viral mutation, epigenetic modification, somatic mutations, and alteration of signaling pathway networks. The synergetic effects of genetic and environmental factors contribute to imbalance of the host immune system, resulting in the activation and maintenance of non-resolving inflammation, thus providing a microenvironment for the process of cancer evolution and development. Under conditions of non-resolving inflammation, activated NF-κB complex and proinflammatory molecules can trans-activate the expression of nucleic acid editing enzymes including APOBEC family of cytidine deaminases, rather than uracil DNA glycosylases (UNG), thus promoting viral and somatic mutations. Actually, the imbalance between mutation-promoting forces like AID/APOBECs and mutation-repairing forces like UNGs is responsible for the generation of somatic and viral mutations[99].Viral mutants facilitate the malignant transformation of normal hepatic cells via inducing EMT. Most mutant cells are eliminated by selective pressures imposed by the weak but active immune response. Although a small proportion of mutant cells survive in the hostile inflammatory microenvironment in precancerous lesions. Those surviving mutant clones later evolve into TICs by altering the original cell signal patterns and promoting EMT through epigenetic regulation possibly by APOBECs. Some established cancer markers such as AFP and SALL4 are usually expressed at the embryonic stage, silenced after birth, and re-expressed in cancer patients. The process of cancer development can be characterized as “backward evolution” and “retro-differentiation”.
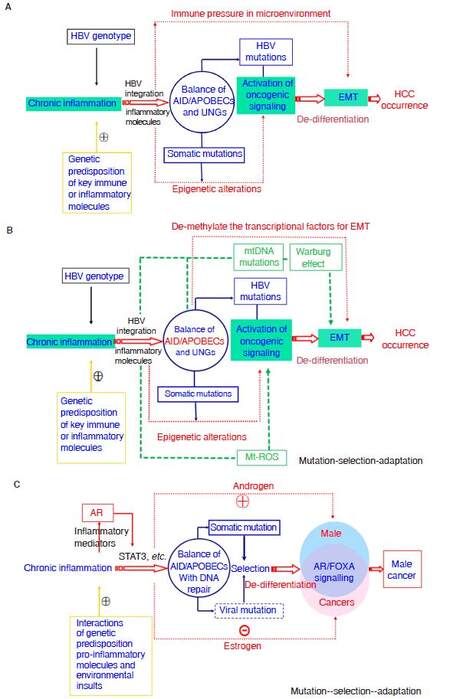
Figure 4. Theoretical framework of Cancer Evo-Dev, as exampled by HBV-induced hepatocarcinogenesis. A: The classic diagram of Cancer Evo-Dev. The imbalance between mutation-promoting forces like AID/APOBECs and mutation-repairing forces like UNGs is responsible for the generation of somatic and viral mutations; B: exposures to some mutagens and HBV infection lead to mitochondria DNA mutations, thus promoting Warburg effect. In addition, AID/APOBECs also demethylates the promoters regions of some transcriptional factors including, thus directly promoting EMT; C: the diagram of Cancer Evo-Dev explaining why HCC is a male sex-predominant cancer. mtDNA: mitochondria DNA; AID/APOBECs: activation-induced cytidine deaminases/the human apolipoprotein B mRNA-editing enzyme catalytic polypeptide; UNG: uracil DNA glycosylase; EMT: epithelial-mesenchymal transition; HBV: hepatitis B virus; AR: androgen receptor; ROS: reactive oxygen species; Mt: mitochondria; HCC: hepatocellular carcinoma
Key issues regarding cancer Evo-Dev
Indispensable role of non-resolving inflammation
It is widely accepted that most solid tumours and some hematologic malignancies are associated with non-resolving inflammation. According to the Darwinian evolutionism and the origin of species, the process of cancer evolution is based on two conditions: the continuous acquisition of somatic mutations and natural selection acting on the resultant phenotypic diversity[101]. A chronic inflammatory microenvironment serves as a niche for that processby inducing endogenous mutagenic factors such as APOBECs and provides selection pressure. During carcinogenesis, cancer cells must overcome four barriers: (1) the cell-cycle checkpoint that regulates cell division; (2) apoptosis, which limits cell proliferation; (3) telomere length, which determines the total number of cell divisions; and (4) the cell adhesion barrier that prevents cell migration. The non-resolving inflammation can alter the “ecologic” conditions in local and/or systematic tissues, weaken the functions of the above barriers, cause genomic instability via inducing the overexpression of AID/APOBECs, and provide opportunities for backward evolution into cancer stem cells in mesenchymal tissues. In inflammatory microenvironment, inflammatory mediators such as prostaglandin E2, leukotrienes, cytokines, and chemokinesare highly induced via autocrine or paracrine modes of action[102], resulting in abnormal transformation of the tissue microenvironment, infiltration of dysfunctional immune cells, and decreased epithelial integrity, thus promoting cancer evolution. Non-resolving inflammation not only promote the occurrence of cancers of the most histotypes, but also facilitate distant metastasis and the recurrence after the treatment[46,103,104], indicating that non-resolving inflammation promotes the development of cancers in the entire course of cancer evolution.
APOBECs bridge inflammation and cancer
The APOBECs, a family of cytidine deaminases, are powerful endogenous mutagenic factors that play critical roles in many biologic processes, especially in innate immunity and humoral immunity. This group of enzymes can specifically catalyze irreversible cytidine and deoxycytidine deamination to convert bases from cytosine to uracil, creating a cytosine-to-uracil mismatch in minus-strand and reverse-transcript guanosine-to-adenosine (G-to-A) transitions in plus-stranded DNA. Activation-induced cytidine deaminase (AID) and APOBEC3 cytidine deaminases were found in the pathways of both the acquired and innate immunities[99]. APOBEC3 cytidine deaminases can also hyper-edit HBV DNA and inhibit HBV replication. APOBEC3 proteins are present at low levels in normal liver, but its gene expression is highly stimulated by both IFN-α and IFN-γ. APOBEC3 cleave amino groups from cytidine bases converting them to uracil in newly synthesized DNA following reverse transcription of pregenomic RNA. This modified HBV DNA is susceptible to degradation, or alternatively, numerous G-to-A nucleotide mutations are incorporated into positive-strand viral DNA[99,105]. This process is counteracted by UNG[106]. Accordingly, APOBECs family members can also increase somatic mutations to a threshold that exceeds the host’s repair ability, thus initiating the cancer evolutionary process. In AID transgenic mouse models, mutations induced in the TP53 and β-catenin genes by constitutive expression of AID can generate HCC (13.75%), lung cancer (8.75%), and gastric cancer (1.25%)[107]. In humans, genetic susceptibility, viral infection, and their interaction contribute to an unbalanced immune system, resulting in chronic inflammation. In the inflammatory microenvironment, the proinflammatory cytokine/chemokine and NF-κB complex are persistently activated, which can significantly increase the expression of APOBECs at the transcription level[108]. The high levels of APOBECs expression can overcome the strength of UNG, APOBECs get the advantage to edit the single-stranded DNAs that are temporarily generated during the transcription and replication process, consequently promoting somatic mutations[109]. If the overall metabolic level exceeds the reserve capacity of the downstream repair pathways, somatic mutations will be further increased. An APOBECs-directed mutagenesis pattern is widespread in human cancers. Significant presence of the APOBEC mutation pattern are evident in bladder, cervical, breast, head and neck, and lung cancers, reaching 68% of all mutations in enrolled tumor samples. The APOBEC mutation pattern also extends to cancer-associated genes, implying that APOBECs-induced mutagenesis is carcinogenic[110]. The spontaneous rate of somatic mutations is not high enough to trigger the evolution process. There must be some mutagenesis-driving forces including defective DNA repair capacity, exogenous or endogenous mutagen exposures, and intrinsic mistakes of DNA replication, which increases the mutation rates in cancer genomes. A distinct mutagenic process generates various mutation combinations termed as “signature”. The APOBECs-related mutation signature is widely prevalent in more than half of all cancer types under investigations, suggesting that the inflammatory response is the common mechanism by which mutations are generated. Even though, the frequencies of somatic mutations in a single gene are not high in the patient population. For example, the rates of mutation in the cording regions of ARID1A and ARID2, two genes with classic HCC-related genetic variations, are 16.8% and 5.6% respectively[111]. Such a low detection rate of each mutation makes it unable to be applied for the prediction, prevention, early diagnosis, and treatment of cancers. However, somatic mutations in different genes with a similar function can alter a specific signal pathway that is related to the stem characteristic of cancers and therefore promote carcinogenesis. For the most malignancies, so-called driver somatic mutations, mostly at low frequencies in tumor tissues, alter a limited number of cellular signaling pathways through which a growth advantage can be incurred. All of the known driver genes can be classified into one or more of 12 pathways of three major functions: cell survival, cell fate, and genome maintenance. Cell survival contains “cell cycle/apoptosis”, “RAS”, “phosphoinositide 3-kinase (PI3K)”, “STAT”, “mitogen-activated protein kinase (MAPK)”, and “transforming growth factor β (TGF-β)”. “NOTCH”, “Hedgehog”, “APC”, “chromatin modification”, and “transcriptional regulation”contribute to cell fate function. Genome maintenance is governed by “DNA damage control”[112]. The combined mutations-affected critical molecules in the signalling pathway networks can be developed as novel diagnostic biomarkers and therapeutic targets. A series of somatic mutations in HBV-induced HCC mainly affect the chromatin remodelling pathways (ARID1A, ARID1B, and ARID2), the p53/RB tumor suppression pathway (IRF2, TP53, and CDKN2A), the Wnt/β-catenin signal pathway (RPS6KA3-AXIN1, NFE2L2-CTNNB1), and the Ras/PI3K pathway (PTEN, PIK3CA, KRAS, NRAS)[111,113-115]. Key genes affecting epigenetic activities including ARID2, encoding a subunit of the polybromo- and BRG1-associated factor (PBAF) chromatin remodeling complex and ARID1A, encoding a component of the SWI/SNF chromatin remodeling complex are most frequent ones. In addition, cell invasion-related factors-coding genes VCAM1 and CDK14, and gene encoding androgen receptor (AR)[113,116]. Both C:G>A:T and T:A>A:T transversions are frequent among the non-silent mutations[114], indicating AID/APOBECs-induced somatic mutation is one of the major mutation patterns. These mutations facilitate the development of HCC via activating some evolutionarily conserved signal pathways, such as PI3K/Akt/mammalian target of rapamycin, NF-κB/TNF-α, Raf/MAPK/ERK, TGF-β1, Jak, Wnt/β-catenin, and STAT3/interleukin 6 (IL-6)[117,118], and also indicate that Wnt/β-catenin signaling may cooperate with both oxidative stress metabolism and Ras/MAPK pathways in hepatocarcinogenesis[113]. ActivatedSTAT3/IL-6 and NF-κB/TNF-α can induce hepatocytes to lose their epithelial characteristics (EMT) and initiate backward evolution. TGF-β1 can facilitate EMT, which can be enhanced by IL-6 and TNF-α. The synergistic effect of those three cytokines can promote the transformation of normal hepatocytes into stem-like cells. Antiviral therapy can significantly reduce the risks of occurrence and postoperative recurrence by HCC via relieving hepatic inflammation[50-53,119], possibly because termination of inflammation can destroy the fertile environment for cancer evolution.
AID/APOBECs-regulated demethylation and EMT are important in malignant transformation
AID/APOBECs not only promote somatic hypermutation but also regulate gene expression epigenetically by directly deaminating 5-methylcytosine (5mC) or 5-hydroxymethylcytosine (5hmC) in concert with base-excision repair to exchange cytosine, thus promoting gene demethylation and removing epigenetic memory to stabilize the pluripotent state in embryonic stem cells[120,121]. EMT, a driving force behind the development of cancers, in its various forms is driven by the transcription factors Snail (SNAI1), Slug (SNAI2), ZEB1 (ZEB1), and ZEB2 (ZEB2). Expression of AID is induced by inflammatory signals that induce the EMT in nontransformed epithelial cells and in cancer cells. AID regulates expression of master regulators (SNAI1, SNAI2, ZEB1, and ZEB2) in the EMT. Knockdown of AID blocks induction of the EMT and prevents cells from acquiring invasive properties, suppresses expression of several key EMT transcriptional regulators and is associated with increased methylation of CpG islands proximal to the promoters of SNAI1, SNAI2, ZEB1, and ZEB2[122]. AID-mediated, CpG-methylation dependent mutagenesis is proven to be a common feature of carcinogenesis[123]. Thus, we have reasons to postulate that re-expression of embryonic factors in cancers as cancer biomarkers might result from epigenetic reprogramming caused by AID/APOBECs, whose expression is induced by proinflammatory factors.
AID/APOBECs promote tumor heterogeneity
There are two kinds of tumor heterogeneity: intertumor heterogeneity and intratumor heterogeneity. First, patients with tumors of the same pathologic type show distinct clinical manifestations, including occurrence, metastasis, therapeutic response to chemo- and radiation-therapies, and postoperative prognosis. This heterogeneity is the basis for the development of biomarkers and therapeutic targets that can predict cancer occurrence, metastasis, and therapeutic response, thus, contributing to personalized medicine. Second, different tumor cells or masses in an individual show significant differences in genomic mutation profile, evolution pathways, and gene expression. This intratumor heterogeneity was discovered and subsequently validated by the application of next-generation sequencing[124,125]. They found that heterogeneous driver alterations that occurred later in evolution were found in more than 75% of the tumors and were common in PIK3CA and NF1 and in genes that are involved in chromatin modification and DNA damage response and repair[125]. An important emerging mechanism fueling tumor diversity and subclonal evolution is genomic DNA cytosine deamination catalyzed by APOBEC3B and at least one other APOBEC family member. Deregulation of APOBEC3 enzymes by different microenvironment causes a general mutator phenotype that manifests as diverse and heterogeneous tumorsubclones. APOBEC mutational signatures may be enriched in tumorsubclones, indicating APOBECs fuel subclonal expansions and tumor heterogeneity. APOBEC family members might represent a new class of drug target aimed at restricting tumor evolution, adaptation, and even chemo-resistance[126]. APOBEC3B-catalysed deamination provides a chronic source of DNA damage in cancers, thus explaining how some tumors evolve rapidly and manifest heterogeneity[127]. Thus, APOBEC3B-catalysed somatic mutations serve as potential drivers in promoting the formation and progression of tumor heterogeneity.
A small proportion of somatic mutations can lead to advantageous phenotypes that are positively selected during the evolutionary process and thus are called “driver” mutations. The remaining mutations are “passengers” that contribute very little to carcinogenesis. Driver mutations are selected at certain phases of carcinogenesis, but might not be detectable at all stages. At the early stage of carcinogenesis, cells with initial driver mutations can survival and multiply rapidly. However, at the later stages, cells with other driver mutations can gain more advantages in the survival competition. They can replace the cells that have only initial mutations and become the dominant subset. For example, in lung cancer patients who continue to be exposed to tobacco smoking, the signatures of the tobacco-related mutations decline over time, accompanied by an increase in the APOBEC-related mutations[128]. Tracing the positive selection of drivers and the patterns of cancer genomic alteration can help in demonstrating the lineage of the malignancy clones and the major mutagenic factors. Exome-sequencing data from solid tumours and hematologic neoplasms confirmed the clonal heterogeneity of primary tumours and metastases, supporting the evolution model at the genetic level[129]. Thus, the APOBEC-related mutations are more likely to be drivers. Tumors in different microenvironments and at different treatment stages might have distinct mutation spectra, thus demonstrating, within a solid tumour, an obvious heterogeneity that is the result of continuously imbalanced evolution that persists under the selection pressure of the microenvironment. Therapies can also serve as selection pressure, bring their own changes in malignant clones, and that evolution-induced heterogeneity will complicate cancer therapeutic regimes. Cancer therapy should therefore be designed as sequential treatments with the specific purpose of targeting critical pathways during cancer evolutionary process.
Energy metabolism and Cancer Evo-Dev
In the 1920s, Otto Warburg and co-workers showed that tumor tissues metabolize approximately tenfold more glucose to lactate in a given time than normal tissues under aerobic conditions, that is, a preferential use of glycolysis for energy production, even in the presence of oxygen, to support rapid growth of cancer cells, a phenomenon known as the Warburg effect[130]. Warburg hypothesized that this phenomenon occurs due to the malfunction of mitochondria in cancer cells. Up to now, there are two conflicting points of view on effects of mitochondria DNA mutations on the Warburg effect. First, the genetic events that drive aberrant cancer cell proliferation also alter biochemical metabolism, including promoting aerobic glycolysis, but do not typically impair mitochondrial function. Mitochondrial biogenesis and quality control are often upregulated in cancers and mitochondria play a central and multifunctional role in malignant tumor progression[131]. Second, mitochondrial mutations could be the origin of the Warburg phenotype by way of hypoxia-inducible factor activation[132]. Pyruvate kinase M2 (PKM2), an alternatively spliced variant of the pyruvate kinase gene that is preferentially expressed during embryonic development and in cancer cells, alters the final rate-limiting step of glycolysis, resulting in the cancer-specific Warburg effect. PKM2 also mediates EMT via interacting with the transcriptional factor TGF-β-induced factor homeobox 2 to induce the deacetylation of histone H3, thus, resulting in repressed E-cadherin expression[133]. In addition, Warburg effect in tumor-associated macrophages (TAMs) promotes vascular network formation, augments extravasation of tumor cells out of blood vessels, and induces higher levels of EMT at inflammatory foci within the tumor[134]. In microenvironment with both hypoxia and hypoglycemia, stem cell-, angiogenic-, and EMT-biomarkers, as well as glycoprotein-P content and invasiveness of cancer cells are enhanced[135]. Thus, we believe that the Warburg effect promotes the evolutionary process of cancer under both hypoxia and hypoglycemia condition. The Warburg effect can provide essential energy for cell survival in hostile microenvironment, furthermore, glycolysis generates the raw material for DNA synthesis of progeny cells.
Roles of Cancer Evo-Dev on specific prophylaxis and targeted therapy of malignancies
Based on studies of HBV-induced hepatocarcinogenesis, we present the hypothesis of Cancer Evo-Dev to elucidate the critical steps of a common evolutionary and developmental process for most malignancies. The framework of Cancer Evo-Dev can be verified in other cancers such as breast cancer, cervical cancer, head and neck cancer, colorectal cancer, gastric cancer, and lung cancers. Key molecular events occur in the critical steps during Cancer Evo-Dev process can be applied for the occurrence and prognosis prediction and specific prophylaxisof malignant diseases. Furthermore, the core molecules in a functional subnetwork that maintains cancer stemness and promotes Cancer Evo-Dev process can be efficiently targeted by the high-efficiency inhibitors to block corresponding signal pathways, thus providing a powerful treatment strategy for advanced cancers. Thus, Cancer Evo-Dev has three major roles in cancer prophylaxis and treatment: first, to identify what kind of precancerous changes or lesions will develop into cancers; second, to testify what kind of prophylactic option or treatment will reduce the cancer incidence and delay its occurrence; and third, to specifically target key pathways that drive the evolution and development of cancer to reduce morbidity and mortality rates. For the control of HBV-induced HCC, a highly fatal malignancy, active prophylaxis should be of top priority [Figure 5]. Our hypothesis can therefore contribute to the realization of "P4 pattern" medicine (predictive, preventive, personalized, and participatory)[2], therefore promote the prophylaxis and control of cancer.
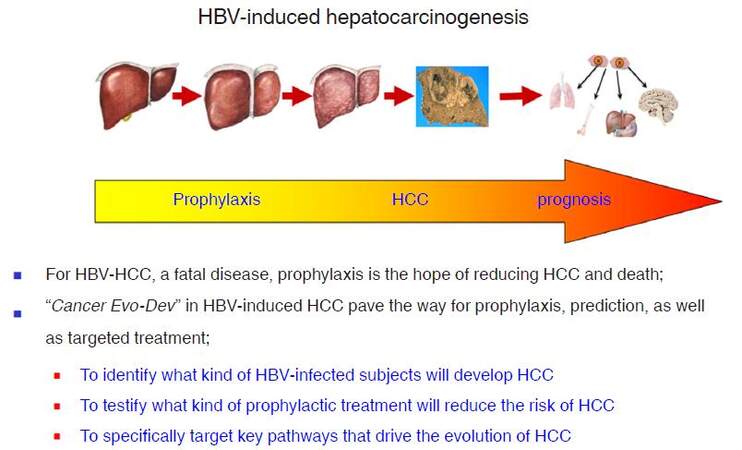
Figure 5. HBV-induced hepatocarcinogenesis. HBV: hepatitis B virus; HCC: hepatocellular carcinoma
Declarations
Authors’ contributionsG.W. Cao contributed solely to the paper.
Financial support and sponsorshipThis work was supported by grants from the National Key Basic Research Program (grant no. 2015CB554000) and the National Natural Scientific Foundation of China (grant no. 91129301). The study sponsors had no role in the writing of the manuscript and in the decision to submit the manuscript for publication.
Conflicts of interestThere are no conflicts of interest.
Patient consentNot applicable.
Ethics approvalNot applicable.
Copyright© The Author(s) 2017.
REFERENCES
1. Cao GW. Non-resolving inflammation. In: Cao GW, editor. Cancer Evo-Dev. Shanghai: Second Military Medical University Press; 2017. pp. 72-110. (in Chinese)
2. Ewald PW. 99th Dahlem conference on infection, inflammation and chronic inflammatory disorders: symbionts and immunopathology in chronic diseases: insights from evolution. Clin Exp Immunol 2010;160:27-34.
3. Du Y, Han X, Ding YB, Yin JH, Cao GW. Prediction and prophylaxis of hepatocellular carcinoma occurrence and postoperative recurrence in chronic hepatitis B virus-infected subjects. World J Gastroenterol 2016;22:6565-72.
4. Cho JY, Paik YH, Sohn W, Cho HC, Gwak GY, Choi MS, Lee JH, Koh KC, Paik SW, Yoo BC. Patients with chronic hepatitis B treated with oral antiviral therapy retain a higher risk for HCC compared with patients with inactive stage disease. Gut 2014;63:1943-50.
5. Fung AKY, Chong CCN, Lee KF, Wong J, Cheung YS, Fong AKW, Lai PBS. Outcomes of emergency and interval hepatectomy for ruptured resectable hepatocellular carcinoma: a single tertiary referral centre experience. Hepatoma Res 2017;3:196-204.
6. Chok KSH. Surgical strategy for huge and advanced hepatocellular carcinoma in Hong Kong. Hepatoma Res 2017;3:189-95.
7. Sarkar J, DeLeon T, Wong LL. MELD score and AST-to-platelet ratio index predict long-term survival in patients with a small hepatocellular carcinoma following non-transplant therapies: a pilot study. Hepatoma Res 2017;3:79-85.
8. Chen L, Zhang Q, Chang W, Du Y, Zhang H, Cao G. Viral and host inflammation-related factors that can predict the prognosis of hepatocellular carcinoma. Eur J Cancer 2012;48:1977-87.
9. Liu WB, Yang F, Shao DY, Cao GW. Novel predictive and prognostic strategies of hepatitis B virus related hepatocellular carcinoma. Hepatoma Res 2016;2:331-40.
10. Aggarwal BB, Vijayalekshmi RV, Sung B. Targeting inflammatory pathways for prevention and therapy of cancer: short-term friend, long-term foe. Clin Cancer Res 2009;15:425-30.
11. He G, Karin M. NF-κB and STAT3 - key players in liver inflammation and cancer. Cell Res 2011;21:159-68.
12. Li Z, Hou X, Cao G. Is mother-to-infant transmission the most important factor for persistent HBV infection? Emerg Microbes Infect 2015;4:e30.
13. Zhang HW, Yin JH, Li YT, Li CZ, Ren H, Gu CY, Wu HY, Liang XS, Zhang P, Zhao JF, Tan XJ, Lu W, Schaefer S, Cao GW. Risk factors for acute hepatitis B and its progression to chronic hepatitis in Shanghai, China. Gut 2008;57:1713-20.
14. Yin J, Zhang H, He Y, Xie J, Liu S, Chang W, Tan X, Gu C, Lu W, Wang H, Bi S, Cui F, Liang X, Schaefer S, Cao G. Distribution and hepatocellular carcinoma-related viral properties of hepatitis B virus genotypes in Mainland China: a community-based study. Cancer Epidemiol Biomarkers Prev 2010;19:777-86.
15. Yin JH, Zhao J, Zhang HW, Xie JX, Li WP, Xu GZ, Shen J, Dong HJ, Zhang J, Wang L, Han JK, Wang HY, Cao GW. HBV genotype C is independently associated with cirrhosis in community-based population. World J Gastroenterol 2010;16:379-83.
16. Chan HL, Wong GL, Tse CH, Chim AM, Yiu KK, Chan HY, Sung JJ, Wong VW. Hepatitis B virus genotype C is associated with more severe liver fibrosis than genotype B. Clin Gastroenterol Hepatol 2009;7:1361-6.
17. Yin J, Zhang H, Li C, Gao C, He Y, Zhai Y, Zhang P, Xu L, Tan X, Chen J, Cheng S, Schaefer S, Cao G. Role of hepatitis B virus genotype mixture, subgenotypes C2 and B2 on hepatocellular carcinoma: compared with chronic hepatitis B and asymptomatic carrier state in the same area. Carcinogenesis 2008;29:1685-91.
18. Kamatani Y, Wattanapokayakit S, Ochi H, Kawaguchi T, Takahashi A, Hosono N, Kubo M, Tsunoda T, Kamatani N, Kumada H, Puseenam A, Sura T, Daigo Y, Chayama K, Chantratita W, Nakamura Y, Matsuda K. A genome-wide association study identifies variants in the HLA-DP locus associated with chronic hepatitis B in Asians. Nat Genet 2009;41:591-5.
19. Guo X, Zhang Y, Li J, Ma J, Wei Z, Tan W, O'Brien SJ. Strong influence of human leukocyte antigen (HLA)-DP gene variants on development of persistent chronic hepatitis B virus carriers in the Han Chinese population. Hepatology 2011;53:422-8.
20. Li S, Qian J, Yang Y, Zhao W, Dai J, Bei JX, Foo JN, McLaren PJ, Li Z, Yang J, Shen F, Liu L, Yang J, Li S, Pan S, Wang Y, Li W, Zhai X, Zhou B, Shi L, Chen X, Chu M, Yan Y, Wang J, Cheng S, Shen J, Jia W, Liu J, Yang J, Wen Z, Li A, Zhang Y, Zhang G, Luo X, Qin H, Chen M, Wang H, Jin L, Lin D, Shen H, He L, de Bakker PI, Wang H, Zeng YX, Wu M, Hu Z, Shi Y, Liu J, Zhou W. GWAS identifies novel susceptibility loci on 6p21.32 and 21q21.3 for hepatocellular carcinoma in chronic hepatitis B virus carriers. PLoS Genet 2012;8:e1002791.
21. Hu Z, Liu Y, Zhai X, Dai J, Jin G, Wang L, Zhu L, Yang Y, Liu J, Chu M, Wen J, Xie K, Du G, Wang Q, Zhou Y, Cao M, Liu L, He Y, Wang Y, Zhou G, Jia W, Lu J, Li S, Liu J, Yang H, Shi Y, Zhou W, Shen H. New loci associated with chronic hepatitis B virus infection in Han Chinese. Nat Genet 2013;45:1499-503.
22. Zhu M, Dai J, Wang C, Wang Y, Qin N, Ma H, Song C, Zhai X, Yang Y, Liu J, Liu L, Li S, Liu J, Yang H, Zhu F, Shi Y, Shen H, Jin G, Zhou W, Hu Z. Fine mapping the MHC region identified four independent variants modifying susceptibility to chronic hepatitis B in Han Chinese. Hum Mol Genet 2016;25:1225-32.
23. Png E, Thalamuthu A, Ong RT, Snippe H, Boland GJ, Seielstad M. A genome-wide association study of hepatitis B vaccine response in an Indonesian population reveals multiple independent risk variants in the HLA region. Hum Mol Genet 2011;20:3893-8.
24. Tan W, Xia J, Dan Y, Li M, Lin S, Pan X, Wang H, Tang Y, Liu N, Tan S, Liu M, He W, Zhang W, Mao Q, Wang Y, Deng G. Genome-wide association study identifies HLA-DR variants conferring risk of HBV-related acute-on-chronic liver failure. Gut 2017;pii:gutjnl-2016-313035.
25. Jiang DK, Ma XP, Wu X, Peng L, Yin J, Dan Y, Huang HX, Ding DL, Zhang LY, Shi Z, Zhang P, Yu H, Sun J, Lilly Zheng S, Deng G, Xu J, Liu Y, Guo J, Cao G, Yu L. Genetic variations in STAT4,C2,HLA-DRB1 and HLA-DQ associated with risk of hepatitis B virus-related liver cirrhosis. Sci Rep 2015;5:16278.
26. Yu L, Cheng YJ, Cheng ML, Yao YM, Zhang Q, Zhao XK, Liu HJ, Hu YX, Mu M, Wang B, Yang GZ, Zhu LL, Zhang S. Quantitative assessment of common genetic variations in HLA-DP with hepatitis B virus infection, clearance and hepatocellular carcinoma development. Sci Rep 2015;5:14933.
27. Jiang DK, Sun J, Cao G, Liu Y, Lin D, Gao YZ, Ren WH, Long XD, Zhang H, Ma XP, Wang Z, Jiang W, Chen TY, Gao Y, Sun LD, Long JR, Huang HX, Wang D, Yu H, Zhang P, Tang LS, Peng B, Cai H, Liu TT, Zhou P, Liu F, Lin X, Tao S, Wan B, Sai-Yin HX, Qin LX, Yin J, Liu L, Wu C, Pei Y, Zhou YF, Zhai Y, Lu PX, Tan A, Zuo XB, Fan J, Chang J, Gu X, Wang NJ, Li Y, Liu YK, Zhai K, Zhang H, Hu Z, Liu J, Yi Q, Xiang Y, Shi R, Ding Q, Zheng W, Shu XO, Mo Z, Shugart YY, Zhang XJ, Zhou G, Shen H, Zheng SL, Xu J, Yu L. Genetic variants in STAT4 and HLA-DQ genes confer risk of hepatitis B virus-related hepatocellular carcinoma. Nat Genet 2013;45:72-5.
28. Schaefer S. Hepatitis B virus taxonomy and hepatitis B virus genotypes. World J Gastroenterol 2007;13:14-21.
29. Yin J, Xie J, Liu S, Zhang H, Han L, Lu W, Shen Q, Xu G, Dong H, Shen J, Zhang J, Han J, Wang L, Liu Y, Wang F, Zhao J, Zhang Q, Ni W, Wang H, Cao G. Association between the various mutations in viral core promoter region to different stages of hepatitis B, ranging of asymptomatic carrier state to hepatocellular carcinoma. Am J Gastroenterol 2011;106:81-92.
30. Yin J, Xie J, Zhang H, Shen Q, Han L, Lu W, Han Y, Li C, Ni W, Wang H, Cao G. Significant association of different preS mutations with hepatitis B–related cirrhosis or hepatocellular carcinoma. J Gastroenterol 2010;45:1063-71.
31. Liu S, Xie J, Yin J, Zhang H, Zhang Q, Pu R, Li C, Ni W, Wang H, Cao G. A matched case–control study of hepatitis B virus mutations in the preS and core promoter regions associated independently with hepatocellular carcinoma. J Med Virol 2011;83:45-53.
32. Liu S, Zhang H, Gu C, Yin J, He Y, Xie J, Cao G. Associations between hepatitis B virus mutations and the risk of hepatocellular carcinoma: a meta-analysis. J Natl Cancer Inst 2009;101:1066-82.
33. Yin J, Wang J, Pu R, Xin H, Li Z, Han X, Ding Y, Du Y, Liu W, Deng Y, Ji X, Wu M, Yu M, Zhang H, Wang H, Thompson TC, Ni W, Cao G. Hepatitis B virus combo mutations improve the prediction and active prophylaxis of hepatocellular carcinoma: a clinic-based cohort study. Cancer Prev Res (Phila) 2015;8:978-88.
34. Fang ZL, Sabin CA, Dong BQ, Ge LY, Wei SC, Chen QY, Fang KX, Yang JY, Wang XY, Harrison TJ. HBV A1762T, G1764A mutations are a valuable biomarker for identifying a subset of male HBsAg carriers at extremely high risk of hepatocellular carcinoma: a prospective study. Am J Gastroenterol 2008;103:2254-62.
35. Chou YC, Yu MW, Wu CF, Yang SY, Lin CL, Liu CJ, Shih WL, Chen PJ, Liaw YF, Chen CJ. Temporal relationship between hepatitis B virus enhancer II/basal core promoter sequence variation and risk of hepatocellular carcinoma. Gut 2008;57:91-7.
36. Bai X, Zhu Y, Jin Y, Guo X, Qian G, Chen T, Zhang J, Wang J, Groopman JD, Gu J, Tu H. Temporal acquisition of sequential mutations in the enhancer II and basal core promoter of HBV in individuals at high risk for hepatocellular carcinoma. Carcinogenesis 2011;32:63-8.
37. Sung FY, Lan CY, Huang CJ, Lin CL, Liu CJ, Chen PJ, Lin SM, Yu MW. Progressive accumulation of mutations in the hepatitis B virus genome and its impact on time to diagnosis of hepatocellular carcinoma. Hepatology 2016;64:720-31.
38. Li Z, Xie Z, Ni H, Zhang Q, Lu W, Yin J, Liu W, Ding Y, Zhao Y, Zhu Y, Pu R, Zhang H, Dong H, Fu Y, Sun Q, Xu G, Cao G. Mother-to-child transmission of hepatitis B virus: evolution of hepatocellular carcinoma-related viral mutations in the post-immunization era. J Clin Virol 2014;61:47-54.
39. Maman Y, Blancher A, Benichou J, Yablonka A, Efroni S, Louzoun Y. Immune-induced evolutionary selection focused on a single reading frame in overlapping hepatitis B virus proteins. J Virol 2011;85:4558-66.
40. Tran A, Kremsdorf D, Capel F, Housset C, Dauguet C, Petit MA, Brechot C. Emergence of and takeover by hepatitis B virus (HBV) with rearrangements in the pre-S/S and pre-C/C genes during chronic HBV infection. J Virol 1991;65:3566-74.
41. Nowak MA, Bonhoeffer S, Hill AM, Boehme R, Thomas HC, McDade H. Viral dynamics in hepatitis B virus infection. Proc Natl Acad Sci U S A 1996;93:4398-402.
42. Orito E, Mizokami M, Ina Y, Moriyama EN, Kameshima N, Yamamoto M, Gojobori T. Host-independent evolution and a genetic classification of the hepadnavirus family based on nucleotide sequences. Proc Natl Acad Sci U S A 1989;86:7059-62.
43. Vartanian JP, Henry M, Marchio A, Suspene R, Aynaud MM, Guetard D, Cervantes-Gonzalez M, Battiston C, Mazzaferro V, Pineau P, Dejean A, Wain-Hobson S. Massive APOBEC3 editing of hepatitis B viral DNA in cirrhosis. PLoS Pathog 2010;6:e1000928.
44. Xu R, Zhang X, Zhang W, Fang Y, Zheng S, Yu XF. Association of human APOBEC3 cytidine deaminases with the generation of hepatitis virus B x antigen mutants and hepatocellular carcinoma. Hepatology 2007;46:1810-20.
45. Yang F, Ma LT, Cao GW. Hepatocellular carcinoma: co-evolution of hepatocytes and hepatitis B virus. Chin J Hepatol 2017;25:321-4. (in Chinese)
46. Chen L, Zhang Q, Chang W, Du Y, Zhang H, Cao G. Viral and host inflammation-related factors that can predict the prognosis of hepatocellular carcinoma. Eur J Cancer 2012;48:1977-87.
47. Yeh CT, So M, Ng J, Yang HW, Chang ML, Lai MW, Chen TC, Lin CY, Yeh TS, Lee WC. Hepatitis B virus-DNA level and basal core promoter A1762T/G1764A mutation in liver tissue independently predict postoperative survival in hepatocellular carcinoma. Hepatology 2010;52:1922-33.
48. Wu TJ, Chan KM, Chou HS, Lee CF, Wu TH, Chen TC, Yeh CT, Lee WC. Liver transplantation in patients with hepatitis B virus-related hepatocellular carcinoma: the influence of viral characteristics on clinical outcome. Ann Surg Oncol 2013;20:3582-90.
49. Mathews P, Lee D, Chung YH, Kim JA, Lee JH, Jin YJ, Park W, Lyu H, Jaffee E, Zheng L, Yu E, Lee YJ. Effects of genomic changes in hepatitis B virus on postoperative recurrence and survival in patients with hepatocellular carcinoma. Ann Surg Oncol 2013;20:1216-22.
50. Wu CY, Chen YJ, Ho HJ, Hsu YC, Kuo KN, Wu MS, Lin JT. Association between nucleoside analogues and risk of hepatitis B virus-related hepatocellular carcinoma recurrence following liver resection. JAMA 2012;308:1906-14.
51. Yin J, Li N, Han Y, Xue J, Deng Y, Shi J, Guo W, Zhang H, Wang H, Cheng S, Cao G. Effect of antiviral treatment with nucleotide/nucleoside analogs on postoperative prognosis of hepatitis B virus-related hepatocellular carcinoma: a two-stage longitudinal clinical study. J Clin Oncol 2013;31:3647-55.
52. Huang S, Xia Y, Lei Z, Zou Q, Li J, Yang T, Wang K, Yan Z, Wan X, Shen F. Antiviral therapy inhibits viral reactivation and improves survival after repeat hepatectomy for hepatitis B virus-related recurrent hepatocellular carcinoma. J Am Coll Surg 2017;224:283-93.e4.
53. Du Y, Su T, Ding Y, Cao G. Effects of antiviral therapy on the recurrence of hepatocellular carcinoma after curative resection or liver transplantation. Hepat Mon 2012;12:e6031.
54. Chen CH, Hung CH, Lee CM, Hu TH, Wang JH, Wang JC, Lu SN, Changchien CS. Pre-S deletion and complex mutations of hepatitis B virus related to advanced liver disease in HBeAg-negative patients. Gastroenterology 2007;133:1466-74.
55. Liu WC, Wu IC, Lee YC, Lin CP, Cheng JH, Lin YJ, Yen CJ, Cheng PN, Li PF, Cheng YT, Cheng PW, Sun KT, Yan SL, Lin JJ, Yang JC, Chang KC, Ho CH, Tseng VS, Chang BC, Wu JC, Chang TT. Hepatocellular carcinoma-associated single-nucleotide variants and deletions identified by the use of genome-wide high-throughput analysis of hepatitis B virus. J Pathol 2017;243:176-92.
56. Huang Y, Tong S, Tai AW, Hussain M, Lok AS. Hepatitis B virus core promoter mutations contribute to hepatocarcinogenesis by deregulating SKP2 and its target, p21. Gastroenterology 2011;141:1412-21, 1421.e1-5.
57. Huang Y, Tai AW, Tong S, Lok AS. HBV core promoter mutations promote cellular proliferation through E2F1-mediated upregulation of S-phase kinase-associated protein 2 transcription. J Hepatol 2013;58:1068-73.
58. Mun HS, Lee SA, Kim H, Hwang ES, Kook YH, Kim BJ. Novel F141L pre-S2 mutation in hepatitis B virus increases the risk of hepatocellular carcinoma in patients with chronic genotype C infections. J Virol 2011;85:123-32.
59. Wang Y, Lau SH, Sham JS, Wu MC, Wang T, Guan XY. Characterization of HBV integrants in 14 hepatocellular carcinomas: association of truncated X gene and hepatocellular carcinogenesis. Oncogene 2004;23:142-8.
60. Sze KM, Chu GK, Lee JM, Ng IO. C-terminal truncated hepatitis B virus x protein is associated with metastasis and enhances invasiveness by C-Jun/matrix metalloproteinase protein 10 activation in hepatocellular carcinoma. Hepatology 2013;57:131-9.
61. Zhang Q, Yin J, Zhang Y, Deng Y, Ji X, Du Y, Pu R, Han Y, Zhao J, Han X, Zhang H, Cao G. HLA-DP polymorphisms affect the outcomes of chronic hepatitis B virus infections, possibly through interacting with viral mutations. J Virol 2013;87:12176-86.
62. Ji X, Zhang Q, Li B, Du Y, Yin J, Liu W, Zhang H, Cao G. Impacts of human leukocyte antigen DQ genetic polymorphisms and their interactions with hepatitis B virus mutations on the risks of viral persistence, liver cirrhosis, and hepatocellular carcinoma. Infect Genet Evol 2014;28:201-9.
63. Xie J, Zhang Y, Zhang Q, Han Y, Yin J, Pu R, Shen Q, Lu W, Du Y, Zhao J, Han X, Zhang H, Cao G. Interaction of signal transducer and activator of transcription 3 polymorphisms with hepatitis B virus mutations in hepatocellular carcinoma. Hepatology 2013;57:2369-77.
64. Zhang Q, Ji XW, Hou XM, Lu FM, Du Y, Yin JH, Sun XY, Deng Y, Zhao J, Han X, Yang GS, Zhang HW, Chen XM, Shen HB, Wang HY, Cao GW. Effect of functional nuclear factor-kappaB genetic polymorphisms on hepatitis B virus persistence and their interactions with viral mutations on the risk of hepatocellular carcinoma. Ann Oncol 2014;25:2413-9.
65. He Y, Zhang H, Yin J, Xie J, Tan X, Liu S, Zhang Q, Li C, Zhao J, Wang H, Cao G. IkappaBalpha gene promoter polymorphisms are associated with hepatocarcinogenesis in patients infected with hepatitis B virus genotype C. Carcinogenesis 2009;30:1916-22.
66. Wang S, Zhang J, Jiao W, Li J, Xun X, Sun Y, Guo X, Huan P, Dong B, Zhang L, Hu X, Sun X, Wang J, Zhao C, Wang Y, Wang D, Huang X, Wang R, Lv J, Li Y, Zhang Z, Liu B, Lu W, Hui Y, Liang J, Zhou Z, Hou R, Li X, Liu Y, Li H, Ning X, Lin Y, Zhao L, Xing Q, Dou J, Li Y, Mao J, Guo H, Dou H, Li T, Mu C, Jiang W, Fu Q, Fu X, Miao Y, Liu J, Yu Q, Li R, Liao H, Li X, Kong Y, Jiang Z, Chourrout D, Li R, Bao Z. Scallop genome provides insights into evolution of bilaterian karyotype and development. Nat Ecol Evol 2017;1:120.
67. Deschamps J, Duboule D. Embryonic timing, axial stem cells, chromatin dynamics, and the Hox clock. Genes Dev 2017;31:1406-16.
68. Hinaux H, Devos L, Blin M, Elipot Y, Bibliowicz J, Alié A, Rétaux S. Sensory evolution in blind cavefish is driven by early embryonic events during gastrulation and neurulation. Development 2016;143:4521-32.
69. Lettice LA, Devenney P, De Angelis C, Hill RE. The conserved Sonic Hedgehog limb enhancer consists of discrete functional elements that regulate precise spatial expression. Cell Rep 2017;20:1396-408.
70. Rodrigues AR, Yakushiji-Kaminatsui N, Atsuta Y, Andrey G, Schorderet P, Duboule D, Tabin CJ. Integration of Shh and Fgfsignaling in controlling Hox gene expression in cultured limb cells. Proc Natl Acad Sci U S A 2017;114:3139-44.
71. Tarazona OA, Slota LA, Lopez DH, Zhang G, Cohn MJ. The genetic program for cartilage development has deep homology within Bilateria. Nature 2016;533:86-9.
72. Zhong C, Zhou YK, Yang SS, Zhao JF, Zhu XL, Chen HH, Chen PC, Huang LQ, Huang X. Developmental expression of the N-myc downstream regulated gene (Ndrg) family during Xenopustropicalis embryogenesis. Int J Dev Biol 2015;59:511-7.
73. Roush W, Pennisi E. Growing pains: evo-devo researchers straddle cultures. Science 1997;277:38-9.
77. Carroll SB. Evo-devo and an expanding evolutionary synthesis: a genetic theory of morphological evolution. Cell 2008;134:25-36.
79. Merlo LM, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat Rev Cancer 2006;6:924-35.
81. Stratton MR. Exploring the genomes of cancer cells: progress and promise. Science 2011;331:1553-8.
82. Xu X, Chang W, Yuan J, Han X, Tan X, Ding Y, Luo Y, Cai H, Liu Y, Gao X, Liu Q, Yu Y, Du Y, Wang H, Ma L, Wang J, Chen K, Ding Y, Fu C, Cao G. Periostin expression in intra-tumoral stromal cells is prognostic and predictive for colorectal carcinoma via creating a cancer-supportive niche. Oncotarget 2016;7:798-813.
83. Yong KJ, Gao C, Lim JS, Yan B, Yang H, Dimitrov T, Kawasaki A, Ong CW, Wong KF, Lee S, Ravikumar S, Srivastava S, Tian X, Poon RT, Fan ST, Luk JM, Dan YY, Salto-Tellez M, Chai L, Tenen DG. Oncofetal gene SALL4 in aggressive hepatocellular carcinoma. N Engl J Med 2013;368:2266-76.
84. Zeng SS, Yamashita T, Kondo M, Nio K, Hayashi T, Hara Y, Nomura Y, Yoshida M, Hayashi T, Oishi N, Ikeda H, Honda M, Kaneko S. The transcription factor SALL4 regulates stemness of EpCAM-positive hepatocellular carcinoma. J Hepatol 2014;60:127-34.
85. Moeini A, Sia D, Zhang Z, Camprecios G, Stueck A, Dong H, Montal R, Torrens L, Martinez-Quetglas I, Fiel MI, Hao K, Villanueva A, Thung SN, Schwartz ME, Llovet JM. Mixed hepatocellular cholangiocarcinomatumors: cholangiolocellular carcinoma is a distinct molecular entity. J Hepatol 2017;66:952-61.
86. Fan H, Cui Z, Zhang H, Mani SK, Diab A, Lefrancois L, Fares N, Merle P, Andrisani O. DNA demethylation induces SALL4 gene re-expression in subgroups of hepatocellular carcinoma associated with Hepatitis B or C virus infection. Oncogene 2017;36:2435-45.
87. Lee JS, Heo J, Libbrecht L, Chu IS, Kaposi-Novak P, Calvisi DF, Mikaelyan A, Roberts LR, Demetris AJ, Sun Z, Nevens F, Roskams T, Thorgeirsson SS. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med 2006;12:410-6.
88. Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H, Lasorella A, Aldape K, Califano A, Iavarone A. The transcriptional network for mesenchymal transformation of brain tumours. Nature 2010;463:318-25.
89. Yu J, Yu J, Rhodes DR, Tomlins SA, Cao X, Chen G, Mehra R, Wang X, Ghosh D, Shah RB, Varambally S, Pienta KJ, Chinnaiyan AM. A polycomb repression signature in metastatic prostate cancer predicts cancer outcome. Cancer Res 2007;67:10657-63.
90. DiMeo TA, Anderson K, Phadke P, Fan C, Perou CM, Naber S, Kuperwasser C. A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Res 2009;69:5364-73.
91. Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, Weinberg RA. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet 2008;40:499-507.
92. Chen H, He X. The convergent cancer evolution toward a single cellular destination. Mol Biol Evol 2016;33:4-12.
93. Qin Y, Tang B, Hu CJ, Xiao YF, Xie R, Yong X, Wu YY, Dong H, Yang SM. An hTERT/ZEB1 complex directly regulates E-cadherin to promote epithelial-to-mesenchymal transition (EMT) in colorectal cancer. Oncotarget 2016;7:351-61.
94. Chen J, Chan AW, To KF, Chen W, Zhang Z, Ren J, Song C, Cheung YS, Lai PB, Cheng SH, Ng MH, Huang A, Ko BC. SIRT2 overexpression in hepatocellular carcinoma mediates epithelial to mesenchymal transition by protein kinase B/glycogen synthase kinase-3β/β-catenin signaling. Hepatology 2013;57:2287-98.
95. Chen T, You Y, Jiang H, Wang ZZ. Epithelial-mesenchymal transition (EMT): a biological process in the development, stem cell differentiation, and tumorigenesis. J Cell Physiol 2017;232:3261-72.
96. Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR. Signatures of mutational processes in human cancer. Nature 2013;500:415-21.
98. Liu WB, Wu JF, Du Y, Cao GW. Cancer Evolution-Development: experience of hepatitis B virus-induced hepatocarcinogenesis. Curr Oncol 2016;23:e49-56.
99. Deng Y, Du Y, Zhang Q, Han X, Cao G. Human cytidine deaminases facilitate hepatitis B virus evolution and link inflammation and hepatocellular carcinoma. Cancer Lett 2014;343:161-71.
100. Ji X, Zhang Q, Du Y, Liu W, Li Z, Hou X, Cao G. Somatic mutations, viral integration and epigenetic modification in the evolution of hepatitis B virus-induced hepatocellular carcinoma. Curr Genomics 2014;15:469-80.
103. Xu X, Chen W, Zhang L, Miao R, Zhou Y, Wan Y, Dong Y, Liu C. Prognostic significance of neutrophil to lymphocyte ratio in patients with hepatocellular carcinoma after transcatheter arterial chemoembolization. Chin Med J (Engl) 2014;127:4204-9.
104. Peng W, Li C, Wen TF, Yan LN, Li B, Wang WT, Yang JY, Xu MQ. Neutrophil to lymphocyte ratio changes predict small hepatocellular carcinoma survival. J Surg Res 2014;192:402-8.
105. Janahi EM, McGarvey MJ. The inhibition of hepatitis B virus by APOBEC cytidine deaminases. J Viral Hepat 2013;20:821-8.
106. Kitamura K, Wang Z, Chowdhury S, Simadu M, Koura M, Muramatsu M. Uracil DNA glycosylase counteracts APOBEC3G-induced hypermutation of hepatitis B viral genomes: excision repair of covalently closed circular DNA. PLoS Pathog 2013;9:e1003361.
107. Morisawa T, Marusawa H, Ueda Y, Iwai A, Okazaki IM, Honjo T, Chiba T. Organ-specific profiles of genetic changes in cancers caused by activation-induced cytidine deaminase expression. Int J Cancer 2008;123:2735-40.
108. Endo Y, Marusawa H, Kinoshita K, Morisawa T, Sakurai T, Okazaki IM, Watashi K, Shimotohno K, Honjo T, Chiba T. Expression of activation-induced cytidine deaminase in human hepatocytes via NF-κB signaling. Oncogene 2007;26:5587-95.
109. Amaddeo G, Cao Q, Ladeiro Y, Imbeaud S, Nault JC, Jaoui D, Gaston Mathe Y, Laurent C, Laurent A, Bioulac-Sage P, Calderaro J, Zucman-Rossi J. Integration of tumour and viral genomic characterizations in HBV-related hepatocellular carcinomas. Gut 2015;64:820-9.
110. Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL, Saksena G, Harris S, Shah RR, Resnick MA, Getz G, Gordenin DA. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet 2013;45:970-6.
111. Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M, Degos F, Clément B, Balabaud C, Chevet E, Laurent A, Couchy G, Letouzé E, Calvo F, Zucman-Rossi J. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet 2012;44:694-8.
112. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science 2013;339:1546-58.
113. Li M, Zhao H, Zhang X, Wood LD, Anders RA, Choti MA, Pawlik TM, Daniel HD, Kannangai R, Offerhaus GJ, Velculescu VE, Wang L, Zhou S, Vogelstein B, Hruban RH, Papadopoulos N, Cai J, Torbenson MS, Kinzler KW. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat Genet 2011;43:828-9.
114. Huang J, Deng Q, Wang Q, Li KY, Dai JH, Li N, Zhu ZD, Zhou B, Liu XY, Liu RF, Fei QL, Chen H, Cai B, Zhou B, Xiao HS, Qin LX, Han ZG. Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nat Genet 2012;44:1117-21.
115. Wong CM, Fan ST, Ng IO. Beta-Catenin mutation and overexpression in hepatocellular carcinoma: clinicopathologic and prognostic significance. Cancer 2001;92:136-45.
116. Yeh SH, Chiu CM, Chen CL, Lu SF, Hsu HC, Chen DS, Chen PJ. Somatic mutations at the trinucleotide repeats of androgen receptor gene in male hepatocellular carcinoma. Int J Cancer 2007;120:1610-7.
117. Han YF, Zhao J, Ma LY, Yin JH, Chang WJ, Zhang HW, Cao GW. Factors predicting occurrence and prognosis of hepatitis-B-virus-related hepatocellular carcinoma. World J Gastroenterol 2011;17:4258-70.
118. Cao GW. Clinical relevance and public health significance of hepatitis B virus genomic variations. World J Gastroenterol 2009;15:5761-9.
119. Chen LP, Zhao J, Du Y, Han YF, Su T, Zhang HW, Cao GW. Antiviral treatment to prevent chronic hepatitis B or C–related hepatocellular carcinoma. World J Virol 2012;1:174-83.
120. Kumar R, DiMenna L, Schrode N, Liu TC, Franck P, Mu-oz-Descalzo S, Hadjantonakis AK, Zarrin AA, Chaudhuri J, Elemento O, Evans T. AID stabilizes stem-cell phenotype by removing epigenetic memory of pluripotency genes. Nature 2013;500:89-92.
121. Nabel CS, Jia H, Ye Y, Shen L, Goldschmidt HL, Stivers JT, Zhang Y, Kohli RM. ID/APOBEC A deaminases disfavor modified cytosines implicated in DNA demethylation. Nat Chem Biol 2012;8:751-8.
122. Mu-oz DP, Lee EL, Takayama S, Coppé JP, Heo SJ, Boffelli D, Di Noia JM, Martin DI. Activation-induced cytidine deaminase (AID) is necessary for the epithelial-mesenchymal transition in mammary epithelial cells. Proc Natl Acad Sci U S A 2013;110:E2977-86.
123. Rogozin IB, Lada AG, Goncearenco A, Green MR, De S, Nudelman G, Panchenko AR, Koonin EV, Pavlov YI. Activation induced deaminase mutational signature overlaps with CpG methylation sites in follicular lymphoma and other cancers. Sci Rep 2016;6:38133.
124. Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012;366:883-92.
125. Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, Shafi S, Johnson DH, Mitter R, Rosenthal R, Salm M, Horswell S, Escudero M, Matthews N, Rowan A, Chambers T, Moore DA, Turajlic S, Xu H, Lee SM, Forster MD, Ahmad T, Hiley CT, Abbosh C, Falzon M, Borg E, Marafioti T, Lawrence D, Hayward M, Kolvekar S, Panagiotopoulos N, Janes SM, Thakrar R, Ahmed A, Blackhall F, Summers Y, Shah R, Joseph L, Quinn AM, Crosbie PA, Naidu B, Middleton G, Langman G, Trotter S, Nicolson M, Remmen H, Kerr K, Chetty M, Gomersall L, Fennell DA, Nakas A, Rathinam S, Anand G, Khan S, Russell P, Ezhil V, Ismail B, Irvin-Sellers M, Prakash V, Lester JF, Kornaszewska M, Attanoos R, Adams H, Davies H, Dentro S, Taniere P, O'Sullivan B, Lowe HL, Hartley JA, Iles N, Bell H, Ngai Y, Shaw JA, Herrero J, Szallasi Z, Schwarz RF, Stewart A, Quezada SA, Le Quesne J, Van Loo P, Dive C, Hackshaw A, Swanton C, TRACERx Consortium. Tracking the evolution of non-small-cell lung cancer. N Engl J Med 2017;376:2109-21.
126. Swanton C, McGranahan N, Starrett GJ, Harris RS. APOBEC enzymes: mutagenic fuel for cancer evolution and heterogeneity. Cancer Discov 2015;5:704-12.
127. Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N, Nikas JB, Yee D, Temiz NA, Donohue DE, McDougle RM, Brown WL, Law EK, Harris RS. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 2013;494:366-70.
128. de Bruin EC, McGranahan N, Mitter R, Salm M, Wedge DC, Yates L, Jamal-Hanjani M, Shafi S, Murugaesu N, Rowan AJ, Grönroos E, Muhammad MA, Horswell S, Gerlinger M, Varela I, Jones D, Marshall J, Voet T, Van Loo P, Rassl DM, Rintoul RC, Janes SM, Lee SM, Forster M, Ahmad T, Lawrence D, Falzon M, Capitanio A, Harkins TT, Lee CC, Tom W, Teefe E, Chen SC, Begum S, Rabinowitz A, Phillimore B, Spencer-Dene B, Stamp G, Szallasi Z, Matthews N, Stewart A, Campbell P, Swanton C. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 2014;346:251-6.
130. Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer 2011;11:325-37.
132. Garcia-Heredia JM, Carnero A. Decoding Warburg's hypothesis: tumor-related mutations in the mitochondrial respiratory chain. Oncotarget 2015;6:41582-99.
133. Hamabe A, Konno M, Tanuma N, Shima H, Tsunekuni K, Kawamoto K, Nishida N, Koseki J, Mimori K, Gotoh N, Yamamoto H, Doki Y, Mori M, Ishii H. Role of pyruvate kinase M2 in transcriptional regulation leading to epithelial-mesenchymal transition. Proc Natl Acad Sci U S A 2014;111:15526-31.
134. Penny HL, Sieow JL, Adriani G, Yeap WH, See Chi Ee P, San Luis B, Lee B, Lee T, Mak SY, Ho YS, Lam KP, Ong CK, Huang RY, Ginhoux F, Rotzschke O, Kamm RD, Wong SC. Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. Oncoimmunology 2016;5:e1191731.
135. Marín-Hernández á, Gallardo-Pérez JC, Hernández-Reséndiz I, Del Mazo-Monsalvo I, Robledo-Cadena DX, Moreno-Sánchez R, Rodríguez-Enríquez S. Hypoglycemia enhances epithelial-mesenchymal transition and invasiveness, and restrains the Warburg phenotype, in hypoxic HeLa cell cultures and microspheroids. J Cell Physiol 2017;232:1346-59.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Cao GW. Cancer Evo-Dev, a novel hypothesis derived from studies on hepatitis B virus-induced carcinogenesis. Hepatoma Res 2017;3:241-59. http://dx.doi.org/10.20517/2394-5079.2017.45
AMA Style
Cao GW. Cancer Evo-Dev, a novel hypothesis derived from studies on hepatitis B virus-induced carcinogenesis. Hepatoma Research. 2017; 3: 241-59. http://dx.doi.org/10.20517/2394-5079.2017.45
Chicago/Turabian Style
Cao, Guang-Wen. 2017. "Cancer Evo-Dev, a novel hypothesis derived from studies on hepatitis B virus-induced carcinogenesis" Hepatoma Research. 3: 241-59. http://dx.doi.org/10.20517/2394-5079.2017.45
ACS Style
Cao, G.W. Cancer Evo-Dev, a novel hypothesis derived from studies on hepatitis B virus-induced carcinogenesis. Hepatoma. Res. 2017, 3, 241-59. http://dx.doi.org/10.20517/2394-5079.2017.45
About This Article
Copyright

Data & Comments
Data


 Cite This Article 17 clicks
Cite This Article 17 clicks

 Like This Article 1
likes
Like This Article 1
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.