
Roles of p53 in extrinsic factor-induced liver carcinogenesis


Abstract
Liver cancer remains one of the most common human cancers with a high mortality rate. Therapies for hepatocellular carcinoma (HCC) remain ineffective, due to the heterogeneity of HCC with regard to both the etiology and mutation spectrum, as well as its chemotherapy resistant nature; thus surgical resection and liver transplantation remain the gold standard of patient care. The most common etiologies of HCC are extrinsic factors. Humans have multiple defense mechanisms against extrinsic factor-induced carcinogenesis, of which tumor suppressors play crucial roles in preventing normal cells from becoming cancerous. The tumor suppressor p53 is one of the most frequently mutated genes in liver cancer. p53 regulates expression of genes involved in cell cycle progression, cell death, and cellular metabolism to avert tumor development due to carcinogens. This review article mainly summarizes extrinsic factors that induce liver cancer and potentially have etiological association with p53, including aflatoxin B1, vinyl chloride, non-alcoholic fatty liver disease, iron overload, and infection of hepatitis viruses.
Keywords
Introduction
Liver cancer is the 6th most common cancer in men and the 9th most common cancer in women with the 3rd highest mortality rate of all cancers globally.[1,2] The majority of these cases (about 80%) occur in Eastern Asia, South-Eastern Asia, Mid-Africa, and West Africa, within the context of viral hepatitis.[2-4] Although there are genetic etiologies for hepatocellular carcinoma (HCC) including hereditary hemochromatosis and α1-antitrypsin deficiency,[5-7] viral hepatitis, as well as exposure to other extrinsic factors, such as aflatoxin B1 (AFB1), polyvinyl chloride (PVC), a poor diet inducing non-alcoholic fatty liver disease (NAFLD), and excess iron exposure, remain among the most common causes of liver cancer.[8,9] Despite vaccinations for hepatitis B virus (HBV), new treatments for hepatitis C virus (HCV), regulations governing PVC production, and restrictions preventing AFB1 contamination of food products, countries still struggle to prevent liver cancer.[9,10]
Surgical resection is currently the preferred treatment, and liver transplantation is ultimately the most effective therapeutic modality of HCC; however, it is limited by the availability of suitable organs.[11,12] Due to a high probability of being diagnosed at advanced stages, as well as poor responses to systematic chemotherapy and radiation therapy, prognosis of HCC is particularly bleak with an incidence to mortality ratio of 0.95 and a 5-year survival rate around 17.5%.[2,13]
Molecular mechanisms involved in liver carcinogenesis remain unclear. The tumor suppressor p53, a transcription factor that regulates many downstream target genes regulating cell cycle progression, apoptosis, DNA repair, senescence, and metabolism,[14,15] is one of the most commonly mutated genes in HCC.[16,17] Indeed, p53 is the most commonly mutated human gene, occurring in > 50% of all human cancers.[18] Additionally, in some HCC cases, proteins such as a 26S proteasome regulatory protein, gankyrin, and a p53-specific ubiquitin ligase, murine double minute 2 (MDM2), are elevated, hence decreasing p53 protein levels.[19,20] MicroRNAs (miRNAs) can also inhibit p53 activity; specifically, miRNA-24, when dysregulated in HCC, is shown to promote invasion and metastasis by decreasing p53 levels.[21] Thus, p53 activity is impaired by multiple mechanisms in HCC, hence contributing to HCC genesis. In this review article, we focus on HCC-inducing extrinsic factors that are etiologically associated with p53 [Table 1].
Extrinsic factors causing liver cancer and their association with p53
| Extrinsic factors | Mechanisms of action | Roles of p53 | References |
|---|---|---|---|
| AFB1 | AFB1 is metabolized to AFB1-8,9-epoxide to form AFB1-N7-guanine adducts, leading to specific mutation at p53 codon 249 (p53R249S) | AFB1 frequently causes p53R249S mutation which enhances IGF-2 expression | [25,29,34] |
| VC | VC activated by CYP2E1 is converted into chloroethylene oxide, which forms bulky DNA adducts, leading to A>T transversions in the genome | It is unclear whether p53 plays protective roles in VC-induced liver cancer | [41,43,44] |
| NAFLD | NAFLD-induced hepatitis leads to cirrhosis and HCC, and dysregulation of NF-κB signaling, the Pl3K-ATK-PTEN pathway, insulin resistance, and expression of certain miRNAs (e.g. miR-34) is suggested; however, the molecular mechanisms behind NAFLD-mediated HCC remain unclear | The miR-34-SIRT1-p53 pathway plays a role in the progression of NAFLD. However, the direct role of p53 in the NAFLD-mediated HCC is unknown | [49,51-57] |
| Iron | Excess iron generates ROS and decreases p53 activity, leading to HCC genesis | Chronic iron overload reduces p53 protein levels by heme-mediated degradation or increased MDM2 levels, which can increase intracellular iron levels via a decrease in ISCUC2, thus further promoting HCC development | [64,68-70] |
| HBV | HBV-induced HCC occurs following repeated inflammation-liver regeneration-cirrhosis process, as well as through oncogenic function of HBx and Ct-HBx in both p53-dependent and -independent manners | Although direct involvement of p53 in HBV-induced HCC is unclear, functional inactivation of p53 by HBx and Ct-HBx may contribute to HCC progression | [76,81,82,85,99,100] |
| HCV | The majority of HCV-mediated HCC is via cirrhosis. But HCV core protein, NS3, and NS5 are implicated in HCC development in both p53- dependent and -independent manners | There is no direct evidence showing dependency of HCV-induced HCC on p53. However, HCV core protein, NS3, and NS5A inhibit p53 activity by binding to p53, altering subcellular localization, or modulating post-translational modifications | [112,118,119,123-125,131,132] |
AFB1
AFB1 is a well-characterized liver mutagen produced by the fungus Aspergillus, and can be ingested by humans from contaminated food products.[22,23] One study estimates the population attributable risk of AFB1-mediated HCC as 17% in some parts of the world.[24] Mechanistically, AFB1 is activated by CYP40s into AFB1-8,9-epoxide, which reacts with DNA, forming 8,9-dihydro-8-(N7-guanyl)-9-hydroxyaflatoxin B1 (AFB1-N7-guanine) adducts; these adducts, if left unrepaired, induce G>T transversions during DNA replication.[25,26]
AFB1 is well-known to generate a specific p53 mutation in the DNA binding domain from an arginine to serine missense mutation at codon 249 (R249S), which is caused by a G>T transversion at the third base of codon 249 [Figure 1A].[27,28] In geographic areas exposed to high levels of AFB1, such as the Qidong City in China, about 50% of HCC cases have the p53R249S mutation,[29] suggesting the involvement of p53 in AFB1-induced HCC. AFB1-8,9-epoxide also reacts with guanines of the p53 gene other than those at codon 249, but these guanine adducts do not form cancer-causing mutations as frequently as p53R249S.[26,28,30] Although AFB1-mediated DNA damages initially activate p53 to induce cell cycle arrest at S to G2/M phases,[31-33] liver cells that gain p53R249S would escape this cellular defense mechanism with a selective advantage for proliferation, which could further proceed toward liver cancer. Indeed, p53R249S is shown to increase transcription of insulin-like growth factor 2 (IGF-2) in Hep3B (p53null) cells, suggesting a possible gain-of-function activity of p53R249S.[34] IGF-2 is over-expressed in 16-40% of human HCC and is implicated in promoting HCC progression.[35] Also, a positive correlation is observed between IGF-2 +3580 AA genotype and the risk of HCC.[36] Intriguingly, silencing of IGF-2 in HepG2 cells leads to decrease in cell survival and proliferation.[37] Thus, AFB1-mediated mutation in p53 plays a crucial role in HCC genesis, possibly through enhanced IGF-2 signaling [Figure 1B].
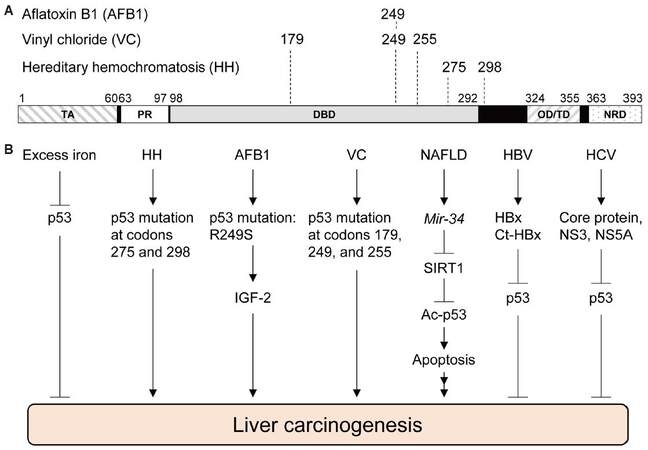
Figure 1. Functional roles of p53 in liver cancer-associated diseases. (A) Functional domains in human p53 and amino acid locations mutated in liver cancer associated with aflatoxin B1 (AFB1), vinyl chloride (VC), and hereditary hemochromatosis (HH). (B) Involvement of p53 in liver carcinogenesis. Multiple hereditary and extrinsic factors cause liver cancer possibly through the p53 pathway. TA: transactivation domain, PR: proline-rich domain, DBD: DNA-binding domain, OD/TD: oligomerization/tetramerization domain, NRD: negative regulatory domain; NAFLD: non-alcoholic fatty liver disease; HBV: hepatitis B virus; HCV: hepatitis C virus; SIRT1: sirtuin 1; IGF-2: insulin-like growth factor 2; Ct-HBx: HBx variants with C-terminal truncations
Vinyl chloride
Vinyl chloride (VC) is a carcinogenic gas used in the manufacture of PVC which induces mainly angiosarcomas of the liver (ASL) and rarely HCC, although it remains controversial whether VC can induce HCC in humans.[38-40] VC is absorbed in the lungs and then metabolized to chloroethylene oxide by CYP2E1 in the liver, which forms bulky DNA adducts, leading to liver cancer.[41,42] There are four VC-associated DNA adducts detected in vivo, including 7-(2-oxoethyl)-deoxyguanosine, 3,N4-etheno-deoxycytidine, 1,N6-etheno-deoxyadenosine, and N2,3-etheno-deoxyguanosine.[41]
VC-induced human ASLs are reported to have an increase in A>T transversions at codons 179, 249, and 255 of the p53 gene [Figure 1A].[43,44] A study using Sprague Dawley rats also indicates that the majority of p53 mutations in ASL and HCC following VC exposure are A>T transversions; the A>T transversions in ASLs are detected at codon 253 of rat p53, which is equivalent with codon 255 in humans.[45] Moreover, serum samples from workers exposed to VC have an increase in the levels of p53 protein with mutant conformation, detected by a conformation-specific p53 antibody PAb240, as well as other antibodies for p53.[44,46] However, it is still unclear whether p53 plays protective roles in VC-induced DNA damages and liver cancer development, and how mutations in p53 contribute to the VC-induced liver cancer [Figure 1B].
NAFLD
NAFLD represents a range of disorders including non-alcoholic fatty liver (NAFL), non-alcoholic steatohepatitis (NASH), fibrosis, cirrhosis, and HCC. NAFLD is associated with metabolic syndrome, type 2 diabetes mellitus, and obesity.[47] It is estimated that 20-30% of individuals in the Western world suffer from NAFLD.[48] However, only 11.5% of patients with NAFLD-induced cirrhosis eventually develop HCC, and about 50% of NASH-induced HCCs occur without cirrhosis.[49,50] These observations indicate the requirement of additional oncogenic events toward NAFLD-associated HCC. However, the molecular mechanisms behind NAFLD-mediated HCC are not fully understood. Several mediators have been implicated in its genesis, including dysregulation of NF-κB signaling, the Pl3K-ATK-PTEN pathway, insulin resistance, and expression of certain miRNAs (e.g. miR-34).[51,52]
p53 has also been implicated in the progression of NAFLD due to multiple mechanisms. In a mouse model for NAFLD where p53+/+ and p53-/- mice are fed a methionine- and choline-deficient diet, p53+/+ mice show increases in histologically observable steatohepatitis, reactive oxygen species (ROS)formation, and fibrosis with increased protein levels of p66Shc, a protein associated with oxidative stress, as compared to p53-/- mice.[53] Human NASH hepatocytes display upregulated p53 activity with increased mRNA levels of p21 and p66Shc, which is positively correlated with fibrosis severity.[53] The miR-34-Sirtuin 1 (SIRT1)-p53 pathway is also implicated in NAFLD pathogenesis; increased miR-34 expression and subsequent decrease in SIRT1 protein levels are detected in human NAFLD liver tissues with increased acetylation of p53, which is correlated with disease severity.[54] Activation of the miR-34a-SIRT1-p53 axis is also shown to contribute to liver fibrosis or NASH by inducing hepatocyte apoptosis.[55,56] Moreover, p53 can upregulate miR-34, which inhibits SIRT1 mRNA expression, leading to increased acetylation of p53, thus forming a positive feedback loop [Figure 1B].[57] These observations indicate that high expression of miR-34 and p53 is associated with NAFLD. However, it should be noted that miR-34a-mediated apoptosis can occur in p53-dependent and p53-independent manners.[58] Nonetheless, surrounding evidence suggests involvement of p53 in the progression of NAFLD and NASH; however, further studies are required to demonstrate whether p53 directly plays a crucial role in the NAFLD-mediated HCC.
Iron overload
Iron is an essential mineral that takes part in numerous metabolic processes, such as heme synthesis, Fe-S cluster biogenesis, and oxygen transport via hemoglobin.[59] However, when iron homeostasis is perturbed, whether due to genetic or environmental causes, there can be severe consequences including cardiomyopathy, hepatic fibrosis, endocrine disorders, and arthropathy.[60,61] Importantly, excess iron is a risk factor for many types of neoplasia, including breast cancer, colorectal cancer, and HCC.[62] In parts of sub-Saharan Africa, dietary iron overload, mainly from beer prepared in iron pots, is strongly associated with an increased risk of HCC.[63] Experimentally, Wistar rats fed a high-iron diet are shown to develop HCC.[64] One mechanism implicated in iron overload-mediated HCC genesis is due to ROS-inducing DNA mutations, as multiple rat models and surveys of human HCCs have linked increased iron levels with increases in 8-oxo-2-deoxyguanosine adducts and oxidizing products such as malondialdehyde.[65-67]
However, there is evidence that iron overload has a direct effect on p53 activity. C57BL/6 mice fed a high-iron diet show a decrease in p53 protein levels in the liver.[68] Also, male Sprague-Dawley rats fed a high-iron diet for prolonged periods of time present with an increase of MDM2, and a subsequent decrease of p53 in the liver.[69] Another molecular mechanism behind decreased levels of p53 due to iron excess includes that p53 is bound by heme, exported to the cytoplasm, and degraded in HepG2 cells via the proteasomal pathway.[68] Thus, both iron excess and dysregulated heme decrease p53 levels, contributing to HCC development [Figure 1B]. Intriguingly, p53 is also involved in reducing intracellular iron levels by transactivating iron-sulfur cluster enzyme 2 which contributes to reduced iron uptake.[70,71] Thus, following chronic iron overload, reduced p53 activity leads to increased intracellular iron levels, further promoting HCC genesis. It should be noted that patients with hereditary hemochromatosis show higher rates of p53 mutations (64-71%), as compared with those in sporadic HCC, supporting a role of p53 in iron overload-induced HCC genesis.[72,73] In HCC tissues from hereditary hemochromatosis, 45% A>C transitions and 33% G>C transversions, including two hotspots at codon 275 and 298, are identified in the p53 gene [Figure 1A].[73] However, in the study using British families with hereditary hemochromatosis, the p53 mutation spectrum consists of 60% A>G transitions and 40% A>T transversions.[72] Nonetheless, it remains to be elucidated whether iron overload indeed induces HCC in a p53-dependent manner in animal models.
HBV
Globally, it is estimated that 248 million individuals have chronic HBV infection and are positive for the hepatitis surface antigen.[74] HBV is the leading cause of HCC, with the majority being attributed to chronic HBV infection.[75] HBV-mediated HCC tumorigenesis can be caused by repeated bouts of immune-mediated hepatocyte death and subsequent tissue repair, with eventual cirrhosis of the liver.[76] Importantly, 10-30% of HBV-related HCCs do not occur in the background of cirrhosis, indicating additional oncogenic mechanisms behind HBV-induced HCC genesis.[77]
HBV, a circular, partially double-stranded DNA virus, consists of four overlapping open reading frames in its genome: a core region, surface region, polymerase region, and X region which produce seven viral proteins named precore, core, polymerase, L, M, HBx, and S.[78-80] Of these, the HBx protein, which plays a pivotal role in viral replication, is most implicated in HCC genesis.[80] Indeed, HBx induces HCC by sequestering p53 to the cytoplasm in transgenic mouse models [Figure 1B].[81,82]
HBx is also implicated in hepatocyte apoptosis.[78] In many contexts, HBx inhibits apoptosis not only by increasing levels of anti-apoptotic protein, survivin, but also by binding to and sequestering p53 to the cytoplasm.[83-86] HBx is also reported to inhibit TGF-β-mediated apoptosis.[87] Conversely, in some contexts, HBx is shown to induce apoptosis in a p53-independent manner.[88-90] Hence, the dual roles of HBx in hepatocyte apoptosis and its association with HCC genesis warrant further investigation.
HBx variants with C-terminal truncations (Ct-HBx) are frequently detected in HCC and might also contribute to HCC development, though there is no direct evidence for it.[91-93] Ct-HBx promotes hepatocyte proliferation and inhibits apoptosis in multiple cell lines.[94-96] Transcriptional downregulation of ubiquitin specific peptidase 16 (USP16) by Ct-HBx is also shown to enhance tumorigenicity and stem-like properties of HCC cells.[97] Moreover, Ct-HBx binds to p53 and inhibits p53-mediated apoptosis similar to HBx [Figure 1B].[85,98,99] Additionally, some Ct-HBx variants have the ability to silence mRNA expression of GAS2, a modulator of p53-mediated apoptosis.[100] Thus, Ct-HBx may contribute to the pathogenesis of HBV-related HCC by downregulating USP16 and inhibiting p53-mediated apoptosis.
Given that p53 is infrequently mutated in HBV-related HCC, p53 mutations are associated with late stage disease, and both HBx and Ct-HBx bind to and inhibit p53 function [Figure 1B],[101-103] inactivation of p53 activity may be favorable for HBV-mediated HCC tumorigenesis, rather than p53 mutation. Importantly, HCC patients with wild-type p53 have better overall survival and an increase in recurrence free survival as compared with those having p53 mutations.[104]
HCV
Hepatitis C is estimated to have a global prevalence of 184 million individuals positive for anti-HCV, and individuals with HCV have a 15 to 20 fold increased risk for HCC.[105,106] HCV is a 9,600 nucleotide positive sense single-stranded RNA virus with a single open reading.[107,108] The HCV genome encodes for a polyprotein that is subsequently cleaved into nine viral proteins, including structural proteins (C, E1, E2), and non-structural proteins (p7, NS2, NS3, NS4A, NS5A, NS5B).[109] Although the vast majority of HCV-related HCCs occur within the context of cirrhosis, there is some evidence showing oncogenic potential for the HCV viral proteins.[77,110,111] Specifically, HCV core, NS3, and NS5 proteins have been implicated in HCC development in both p53-dependent and -independent manners.[112]
Transgenic mice expressing the HCV core protein indeed spontaneously develop HCC, without the background of cirrhosis.[113,114] HCV core protein also increases ROS, inhibits Fas- and TNF-mediated apoptosis, and upregulates the Wnt-β-catenin pathway.[115-117] Importantly, the core protein inhibits p53 activity by altering its subcellular localization to the perinuclear region and nuclear granular structures, as well as its post-translational modifications such as phosphorylation and acetylation of p53 in HeLa and HepG2 cell lines [Figure 1B].[118] Moreover, the core protein upregulates SIRT1, a deacetylation enzyme for p53, leading to impaired p53-dependent apoptosis in HepG2 cells [Figure 1B].[119] Thus, HCV core protein likely causes HCC in both p53-dependent and -independent manners.
A non-structural HCV protein, NS3, is another HCV protein that can transform human hepatocytes with an increase in cyclooxygenase-2 and activation of mitogen-activated protein kinase.[120-122] NS3 also complexes with p53 in HeLa and NIH3T3 cells[123,124] and inhibit p53’s transcriptional activity in NIH3T3 and Huh7 cells [Figure 1B].[124,125] Moreover, NIH3T3 cells transformed by overexpression of NS3 can form tumors in mice.[126] However, it remains unclear whether transformation by NS3 is p53-dependent or not.
Another non-structural HCV protein, NS5A, can cause steatosis and HCC in transgenic mouse models.[127] NS5A is shown to inhibit TNFα-mediated apoptosis, transactivate c-fos, and inhibit Bax-mediated apoptosis independent of p53.[128-130] However, NS5A can also bind to and colocalize with p53 to the perinuclear membrane, leading to inhibition of p53 transcriptional activity [Figure 1B].[131,132] Moreover, NS5A binds with hTAFII32 at the nucleoplasm membrane and inhibits its ability to stabilize p53, resulting in abrogation of p53-mediated apoptosis in Hep3B cells.[132] Thus, NS5A contributes to HCC development and progression through p53-dependent and -independent mechanisms.
Conclusion
In summary, there is a large body of data indicating p53’s involvement in extrinsic factor-induced liver carcinogenesis. Nonetheless, demonstrating in vivo evidence for the protective role of p53 in HCC genesis is crucial. While many of the aforementioned risk factors for liver cancer have become preventable or treatable, efficient therapeutic strategies are still limited. Hence, understanding the role of p53 in the molecular pathogenesis of HCC and restoring p53 activity in tumors would significantly help accelerate the development of new therapies for this therapy-resistant disease.
Acknowledgments
We thank Atul Ranjan for editing the manuscript and helpful discussion.
Authors’ contributions
Collection of literature data: T. Link, T. Iwakuma
Design, writing and revision of the paper: T. Link, T. Iwakuma
Financial support and sponsorship
This project is supported by NIH 1-R01-CA174735-03 (to T. Iwakuma) grant.
Conflicts of interest
There are no conflicts of interest.
Patient consent
Not applicable.
Ethics approval
Not applicable.
REFERENCES
1. Bray F, Ren JS, Masuyer E, Ferlay J. Global estimates of cancer prevalence for 27 sites in the adult population in 2008. Int J Cancer 2013;132:1133-45.
2. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015;136:E359-86.
3. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011;61:69-90.
4. Dhanasekaran R, Limaye A, Cabrera R. Hepatocellular carcinoma: current trends in worldwide epidemiology, risk factors, diagnosis, and therapeutics. Hepat Med 2012;4:19-37.
5. Strohmeyer G, Niederau C, Stremmel W. Survival and causes of death in hemochromatosis. Observations in 163 patients. Ann N Y Acad Sci 1988;526:245-57.
6. Elzouki AN, Eriksson S. Risk of hepatobiliary disease in adults with severe alpha 1-antitrypsin deficiency (PiZZ): is chronic viral hepatitis B or C an additional risk factor for cirrhosis and hepatocellular carcinoma? Eur J Gastroenterol Hepatol 1996;8:989-94.
7. Fracanzani AL, Conte D, Fraquelli M, Taioli E, Mattioli M, Losco A, Fargion S. Increased cancer risk in a cohort of 230 patients with hereditary hemochromatosis in comparison to matched control patients with non-iron-related chronic liver disease. Hepatology 2001;33:647-51.
8. Chuang SC, La Vecchia C, Boffetta P. Liver cancer: descriptive epidemiology and risk factors other than HBV and HCV infection. Cancer Lett 2009;286:9-14.
9. Magnussen A, Parsi MA. Aflatoxins, hepatocellular carcinoma and public health. World J Gastroenterol 2013;19:1508-12.
11. Wertheim JA, Petrowsky H, Saab S, Kupiec-Weglinski JW, Busuttil RW. Major challenges limiting liver transplantation in the United States. Am J Transplant 2011;11:1773-84.
12. Cillo U, Giuliani T, Polacco M, Herrero Manley LM, Crivellari G, Vitale A. Prediction of hepatocellular carcinoma biological behavior in patient selection for liver transplantation. World J Gastroenterol 2016;22:232-52.
13. Howlader N, Noone AM, Krapcho M, Miller D, Bishop K, Altekruse SF, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA (editors). SEER Cancer Statistics Review, 1975-2013. Bethesda, MD: National Cancer Institute. Available from: http://seer.cancer.gov/csr/1975_2013/. [Based on November 2015 SEER data submission; posted to the SEER web site on April 2016; last accessed on May 26, 2017].
15. Lane D, Levine A. p53 Research: the past thirty years and the next thirty years. Cold Spring Harb Perspect Biol 2010;2:a000893.
16. Cleary SP, Jeck WR, Zhao X, Chen K, Selitsky SR, Savich GL, Tan TX, Wu MC, Getz G, Lawrence MS, Parker JS, Li J, Powers S, Kim H, Fischer S, Guindi M, Ghanekar A, Chiang DY. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology 2013;58:1693-702.
17. Schulze K, Imbeaud S, Letouzé E, Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C, Shinde J, Soysouvanh F, Calatayud AL, Pinyol R, Pelletier L, Balabaud C, Laurent A, Blanc JF, Mazzaferro V, Calvo F, Villanueva A, Nault JC, Bioulac-Sage P, Stratton MR, Llovet JM, Zucman-Rossi J. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet 2015;47:505-11.
18. Parrales A, Iwakuma T. Targeting oncogenic mutant p53 for cancer therapy. Front Oncol 2015;5:288.
19. Iakova P, Timchenko L, Timchenko NA. Intracellular signaling and hepatocellular carcinoma. Semin Cancer Biol 2011;21:28-34.
20. Ranjan A, Bera K, Iwakuma T. Murine double minute 2, a potential p53-independent regulator of liver cancer metastasis. Hepatoma Res 2016;2:114-21.
21. Chen L, Luo L, Chen W, Xu HX, Chen F, Chen LZ, Zeng WT, Chen JS, Huang XH. MicroRNA-24 increases hepatocellular carcinoma cell metastasis and invasion by targeting p53: miR-24 targeted p53. Biomed Pharmacother 2016;84:1113-8.
22. Shank RC. Metabolic activation of mycotoxins by animals and humans: an overview. J Toxicol Environ Health 1977;2:1229-44.
23. Hsieh DP, Atkinson DN. Bisfuranoid mycotoxins: their genotoxicity and carcinogenicity. Adv Exp Med Biol 1991;283:525-32.
24. Liu Y, Chang CC, Marsh GM, Wu F. Population attributable risk of aflatoxin-related liver cancer: systematic review and meta-analysis. Eur J Cancer 2012;48:2125-36.
25. Gross-Steinmeyer K, Eaton DL. Dietary modulation of the biotransformation and genotoxicity of aflatoxin B(1). Toxicology 2012;299:69-79.
26. Smela ME, Currier SS, Bailey EA, Essigmann JM. The chemistry and biology of aflatoxin B(1): from mutational spectrometry to carcinogenesis. Carcinogenesis 2001;22:535-45.
27. Bressac B, Kew M, Wands J, Ozturk M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature 1991;350:429-31.
28. Aguilar F, Hussain SP, Cerutti P. Aflatoxin B1 induces the transversion of G-->T in codon 249 of the p53 tumor suppressor gene in human hepatocytes. Proc Natl Acad Sci U S A 1993;90:8586-90.
29. Ming L, Thorgeirsson SS, Gail MH, Lu P, Harris CC, Wang N, Shao Y, Wu Z, Liu G, Wang X, Sun Z. Dominant role of hepatitis B virus and cofactor role of aflatoxin in hepatocarcinogenesis in Qidong, China. Hepatology 2002;36:1214-20.
30. Puisieux A, Lim S, Groopman J, Ozturk M. Selective targeting of p53 gene mutational hotspots in human cancers by etiologically defined carcinogens. Cancer Res 1991;51:6185-9.
31. Hussain SP, Harris CC. p53 mutation spectrum and load: the generation of hypotheses linking the exposure of endogenous or exogenous carcinogens to human cancer. Mutat Res 1999;428:23-32.
32. Ricordy R, Gensabella G, Cacci E, Augusti-Tocco G. Impairment of cell cycle progression by aflatoxin B1 in human cell lines. Mutagenesis 2002;17:241-9.
33. Kim J, Park SH, Do KH, Kim D, Moon Y. Interference with mutagenic aflatoxin B1-induced checkpoints through antagonistic action of ochratoxin A in intestinal cancer cells: a molecular explanation on potential risk of crosstalk between carcinogens. Oncotarget 2016;7:39627-39.
34. Lee YI, Lee S, Das GC, Park US, Park SM, Lee YI. Activation of the insulin-like growth factor II transcription by aflatoxin B1 induced p53 mutant 249 is caused by activation of transcription complexes; implications for a gain-of-function during the formation of hepatocellular carcinoma. Oncogene 2000;19:3717-26.
35. Whittaker S, Marais R, Zhu AX. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene 2010;29:4989-5005.
36. Weng CJ, Hsieh YH, Tsai CM, Chu YH, Ueng KC, Liu YF, Yeh YH, Su SC, Chen YC, Chen MK, Yang SF. Relationship of insulin-like growth factors system gene polymorphisms with the susceptibility and pathological development of hepatocellular carcinoma. Ann Surg Oncol 2010;17:1808-15.
37. Yao N, Yao D, Wang L, Dong Z, Wu W, Qiu L, Yan X, Yu D, Chen J, Sai W, Zhang H, Yang J. Inhibition of autocrine IGF-II on effect of human HepG2 cell proliferation and angiogenesis factor expression. Tumour Biol 2012;33:1767-76.
38. Mastrangelo G, Fedeli U, Fadda E, Valentini F, Agnesi R, Magarotto G, Marchì T, Buda A, Pinzani M, Martines D. Increased risk of hepatocellular carcinoma and liver cirrhosis in vinyl chloride workers: synergistic effect of occupational exposure with alcohol intake. Environ Health Perspect 2004;112:1188-92.
39. Dogliotti E. Molecular mechanisms of carcinogenesis by vinyl chloride. Ann Ist Super Sanita 2006;42:163-9.
40. Dragani TA, Zocchetti C. Occupational exposure to vinyl chloride and risk of hepatocellular carcinoma. Cancer Causes Control 2008;19:1193-200.
42. el Ghissassi F, Barbin A, Bartsch H. Metabolic activation of vinyl chloride by rat liver microsomes: low-dose kinetics and involvement of cytochrome P450 2E1. Biochem Pharmacol 1998;55:1445-52.
43. Hollstein M, Marion MJ, Lehman T, Welsh J, Harris CC, Martel-Planche G, Kusters I, Montesano R. p53 mutations at A:T base pairs in angiosarcomas of vinyl chloride-exposed factory workers. Carcinogenesis 1994;15:1-3.
44. Trivers GE, Cawley HL, DeBenedetti VM, Hollstein M, Marion MJ, Bennett WP, Hoover ML, Prives CC, Tamburro CC, Harris CC. Anti-p53 antibodies in sera of workers occupationally exposed to vinyl chloride. J Natl Cancer Inst 1995;87:1400-7.
45. Barbin A, Froment O, Boivin S, Marion MJ, Belpoggi F, Maltoni C, Montesano R. p53 gene mutation pattern in rat liver tumors induced by vinyl chloride. Cancer Res 1997;57:1695-8.
46. Smith SJ, Li Y, Whitley R, Marion MJ, Partilo S, Carney WP, Brandt-Rauf PW. Molecular epidemiology of p53 protein mutations in workers exposed to vinyl chloride. Am J Epidemiol 1998;147:302-8.
47. Calzadilla Bertot L, Adams LA. The natural course of non-alcoholic fatty liver disease. Int J Mol Sci 2016;17:774.
48. Bedogni G, Nobili V, Tiribelli C. Epidemiology of fatty liver: an update. World J Gastroenterol 2014;20:9050-4.
49. Baffy G. Hepatocellular carcinoma in non-alcoholic fatty liver disease: epidemiology, pathogenesis, and prevention. J Clin Transl Hepatol 2013;1:131-7.
50. Ertle J, Dechene A, Sowa JP, Penndorf V, Herzer K, Kaiser G, Schlaak JF, Gerken G, Syn WK, Canbay A. Non-alcoholic fatty liver disease progresses to hepatocellular carcinoma in the absence of apparent cirrhosis. Int J Cancer 2011;128:2436-43.
51. Sanyal AJ, Yoon SK, Lencioni R. The etiology of hepatocellular carcinoma and consequences for treatment. Oncologist 2010;15 Suppl 4:14-22.
52. Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol 2013;10:656-65.
53. Tomita K, Teratani T, Suzuki T, Oshikawa T, Yokoyama H, Shimamura K, Nishiyama K, Mataki N, Irie R, Minamino T, Okada Y, Kurihara C, Ebinuma H, Saito H, Shimizu I, Yoshida Y, Hokari R, Sugiyama K, Hatsuse K, Yamamoto J, Kanai T, Miura S, Hibi T. p53/p66Shc-mediated signaling contributes to the progression of non-alcoholic steatohepatitis in humans and mice. J Hepatol 2012;57:837-43.
54. Castro RE, Ferreira DM, Afonso MB, Borralho PM, Machado MV, Cortez-Pinto H, Rodrigues CM. miR-34a/SIRT1/p53 is suppressed by ursodeoxycholic acid in the rat liver and activated by disease severity in human non-alcoholic fatty liver disease. J Hepatol 2013;58:119-25.
55. Tian XF, Ji FJ, Zang HL, Cao H. Activation of the miR-34a/SIRT1/p53 signaling pathway contributes to the progress of liver fibrosis via inducing apoptosis in hepatocytes but not in HSCs. PLoS One 2016;11:e0158657.
56. Ferreira DM, Afonso MB, Rodrigues PM, Simao AL, Pereira DM, Borralho PM, Rodrigues CM, Castro RE. c-Jun N-terminal kinase 1/c-Jun activation of the p53/microRNA 34a/sirtuin 1 pathway contributes to apoptosis induced by deoxycholic acid in rat liver. Mol Cell Biol 2014;34:1100-20.
58. Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ, Arking DE, Beer MA, Maitra A, Mendell JT. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell 2007;26:745-52.
59. Chifman J, Laubenbacher R, Torti SV. A systems biology approach to iron metabolism. Adv Exp Med Biol 2014;844:201-25.
60. Siddique A, Kowdley KV. Review article: the iron overload syndromes. Aliment Pharmacol Ther 2012;35:876-93.
61. Heeney MM. Iron clad: iron homeostasis and the diagnosis of hereditary iron overload. Hematology Am Soc Hematol Educ Program 2014;2014:202-9.
63. Kew MC. Hepatocellular carcinoma in African Blacks: recent progress in etiology and pathogenesis. World J Hepatol 2010;2:65-73.
64. Asare GA, Paterson AC, Kew MC, Khan S, Mossanda KS. Iron-free neoplastic nodules and hepatocellular carcinoma without cirrhosis in Wistar rats fed a diet high in iron. J Pathol 2006;208:82-90.
65. Kang JO, Jones C, Brothwell B. Toxicity associated with iron overload found in hemochromatosis: possible mechanism in a rat model. Clin Lab Sci 1998;11:350-4.
66. Jungst C, Cheng B, Gehrke R, Schmitz V, Nischalke HD, Ramakers J, Schramel P, Schirmacher P, Sauerbruch T, Caselmann WH. Oxidative damage is increased in human liver tissue adjacent to hepatocellular carcinoma. Hepatology 2004;39:1663-72.
67. Asare GA, Mossanda KS, Kew MC, Paterson AC, Kahler-Venter CP, Siziba K. Hepatocellular carcinoma caused by iron overload: a possible mechanism of direct hepatocarcinogenicity. Toxicology 2006;219:41-52.
68. Shen J, Sheng X, Chang Z, Wu Q, Wang S, Xuan Z, Li D, Wu Y, Shang Y, Kong X, Yu L, Li L, Ruan K, Hu H, Huang Y, Hui L, Xie D, Wang F, Hu R. Iron metabolism regulates p53 signaling through direct heme-p53 interaction and modulation of p53 localization, stability, and function. Cell Rep 2014;7:180-93.
69. Bloomer SA, Brown KE. Tumour promotion versus tumour suppression in chronic hepatic iron overload. Cell Biochem Funct 2015;33:241-8.
70. Zhang F, Wang W, Tsuji Y, Torti SV, Torti FM. Post-transcriptional modulation of iron homeostasis during p53-dependent growth arrest. J Biol Chem 2008;283:33911-8.
71. Funauchi Y, Tanikawa C, Yi Lo PH, Mori J, Daigo Y, Takano A, Miyagi Y, Okawa A, Nakamura Y, Matsuda K. Regulation of iron homeostasis by the p53-ISCU pathway. Sci Rep 2015;5:16497.
72. Vautier G, Bomford AB, Portmann BC, Metivier E, Williams R, Ryder SD. p53 mutations in british patients with hepatocellular carcinoma: clustering in genetic hemochromatosis. Gastroenterology 1999;117:154-60.
73. Marrogi AJ, Khan MA, van Gijssel HE, Welsh JA, Rahim H, Demetris AJ, Kowdley KV, Hussain SP, Nair J, Bartsch H, Okby N, Poirier MC, Ishak KG, Harris CC. Oxidative stress and p53 mutations in the carcinogenesis of iron overload-associated hepatocellular carcinoma. J Natl Cancer Inst 2001;93:1652-5.
74. Ott JJ, Stevens GA, Groeger J, Wiersma ST. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine 2012;30:2212-9.
75. Chan SL, Wong VW, Qin S, Chan HL. Infection and cancer: the case of hepatitis B. J Clin Oncol 2016;34:83-90.
76. Tarocchi M, Polvani S, Marroncini G, Galli A. Molecular mechanism of hepatitis B virus-induced hepatocarcinogenesis. World J Gastroenterol 2014;20:11630-40.
77. El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 2007;132:2557-76.
78. Kew MC. Hepatitis B virus x protein in the pathogenesis of hepatitis B virus-induced hepatocellular carcinoma. J Gastroenterol Hepatol 2011;26:144-52.
79. Datta S, Chatterjee S, Veer V, Chakravarty R. Molecular biology of the hepatitis B virus for clinicians. J Clin Exp Hepatol 2012;2:353-65.
80. Seeger C, Mason WS. Molecular biology of hepatitis B virus infection. Virology 2015;479–480:672-86.
81. Kim CM, Koike K, Saito I, Miyamura T, Jay G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 1991;351:317-20.
82. Ueda H, Ullrich SJ, Gangemi JD, Kappel CA, Ngo L, Feitelson MA, Jay G. Functional inactivation but not structural mutation of p53 causes liver cancer. Nat Genet 1995;9:41-7.
83. Wang XW, Forrester K, Yeh H, Feitelson MA, Gu JR, Harris CC. Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding, transcriptional activity, and association with transcription factor ERCC3. Proc Natl Acad Sci U S A 1994;91:2230-4.
84. Truant R, Antunovic J, Greenblatt J, Prives C, Cromlish JA. Direct interaction of the hepatitis B virus HBx protein with p53 leads to inhibition by HBx of p53 response element-directed transactivation. J Virol 1995;69:1851-9.
85. Elmore LW, Hancock AR, Chang SF, Wang XW, Chang S, Callahan CP, Geller DA, Will H, Harris CC. Hepatitis B virus X protein and p53 tumor suppressor interactions in the modulation of apoptosis. Proc Natl Acad Sci U S A 1997;94:14707-12.
86. Kuo TC, Chao CC. Hepatitis B virus X protein prevents apoptosis of hepatocellular carcinoma cells by upregulating SATB1 and HURP expression. Biochem Pharmacol 2010;80:1093-102.
87. Shih WL, Kuo ML, Chuang SE, Cheng AL, Doong SL. Hepatitis B virus X protein inhibits transforming growth factor-beta -induced apoptosis through the activation of phosphatidylinositol 3-kinase pathway. J Biol Chem 2000;275:25858-64.
88. Kim H, Lee H, Yun Y. X-gene product of hepatitis B virus induces apoptosis in liver cells. J Biol Chem 1998;273:381-5.
89. Terradillos O, Pollicino T, Lecoeur H, Tripodi M, Gougeon ML, Tiollais P, Buendia MA. p53-independent apoptotic effects of the hepatitis B virus HBx protein in vivo and in vitro. Oncogene 1998;17:2115-23.
90. Schuster R, Gerlich WH, Schaefer S. Induction of apoptosis by the transactivating domains of the hepatitis B virus X gene leads to suppression of oncogenic transformation of primary rat embryo fibroblasts. Oncogene 2000;19:1173-80.
91. Wang Y, Lau SH, Sham JS, Wu MC, Wang T, Guan XY. Characterization of HBV integrants in 14 hepatocellular carcinomas: association of truncated X gene and hepatocellular carcinogenesis. Oncogene 2004;23:142-8.
92. Ma NF, Lau SH, Hu L, Xie D, Wu J, Yang J, Wang Y, Wu MC, Fung J, Bai X, Tzang CH, Fu L, Yang M, Su YA, Guan XY. COOH-terminal truncated HBV X protein plays key role in hepatocarcinogenesis. Clin Cancer Res 2008;14:5061-8.
93. Liu LP, Hu BG, Ye C, Ho RL, Chen GG, Lai PB. HBx mutants differentially affect the activation of hypoxia-inducible factor-1alpha in hepatocellular carcinoma. Br J Cancer 2014;110:1066-73.
94. Tu H, Bonura C, Giannini C, Mouly H, Soussan P, Kew M, Paterlini-Bréchot P, Bréchot C, Kremsdorf D. Biological impact of natural COOH-terminal deletions of hepatitis B virus X protein in hepatocellular carcinoma tissues. Cancer Res 2001;61:7803-10.
95. Sirma H, Giannini C, Poussin K, Paterlini P, Kremsdorf D, Brechot C. Hepatitis B virus X mutants, present in hepatocellular carcinoma tissue abrogate both the antiproliferative and transactivation effects of HBx. Oncogene 1999;18:4848-59.
96. Zhang H, Shan CL, Li N, Zhang X, Zhang XZ, Xu FQ, Zhang S, Qiu LY, Ye LH, Zhang XD. Identification of a natural mutant of HBV X protein truncated 27 amino acids at the COOH terminal and its effect on liver cell proliferation. Acta Pharmacol Sin 2008;29:473-80.
97. Qian Y, Wang B, Ma A, Zhang L, Xu G, Ding Q, Jing T, Wu L, Liu Y, Yang Z, Liu Y. USP16 downregulation by carboxyl-terminal truncated HBx promotes the growth of hepatocellular carcinoma cells. Sci Rep 2016;6:33039.
98. Huo TI, Wang XW, Forgues M, Wu CG, Spillare EA, Giannini C, Brechot C, Harris CC. Hepatitis B virus X mutants derived from human hepatocellular carcinoma retain the ability to abrogate p53-induced apoptosis. Oncogene 2001;20:3620-8.
99. Iyer S, Groopman JD. Interaction of mutant hepatitis B X protein with p53 tumor suppressor protein affects both transcription and cell survival. Mol Carcinog 2011;50:972-80.
100. Zhu R, Mok MT, Kang W, Lau SS, Yip WK, Chen Y, Lai PB, Wong VW, To KF, Sung JJ, Cheng AS, Chan HL. Truncated HBx-dependent silencing of GAS2 promotes hepatocarcinogenesis through deregulation of cell cycle, senescence and p53-mediated apoptosis. J Pathol 2015;237:38-49.
101. Long J, Wang Y, Li M, Tong WM, Jia JD, Huang J. Correlation of TP53 mutations with HCV positivity in hepatocarcinogenesis: identification of a novel TP53 microindel in hepatocellular carcinoma with HCV infection. Oncol Rep 2013;30:119-24.
102. Saitta C, Lanza M, Bertuccio A, Lazzara S, Navarra G, Raimondo G, Pollicino T. Evaluation of CTNNB1 and TP53 variability in patients with hepatocellular carcinoma and occult hepatitis B virus infection. Cancer Genet 2015;208:513-6.
103. Amaddeo G, Cao Q, Ladeiro Y, Imbeaud S, Nault JC, Jaoui D, Gaston Mathe Y, Laurent C, Laurent A, Bioulac-Sage P, Calderaro J, Zucman-Rossi J. Integration of tumour and viral genomic characterisations in HBV-related hepatocellular carcinomas. Gut 2015;64:820-9.
104. Liu J, Ma Q, Zhang M, Wang X, Zhang D, Li W, Wang F, Wu E. Alterations of TP53 are associated with a poor outcome for patients with hepatocellular carcinoma: evidence from a systematic review and meta-analysis. Eur J Cancer 2012;48:2328-38.
105. El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012;142:1264-73.e1.
106. Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology 2013;57:1333-42.
107. Choo QL, Richman KH, Han JH, Berger K, Lee C, Dong C, Gallegos C, Coit D, Medina-Selby R, Barr PJ. Genetic organization and diversity of the hepatitis C virus. Proc Natl Acad Sci U S A 1991;88:2451-5.
109. Bukh J. The history of hepatitis C virus (HCV): basic research reveals unique features in phylogeny, evolution and the viral life cycle with new perspectives for epidemic control. J Hepatol 2016;65:S2-21.
110. Niederau C, Lange S, Heintges T, Erhardt A, Buschkamp M, Hürter D, Nawrocki M, Kruska L, Hensel F, Petry W, Häussinger D. Prognosis of chronic hepatitis C: results of a large, prospective cohort study. Hepatology 1998;28:1687-95.
111. Grebely J, Page K, Sacks-Davis R, van der Loeff MS, Rice TM, Bruneau J, Morris MD, Hajarizadeh B, Amin J, Cox AL, Kim AY, McGovern BH, Schinkel J, George J, Shoukry NH, Lauer GM, Maher L, Lloyd AR, Hellard M, Dore GJ, Prins M; InC3 Study Group. The effects of female sex, viral genotype, and IL28B genotype on spontaneous clearance of acute hepatitis C virus infection. Hepatology 2014;59:109-20.
112. Bartosch B, Thimme R, Blum HE, Zoulim F. Hepatitis C virus-induced hepatocarcinogenesis. J Hepatol 2009;51:810-20.
113. Moriya K, Fujie H, Shintani Y, Yotsuyanagi H, Tsutsumi T, Ishibashi K, Matsuura Y, Kimura S, Miyamura T, Koike K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med 1998;4:1065-7.
114. Lemon SM, Lerat H, Weinman SA, Honda M. A transgenic mouse model of steatosis and hepatocellular carcinoma associated with chronic hepatitis C virus infection in humans. Trans Am Clin Climatol Assoc 2000;111:146-56; discussion 156-7.
115. Ivanov AV, Smirnova OA, Petrushanko IY, Ivanova ON, Karpenko IL, Alekseeva E, Sominskaya I, Makarov AA, Bartosch B, Kochetkov SN, Isaguliants MG. HCV core protein uses multiple mechanisms to induce oxidative stress in human hepatoma Huh7 cells. Viruses 2015;7:2745-70.
116. Marusawa H, Hijikata M, Chiba T, Shimotohno K. Hepatitis C virus core protein inhibits Fas- and tumor necrosis factor alpha-mediated apoptosis via NF-kappaB activation. J Virol 1999;73:4713-20.
117. Liu J, Wang Z, Tang J, Tang R, Shan X, Zhang W, Chen Q, Zhou F, Chen K, Huang A, Tang N. Hepatitis C virus core protein activates Wnt/beta-catenin signaling through multiple regulation of upstream molecules in the SMMC-7721 cell line. Arch Virol 2011;156:1013-23.
118. Kao CF, Chen SY, Chen JY, Wu Lee YH. Modulation of p53 transcription regulatory activity and post-translational modification by hepatitis C virus core protein. Oncogene 2004;23:2472-83.
119. Feng S, Li M, Zhang J, Liu S, Wang Q, Quan M, Zhang M, Cheng J. Regulation of HepG2 cell apoptosis by hepatitis C virus (HCV) core protein via the sirt1-p53-bax pathway. Virus Genes 2015;51:338-46.
120. He QQ, Cheng RX, Sun Y, Feng DY, Chen ZC, Zheng H. Hepatocyte transformation and tumor development induced by hepatitis C virus NS3 c-terminal deleted protein. World J Gastroenterol 2003;9:474-8.
121. Lu L, Wei L, Peng G, Mu Y, Wu K, Kang L, Yan X, Zhu Y, Wu J. NS3 protein of hepatitis C virus regulates cyclooxygenase-2 expression through multiple signaling pathways. Virology 2008;371:61-70.
122. Feng DY, Sun Y, Cheng RX, Ouyang XM, Zheng H. Effect of hepatitis C virus nonstructural protein NS3 on proliferation and MAPK phosphorylation of normal hepatocyte line. World J Gastroenterol 2005;11:2157-61.
123. Ishido S, Hotta H. Complex formation of the nonstructural protein 3 of hepatitis C virus with the p53 tumor suppressor. FEBS Lett 1998;438:258-62.
124. Kwun HJ, Jung EY, Ahn JY, Lee MN, Jang KL. p53-dependent transcriptional repression of p21(waf1) by hepatitis C virus NS3. J Gen Virol 2001;82:2235-41.
125. Deng L, Nagano-Fujii M, Tanaka M, Nomura-Takigawa Y, Ikeda M, Kato N, Sada K, Hotta H. NS3 protein of hepatitis C virus associates with the tumour suppressor p53 and inhibits its function in an NS3 sequence-dependent manner. J Gen Virol 2006;87:1703-13.
126. Sakamuro D, Furukawa T, Takegami T. Hepatitis C virus nonstructural protein NS3 transforms NIH 3T3 cells. J Virol 1995;69:3893-6.
127. Wang AG, Lee DS, Moon HB, Kim JM, Cho KH, Choi SH, Ha HL, Han YH, Kim DG, Hwang SB, Yu DY. Non-structural 5A protein of hepatitis C virus induces a range of liver pathology in transgenic mice. J Pathol 2009;219:253-62.
128. Ghosh AK, Majumder M, Steele R, Meyer K, Ray R, Ray RB. Hepatitis C virus NS5A protein protects against TNF-alpha mediated apoptotic cell death. Virus Res 2000;67:173-8.
129. Yeh CT, Chang MH, Shyu WC, Chang ML, Yang PY, Tsao ML, Lai HY. Characterization of a HCV NS5A protein derived from a patient with hepatoma. Biochem Biophys Res Commun 2005;327:516-22.
130. Chung YL, Sheu ML, Yen SH. Hepatitis C virus NS5A as a potential viral Bcl-2 homologue interacts with Bax and inhibits apoptosis in hepatocellular carcinoma. Int J Cancer 2003;107:65-73.
131. Majumder M, Ghosh AK, Steele R, Ray R, Ray RB. Hepatitis C virus NS5A physically associates with p53 and regulates p21/waf1 gene expression in a p53-dependent manner. J Virol 2001;75:1401-7.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Link T, Iwakuma T. Roles of p53 in extrinsic factor-induced liver carcinogenesis. Hepatoma Res 2017;3:95-104. http://dx.doi.org/10.20517/2394-5079.2017.07
AMA Style
Link T, Iwakuma T. Roles of p53 in extrinsic factor-induced liver carcinogenesis. Hepatoma Research. 2017; 3: 95-104. http://dx.doi.org/10.20517/2394-5079.2017.07
Chicago/Turabian Style
Link, Tim, Tomoo Iwakuma. 2017. "Roles of p53 in extrinsic factor-induced liver carcinogenesis" Hepatoma Research. 3: 95-104. http://dx.doi.org/10.20517/2394-5079.2017.07
ACS Style
Link, T.; Iwakuma T. Roles of p53 in extrinsic factor-induced liver carcinogenesis. Hepatoma. Res. 2017, 3, 95-104. http://dx.doi.org/10.20517/2394-5079.2017.07
About This Article
Copyright

Author Biographies

Data & Comments
Data


 Cite This Article 2 clicks
Cite This Article 2 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.