
Immunotherapy for hepatocellular carcinoma: the force awakens in HCC?


Abstract
Systemic therapy for hepatocellular carcinoma (HCC) has been disappointing. The only drug approved by Food and Drug Administration recently has been sorafenib. Sorafenib has modest benefits with a low response rate and an improvement in time to progression of only 2-3 months. Multiple randomized trials, which compare the new agent to sorafenib as either first line or second line therapy, have been negative, showing no improved clinical benefit. Recently, in a large phase III randomized trial, regorafenib has shown superiority to placebo as a second line treatment for HCC. However, this drug has multiple side effects and is not well tolerated by many patients. The clinical benefit is also modest. Clearly, new approaches to treat advanced HCC are still needed. There is data showing that HCC is immunogenic and the immune system can be stimulated to attack these cancer cells. This article will briefly review immunotherapy as a promising treatment for HCC.
Keywords
Introduction
There is a strong rationale to evaluate immunotherapy in this disease. Hepatocellular carcinoma (HCC) is typically an inflammation-associated cancer and can be immunogenic. Both hepatitis B virus (HBV) and hepatitis C virus (HCV) are known to be risk factors for the development of HCC. The HCC from HCV typically develops in a setting of long standing liver cirrhosis. On the other hand, HCC may develop from HBV even in the absence of liver cirrhosis. Increasingly, HCC appears to be developing from nonalcoholic steatohepatitis (NASH) with obesity being a risk factor. Studies in mice have shown that dietary factors and genetic obesity can promote liver inflammation and tumorigenesis. This appears to be mediated by enhancing interleukin (IL)-6 and tumor necrosis factor (TNF) expression. With increasing incidence of obesity in the western countries, NASH will become a greater risk factor for HCC.
Another supporting factor is that spontaneous regression have been reported in HCC.[1] Spontaneous regression has been reported in other cancers as well and is thought to be mediated by immune response in the host. Although infrequent, these occurrences suggest it may be possible to enhance the immune system against certain malignancies.
In addition, more than 50% HCC patients develop spontaneous cellular or humoral immune response against NY-ESO-1.[2] Furthermore, tumor-associated antigens (TAA)-specific CD8+ T-cell immune responses have been described [most studied include AFP, glypican-3, NY-ESO-1, SSX-2, MAGE-A, human telomerase-reverse transcriptase (h-TERT)].[3] More than 50% HCC patients had HCC-infiltrating TAA-specific CD8+ T-cells and these cell numbers correlated with progression-free survival.[4]
In HCC, the number of T regulatory cells increased both in peripheral blood and inside tumor itself.[5] Intratumoral T regs correlated with disease progression and poor prognosis.[6] Activation of T-cell infiltration (CD4+, CD8+, natural killer cells) has been observed after liver ablation.[7,8] Together the data shows that inflammation is a common feature seen in HCC and this tumor can elicit an immune response.
Reasons for HCC immune tolerance
If HCC is typically an inflammation-associated cancer and can be immunogenic, why is there immune tolerance? There are several factors which may be involved.
On a cellular level, the liver is a site for myeloid-derived suppressor cells (MDSC) which can inhibit effector T-cell function and decrease natural killer (NK) cell cytotoxicity and cytokine production. The frequency of MDSCs correlates with progression-free survival in HCC after radiofrequency ablation.[9] It has been suggested that MDSCs interact with Kupffer cells to induce programmed death-1 (PD-1) expression and MDSCs may help expand T regs.[10] Depletion of T regs or MDSCs may prompt spontaneous immune responses against α-fetoprotein (AFP).[11,12]
A new subset of immune suppressive cells has been described in HCC called regulatory dendritic cells (DCs) which can suppress T-cell activation via IL-10 and indoleamine 2,3-dioxygenase (IDO) production.[13] Unexpectedly, it was found that these dendritic cells expressed high levels of cytotoxic T-lymphocyte antigen-4 (CTLA-4) and PD-1. CTLA-4 was discovered to be essential for IL-10 and IDO production. This finding represents a target for immunotherapy as well as one possible explanation for immune tolerance.
Human HCC tumor-infiltrating CD4+ CD69+ T regs are higher than conventional CD4+ CD25+ Foxp3+ T regs and correlates with tumor progression.[14] These T regs do not express CD25 or Foxp3 but express high levels of mouse transforming growth factor beta 1 (mTGF-β1), PD-1, CTLA-4 and could suppress CD4 T-cell proliferation via mTGF-β1.[14] The percentage of these T regs in tumors correlated significantly with tumor progression.
Failure of HCC-associated antigen production presentation by antigen presenting cells is due to decreased expression of HLA class 1 molecules and ineffective antigen processing.[15,16]
Increase in T regulatory cells, invariant NK T-cells, MDSC and tumor-associated macrophages may play a role and a decrease in CD4+ T helper cells has been reported.[4,11,17-20]
There is an increase in CD4+, CD25+ T regs within tumor infiltrating lymphocytes (TILs) which is associated with decrease in number and function of CD8+ T-cells.[5,17]
T-cell (CD4+) exhaustion and apoptosis have been associated with chronic HCV infection.[21] The CD4+ T-cells of chronic HCV-infected patients displayed increased surface expression of TRAIL and expression of other immune exhaustion molecules. In addition, indoleamine 2,3-dioxygenase activity in increased and IDO is a T-cell proliferation-limiting enzyme. Other molecules associated with T-cell exhaustion and apoptosis signaling in peripheral blood mononucleocytes from chronically infected HCV patients have been described.[21]
LAG-3 expression has been found to be significantly up-regulated in tumor infiltrating CD8+ T-cells in HCC patients and a severe functional defect was detected in tumor infiltrating HBV-specific CD8+ T-cells at the tumor site.[22] Because LAG-3 is an inhibitory molecule that helps to downregulate T-cell responses, there was a correlation between LAG-3 expression and HBV-specific CD8+ T-cell dysfunction.
Thus, the immunosuppression that is seen in HCC is complex and involves a balance of multiple factors [Figure 1]. The factors which are involved in tumor progression include hepatic tolerance to various antigens, chronic inflammation which predisposes to immunosuppression and liver cancer dependent immune tolerance. Poorly counter-balancing this involves antitumor immune response.
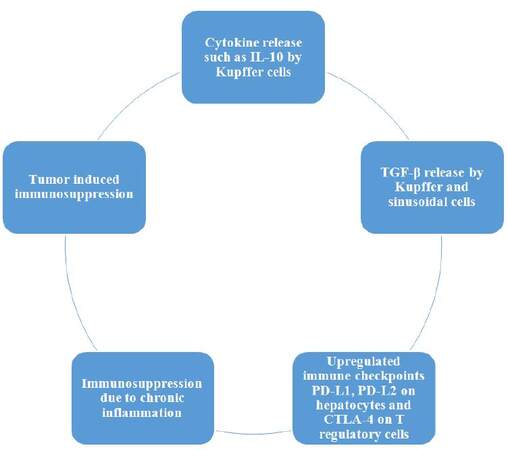
Figure 1. Selected known mechanisms of immune tolerance in HCC. HCC: hepatocellular carcinoma; IL-10: interleukin-10; TGF-β: transforming growth factor beta; PD: programmed death; CTLA: cytotoxic T-lymphocyte antigen
Rationale for immunotherapy for HCC
NK cells have been divided into two major groups, one mainly cytotoxic and the other mainly involved in cytokine secretion. In HCV-related hepatocellular carcinoma a prevalent cytotoxic NK phenotype was found in HCC patients with longer time to tumor recurrence and overall survival.[23] This suggests a role for NK cells in the immune response against HCC and a rationale for immunotherapy using NK enhancing therapy.
Pim kinases are downstream effector molecules of certain oncogenes. Pim-3 expression occurs in certain solid tumors and is highly expressed in HCC tissues and cell lines. In HCC Pim-3 is associated with acceleration of HCC development and has been found to inhibit apoptosis by phosphorylating the proapoptotic BH3-only protein BAD. A dual-function vector with both immunostimulatory and Pim-3-silencing effects inhibited Hepa1-6 cell growth by regulating expression of apoptosis-related proteins and inducing secretion of type I interferons.[24] NK cells, CD4+ T and CD8+ T-cells and macrophages were required for effective tumor inhibition and CD4+ T-cells were demonstrated to play helper role in NK cell activation. This novel bi-functional vector represents a novel immunotherapy approach to treat HCC.
PD-1/PD-L1 pathway has been associated with T-cell exhaustion in chronic hepatitis virus infection.[25] Inhibition of T-cell activation is due to PD-1 ligation, which causes recruitment of SHP-2 phosphatase. This in turn inhibits PI3K activity and downstream activation of AKt, with dampening of T-cell receptor signaling. PD-1/PD-L-1 involvement in chronic viral infection-associated T-cell exhaustion was first shown in a murine lymphocytic choriomeningitis model.[26] PD-1/PD-L-1 blockade can produce functional recovery of T-cells, cytokine secretion, cytotoxic capability and decreased viral load.
Immunotherapy trials in HCC
Interferons
Immunotherapy has been used for HCC in the past with limited success. Interferon alpha was combined with systemic chemotherapy and modest activity was observed. In one study, 26 patients with advanced HCC received intravenous cisplatin, doxorubicin, and 5-fluorouracil combined with subcutaneous administered human recombinant α-interferon-2a (PIAF regimen).[27] The disease control rate was 50% (4 partial responses and 9 stable disease). The 1-year survival rate was 24.3% and median survival time was 6.0 months. A modified PIAF showed superior response rate and survival compared to standard dose PIAF.[28] A randomized trial of PIAF versus standard dose doxorubicin showed that PIAF had more activity and greater toxicity, particularly myelosuppression.[29] Since myelosuppression was greater than single agent doxorubicin, this treatment regimen is best for younger patients with good performance status and adequate bone marrow reserve.
Checkpoint inhibitors
Checkpoint inhibitors have demonstrated activity in a number of cancers including HCC. CTLA-4 blockade has been evaluated using Tremelimumab.[30] In this study the drug was given at 15 mg/kg intravenously every 90 days. Twenty patients were treated and 17 patients were assessable for response. In these 17 patients, the overall response rate was 17.6% and disease control rate was 76.4%. The median time to tumor progression was 6.48 months. Toxicity was mainly hepatic. There were > grade 3 AST and ALT elevation in 45% patients and 25%, respectively. Serum bilirubin elevated occurred in 10%. Skin rash occurred in 5%. An antiviral effect was noted. FoxP3+ natural T reg expanded and viral hepatitis C load decreased.
A dose escalation study of the PD-1 inhibitor Nivolumab was initially reported at ASCO in 2015 and subsequently updated at ASCO 2016.[31] In the initial report, 43 patients were treated in dose-escalation phase. Median of 6 (range 1-42) doses of Nivolumab administered. One dose-limiting toxicity (DLT) with grade 2 hepatic decompensation occurred at 10 mg/kg in uninfected cohort. No maximum tolerated dose was identified. The 3 mg/kg dose was selected for dose expansion in uninfected and HCV-infected cohorts. Thirty patients had discontinued therapy (26 patients due to progressive disease, 2 due to complete response (CR), 2 due to adverse events (1 due to increase in bilirubin, 1 due to treatment-related increase in AST/ALT and hepatitis). Responses usually occurred within 3 months of drug initiation. The overall response rate was 15% (10% CR and 5% partial response, PR). The preliminary overall 1-year survival was 62%. This is superior to the 1-year overall survival of 30% reported in phase III trials after sorafenib failure.
An update at ASCO 2016 showed the following: the dose escalation part involved 48 patients and the expansion part involved 214 patients.[32] The dose of nivolumab for the dose escalation ranged from 0.1 mg/kg to 10 mg/kg, while in the dose expansion study nivolumab was given at 3 mg/kg every 2 weeks.
A total of 10 patients had HCV, 15 patients had HBV and 23 patients were uninfected. Safety data was presented for 48 patients. Of these patients 79% had treatment-related adverse events (TRAEs) of any grade, including 25 % with grade 3-4. The most common TRAEs were rash, pruritus and elevation of AST, ALT, lipase and amylase. AST and ALT and lipase/amylase occurred more frequently in this group of patients compared to other nivolumab-treated patient populations. Most of these TRAEs were noted to be asymptomatic and reversible. Five of 7 patients responded within 3 months of start of treatment.
Response was ongoing beyond 24 months in 1 patient who stopped treatment with a complete response. The median duration of response was 17 months, range 6-24 months. Stable disease occurred out to 12-18 months. Altogether, 3 patients had a complete response and four had a partial response; the disease control rate was 65%. The expansion phase was continuing with nivolumab at 3 mg/kg. In conclusion, Nivolumab has shown manageable safety and toxicity in HCC patients, including HBV and HCV infected patients. Antitumor activity was observed across all dose levels and cohorts. The response was durable in many patients and the 1-year overall survival rate was encouraging.
An investigator-initiated, first in-human phase II trial of the PD-1 inhibitor pembrolizumab was started at the University of Miami (ClinicalTrials.gov NCT02658019). The dose of pembrolizumab is 200 mg intravenously every 3 weeks. So far, 10 patients have been treated. It is too early yet to assess response. Correlative studies include tumor staining for PD-L-1, and changes in hepatitis B and hepatitis C viral titers. In addition, several representative cytokines associated with T-cell activation and suppression in serum and peripheral blood mononuclear cells will also be analyzed pre- and post-treatment. T-cell proliferation and activation in response to T cell receptor (TCR) and CD28 signals require IL-2, IL-12, IFN-γ stimulation, but can be suppress by immunosuppressive cytokines such as IL-10 and TGF-β. Changes in these cytokines may be important to predict response/toxicity to therapy.
Dendritic cell vaccine
Another approach to immunotherapy is by infusion of autologous dendritic cell vaccine.[33] Patients with advanced HCC were infused with mature autologous dendritic cells pulsed ex vivo with a liver tumor cell lysate and compared to a control group. There were 15 patients in each group. In terms of response for the treated group, 2 patients (13.3%) had partial response, 9 (60%) had stable disease and 4 (26.7%) patients had progressive disease. Serum gamma interferon and CD8+ T-cells both were increased after dendritic cell vaccination. The median survival time for the treated group was 7 months versus 4 months for the untreated group. The side effects of the dendritic cell vaccine were minimal with low-grade fever and mild bone aches.
Viral oncolytics
A viral oncolytic immunotherapy approach has been studied using a vaccinia virus.[34-36] These therapies have been designed to replicate selectively within HCC cells and produce cell lysis, while inducing tumor-specific immunity. JX-594 (Pexa-Vec) is a vaccinia virus with a disrupted viral thymidine kinase gene with insertion of human granulocytic-macrophage colony-stimulating factor and β-galactosidase transgenes both for immune stimulation and replication assessment. A phase I trial of JX-594 demonstrated feasibility and tolerability and produced some responses.[35,36] A randomized, dose finding trial was performed with direct infusion of the vaccinia virus into liver tumors (days 1, 15 and 29). Patient survival duration was significantly longer with high dose (median 14.1 months) compared to low dose (6.7 months). Responses were observed in both injected and noninjected tumors within both dose groups.
Future direction
Immunotherapy, especially checkpoint inhibitors, has recently shown promise in a number of cancers, including HCC. How to improve current immunotherapy for HCC? Understanding the mechanisms of resistance and methods to potentiate response remain a challenge. It has been shown that intrahepatic HCV-specific CD8 T-cells from patients with chronic HCV infection were highly PD-1 positive, very dysfunctional, and unexpectedly refractory to PD1/PD L-1 blockade.[37] This functional impairment was HCV-specific and directly correlated with the level of PD-1 expression. The highly PD-1 positive intrahepatic CD8 T-cells were more exhausted with increased CTLA-4 and with reduced CD28 and CD 127 than circulating T-cells. Thus, a novel therapeutic approach is to combine CTLA-4 with PD-1 blockade. Indeed, studies have shown that there is a synergistic reversal of intrahepatic HCV-specific CD8 T-cell exhaustion by combining anti-CTLA-4 antibody with PD-1 inhibitor.[38] This has therapeutic implications as both anti-CTLA-4 antibody and PD-1 inhibitors alone have shown activity in HCC.[30-32] As demonstrated in the treatment of malignant melanoma, the combination has been tolerable and antitumor response may be greater than single agent used alone. However, toxicities including more autoimmune reactions such as diarrhea, hypophysitis and hepatitis may be more frequent and the dose and tolerability of combination therapy will need to be carefully defined. Drug-induced hepatitis is the main concern and may limit patients with significant liver impairment including those with a history of autoimmune diseases such as autoimmune hepatitis.
Currently, a study is being planned to combine anti-CTLA-4 antibody with anti-PD-1 inhibitor in advanced, unresectable HCC.
Another approach is to apply different modalities together. There is a rationale to evaluate combinatorial therapy for HCC. Adding checkpoint inhibitor to TACE or liver ablation seems reasonable. TACE and liver ablation can both increase T-cell infiltration including NK cells.[7,39] Another study showed that CD4/CD8 ratio and number of B cells and natural killer cells were significantly decreased in HCC patients prior to treatment.[40] When compared to pretreatment levels, the CD4+ and CD4/CD8 ratio decreased but the CD8+ cells increased in the TACE group. In the TACE + RFA group, the CD4/CD8 ratio and the natural killer cells and the CD8+ cells increased. On the other hand, the CD3+, CD8+, CD4/CD8 ratio and natural killer cell increased in the RFA group. More recently, tremelimumab has been combined with subtotal ablation (TACE, RFA or cryoablation) in patients with either HCC or biliary tract carcinomas.[41] In this study 14 patients had TACE, 19 had RFA, including 9 with biliary tract cancers and 5 had cryoablation. No dose limiting toxicity was noted during the trial. Seventeen patients were evaluable for response for lesions outside of TACE/RFA-treated lesions. Of these 4 patients or 23.5% had confirmed partial response. This is noteworthy that 10 of 12 patients with quantifiable HCV had marked reduction in viral load. Liver biopsies were done at week 6. These showed increase in CD8+ T-cells only in patients with clinical response. Furthermore, in peripheral blood mononuclear cells, there was a statistically significant change in CD4/T reg and CD8/T reg ratio in clinical responders. The median time to tumor progression for evaluable HCC patients was 5.7 months.[41]
Chemotherapy has been used for lymphodepleting prior to adoptive T-cell therapy. This lymphodepletion has been shown to enhance immune reconstitution by the transferred cells and increase tumor specific responses. Low dose cyclophosphamide can impair T regulatory cells and can unmask AFP- specific CD4+ T-cell responses in patients with advanced HCC.[12] Adoptive T-cell therapy itself, which uses a patient’s own T lymphocytes genetically altered to enhance anti-tumor activity, expanded in vivo and then infused into the patient, has a number of problems. Problems include the need for surgery to obtain tumor-reactive TIL cells and the expansion of the TIL cells from tumors. Alternatively, adoptive transfer of bulk T lymphocytes can be procured from peripheral blood and expanded in vivo to generate large number of T lymphocytes before infusing back into the patient. Problems with this approach include tumor cells can have low antigen presentation and most tumor antigens are normally expressed as self-antigens. Thus, the T-cell receptor (TCR) may have low affinity for these self-tumor antigens.
To overcome these obstacles, T-cells have been genetically engineered to stably express transgenes using viral transduction, often with vectors from gamma retroviruses or lentiviruses. Molecularly engineered TCRs have certain advantages including the ability to target all cellular proteins and not just cell surface epitopes. Genetically altering the T-cells to express a tumor antigen specific TCR is one way to target a specific tumor antigen (chimeric antigen receptor-T-cells or car-T-cells). TCR engineered T-cell transfer using human TCRs targeting AFP is currently in clinical trial in several institutions, including our Center. Known toxicities to this approach include side effects from lymphodepleting chemotherapy, cytokine release syndrome, and possible autoimmune toxicities. Car-T-cell treatment has been successful to treat various hematologic malignancies including lymphoid leukemia and acute myeloid leukemia.
Adding checkpoint inhibitor to sorafenib therapy is also reasonable. Sorafenib reduces hepatic infiltrated T regs by suppressing TGF-β signal.[42] However, sorafenib has other effects which may inhibit the immune system. For example, sorafenib but not sunitinib, appears to have a detrimental effect on dendritic cell phenotype and can inhibit cytokine secretion, migration ability and T-cell stimulatory capacity, while not affecting the function and phenotype of T-cells.[43] Sorafenib has been shown to inhibit JAK-STAT signal transduction in human immune cells.[44] The immune effects of sorafenib were dose dependent. At pharmacologic doses of sorafenib, the drug decreased T effector cell activation by down regulating CD25 surface expression. At low doses, sorafenib produced T effector cell activation, with significant increase in T effector cell proliferation, IL2 secretion and up regulation of CD25 cell surface expression and could reduce T regulatory cell suppression. Thus, the dose of sorafenib used may be critical for the desired immune effects. Other actions of sorafenib on the immune system include: (1) inhibit NK function; (2) increase MDSC; and (3) upregulate PD-L1 expression. In mice bearing orthotopic HCC, sorafenib upregulated tumor-specific T effector cell function, while the proportion of PD-1 expressing CD8+ T-cells and regulatory T-cells were reduced.[45] In addition, the function of T regulatory cells was inhibited. In another study mouse and human HCC tumor samples expressing low pERK showed intense inflammatory infiltrating cells and significant enrichment of CD8+ that expressed PD-1.[46] Patients with pERK PD-1 positive tumors had worse prognosis than pERK PD-1 negative tumors. PD-1 immunotherapy could complement sorafenib by targeting tumor cells resistant to sorafenib.
Recently, PD-L1 expression in HCC was shown to be significantly associated with markers of tumor aggressiveness including high AFP serum levels, satellite nodules, macrovascular invasion, microvascular invasion, poor histologic differentiation progenitor subtype (cytokeratin 19 expression).[47] High PD-L1 expression in immune cells in the tumor microenvironment also correlated with high serum AFP levels, macrovascular invasion, poor differentiation, high PD-1 expression and lymphoepithelioma subtype.
Targeting glypican-3 (GPC3) immunologically is a novel approach. GPC3 is a member of the glypican family of heparin sulfate proteoglycans on the cell surface. This molecule is overexpressed in 80% of HCC and when present, carries a poor prognosis.[48,49] Not only is it a prognostic factor and important for cell proliferation by stimulating Wnt signaling, but it is a tumor specific and becomes an attractive target as a tumor antigen.[50] Clinical trials have started with both peptide-based vaccine[51] or anti-GPC3 antibodies.[52,53] The phase I trial of peptide-based vaccine showed the treatment was well tolerated and one partial response and several stable disease responses were observed.[51] There was a correlation between overall survival and GPC-3 specific CTL response. Two recent phase I studies of antibody therapy against glypican-3 were reported recently.[52,53] In the first study, a recombinant, humanized monoclonal antibody against GPC3 was performed in advanced HCC.[52] There were no dose-limiting toxicities (DLT) noted. The most common side effects include transfusion reactions (35%), fatigue and pyrexia and diarrhea. Stable disease was seen in several patients. A phase I study of this antibody in Japanese patients with advanced HCC was performed.[53] The most common toxicity was lymphocytopenia, natural killer cell decrease, C-reactive protein increase and pyrexia. Infusion reactions were observed in 62% patients. No DLT or maximum tolerated dose (MTD) was noted. There were no partial or complete responses but stable disease was noted in several patients. A phase I trial of combination of anti GC33 antibody with sorafenib in 40 patients was reported.[54] There were 3 DLTs seen (grade 3 hyponatremia, grade 3 hyponatremia and hypoglycemia and grade 3 ALT increase). One partial response and 6 stable diseases were reported. No MTD was obtained with combining the antibody to GC33 with sorafenib at a dose of 400 mg bid daily. A phase I trial of T-cell redirecting bispecific antibody against glypican-3 is another approach.[55]
Another clinical trial for HCC starting at University of Miami is the study of a bifunctional fusion protein that will combine PD-L-1 antibody with the soluble extracellular domain of TGF-β receptor type II as a TGF-β neutralizing "trap". This compound (developed by Merck) will target two major mechanisms of immunosuppression, PD-1/PD-L-1 axis as described previously and TGF-β. TGF-β is known to have growth inhibitory effects on normal epithelial cells and can act as tumor suppressor in early stage cancer development. Later as the tumor advances, TGF-β loses its ability to suppress cancer and various cancers can actually produce this molecule which acts as a stimulatory molecule for cell growth and division. Then, TGF-β can downregulate the effector function of T cytotoxic cells and natural killer cells, while inducing differentiation of CD4+ T-cells to T reg cells. In mice, the blocking of TGF-β signaling in T-cells or deletion of TGF-β from T-cells resulted in decreased PD-1 expression in tumor-infiltrating CD8+ T-cells.[56] Further studies in mice model of HCC showed that TGF-β increased the PD-L1 expression in dendritic cells which led to T-cell apoptosis and increased number of CD25+, Foxp3+ T regulatory cells.[56,57] Thus, there is a strong rationale to target two pathways of immunosuppression in HCC simultaneously.
In summary, immunotherapy represents a novel approach to treat advanced hepatocellular carcinoma. Immune cells have been found in HCC specimens that appear "fatigued" or suppressed. New checkpoint inhibitors and other agents may awaken these immune cells to attack the tumor. Early results from clinical trials suggest it is feasible to do so and encouraging antitumor responses were been observed. The challenge is to further define what patients may benefit from this approach, how to predict or reduce toxicities, particularly liver toxicity, and how best to combine this therapy with other known modalities of treatment.
Authors’ contributions
Manuscript writing and editing: L.G. Feun
Manuscript review, participating in the phase II trial of Pembrolizumab in HCC, and performing the laboratory correlates: Y.Y. Li, M. Wangpaichitr, C.J. Wu, N. Savaraj
Financial support and sponsorship
None.
Conflicts of interest
There are no conflicts of interest.
Patient consent
There is no patient involved.
Ethics approval
This review paper is waived for ethics approval.
REFERENCES
1. Huz JI, Melis M, Sarpel U. Spontaneous regression of hepatocellular carcinoma is most often associated with tumor hypoxia or a systemic inflammatory response. HPB (Oxford) 2012;14:500-5.
2. Korangy F, Ormandy LA, Bleck JS, Klempnauer J, Wilkens L, Manns MP, Greten TF. Spontaneous tumor-specific humoral and cellular immune responses to NY-ESO-1 in hepatocellular carcinoma. Clin Cancer Res 2004;10:4332-41.
3. Hato T, Goyal L, Greten TF, Duda DG, Zhu AX. Immune checkpoint blockade in hepatocellular carcinoma: current progress and future directions. Hepatology 2014;60:1776-82.
4. Flecken T, Schmidt N, Hild S, Gostick E, Drognitz O, Zeiser R, Schemmer P, Bruns H, Eiermann T, Price DA, Blum HE, Neumann-Haefelin C, Thimme R. Immunodominance and functional alterations of tumor-associated antigen-specific CD8+ T-cell responses in hepatocellular carcinoma. Hepatology 2014;59:1415-26.
5. Ormandy LA, Hillemann T, Wedemeyer H, Manns MP, Greten TF, Korangy F. Increased populations of regulatory T cells in peripheral blood of patients with hepatocellular carcinoma. Cancer Res 2005;65:2457-64.
6. Gao Q, Qiu SJ, Fan J, Zhou J, Wang XY, Xiao YS, Xu Y, Li YW, Tang ZY. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol 2007;25:2586-93.
7. Zerbini A, Pilli M, Penna A, Pelosi G, Schianchi C, Molinari A, Schivazappa S, Zibera C, Fagnoni FF, Ferrari C, Missale G. Radiofrequency thermal ablation of hepatocellular carcinoma liver nodules can activate and enhance tumor-specific T-cell responses. Cancer Res 2006;66:1139-46.
8. Wissniowski TT, Hänsler J, Neureiter D, Frieser M, Schaber S, Esslinger B, Voll R, Strobel D, Hahn EG, Schuppan D. Activation of tumor-specific T lymphocytes by radiofrequency ablation of the VX2 hepatoma in rabbits. Cancer Res 2003;63:6496-500.
9. Arihara F, Mizukoshi E, Kitahara M, Takata Y, Arai K, Yamashita T, Nakamoto Y, Kaneko S. Increase in CD14+HLA-DR -/low myeloid-derived suppressor cells in hepatocellular carcinoma patients and its impact on prognosis. Cancer Immunol Immunother 2013;62:1421-30.
10. Ilkovitch D, Lopez DM. The liver is a site for tumor-induced myeloid-derived suppressor cell accumulation and immuno-suppression. Cancer Res 2009;69:5514-21.
11. Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Krüger C, Manns MP, Greten TF, Korangy F. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology 2008;135:234-43.
12. Greten TF, Ormandy LA, Fikuart A, Hochst B, Henschen S, Horning M, Manns MP, Korangy F. Low-dose cyclophosphamide treatment impairs regulatory T cells and unmasks AFP-specific CD4+ T-cell responses in patients with advanced HCC. J Immunother 2010;33:211-8.
13. Han Y, Chen Z, Yang Y, Jiang Z, Gu Y, Liu Y, Lin C, Pan Z, Yu Y, Jiang M, Zhou W, Cao X. Human CD14+CTLA4+ regulatory dendritic cells suppress T-cell response by cytotoxic T-lymphocyte antigen-4-dependent IL-10 and indoleamine-2-3-dioxgenase production in hepatocellular carcinoma. Hepatology 2014;59:567-79.
14. Han Y, Yang Y, Chen Z, Jiang Z, Gu Y, Liu Y, Xu S, Lin C, Pan Z, Zhou W, Cao X. Human hepatocellular carcinoma-infiltrating CD4+CD69+Foxp3- regulatory T cell suppresses T cell response via membrane-bound TGF-β1. J Mol Med (Berl) 2014;92:539-50.
15. Kurokohchi K, Carrington M, Mann DL, Simonis TB, Alexander-Miller MA, Feinstone SM, Akatsuka T, Berzofsky JA. Expression of HLA class I molecules and the transporter associated with antigen processing in hepatocellular carcinoma. Hepatology 1996;23:1181-8.
16. Matsui M, Machida S, Itani-Yohda T, Akatsuka T. Downregulation of the proteasome subunits, transporter, and antigen presentation in hepatocellular carcinoma, and their restoration by interferon-gamma. J Gastroenterol Hepatol 2002;17:897-907.
17. Unitt E, Rushbrook SM, Marshall A, Davies S, Gibbs P, Morris LS, Coleman N, Alexander GJ. Compromised lymphocytes infiltrate hepatocellular carcinoma: the role of T regulatory cells. Hepatology 2005;41:722-30.
18. Cariani E, Pilli M, Zerbini A, Rota C, Olivani A, Pelosi G, Schianchi C, Soliani P, Campanini N, Silini EM, Trenti T, Ferrari C, Missale G. Immunological and molecular correlates of disease recurrence after liver resection for hepatocellular carcinoma. PLos One 2012:7:es32493.
19. Zhu XD, Zhang JB, Zhuang PY, Zhu HG, Zhang W, Xiong YQ, Wu WZ, Wang L, Tang ZY, Sun HC. High expression of macrophage colony-stimulating factor in peritumoral liver tissue is associated with poor survival after curative resection of hepatocellular carcinoma. J Clin Oncol 2008;26:2707-16.
20. Yeung OW, Lo C, Ling C, Qi X, Geng W, Li CX, Ng KT, Forbes SJ, Guan XY, Poon RT, Fan ST, Man K. Alternatively activated (M2) macrophages promote tumor growth and invasiveness in hepatocellular carcinoma. J Hepatol 2015;62:607-16.
21. Barathan M, Gopal K, Mohamed R, Ellegard R, Saeidi A, Vadivelu J, Ansari AW, Rothan HA, Ram MR, Zandi K, Chang LY, Vignesh R, Che KF, Kamarulzaman A, Velu V, Larsson M, Kamarul T, Shankar EM. Chronic hepatitis C virus infection triggers spontaneous differential expression of biosignatures associated with T cell exhaustion and apoptosis signaling in peripheral blood mononucleocytes. Apoptosis 2015;20:466-80.
22. Li FJ, Zhang Y, Jin GX, Yao L, Wu EQ. Expression of LAG-3 is coincident with the impaired effector function of HBV-specific CD8(+) T cell in HCC patients. Immunol Lett 2013;150:116-22.
23. Cariani E, PilliM, Zerbini A, Rota C, Olivani A, Zanelli P, Zanetti A, Trenti T, Ferrari C, Missale G. HLA and killer immunoglobulin-like receptor gene as outcome predictors of hepatitis C virus-related hepatocellular carcinoma. Clin Can Res 2013;19:5465-73.
24. Guo Q, Lan P, Yu X, Han Q, Zhang J, Tian Z, Zhang C. Immunotherapy for hepatoma using a dual-function vector with both immunostimulatory and Pim-3-silencing effects. Mol Cancer Ther 2014;13:1503-13.
25. Watanabe T, Bertoletti, A, Tanoto TA. PD-1/PD-L1 pathway and T-cell exhaustion in chronic hepatitis virus infection. J Viral Hepat 2010;17:453-8.
26. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006;439:682-7.
27. Yin XY, Lü MD, Liang LJ, Lai JM, Li DM, Kuang M. Systemic chemo-immunotherapy for advanced-stage hepatocellular carcinoma. World J Gastroenterol 2005;11:2526-9.
28. Kaseb AO, Shindoh J, Patt YZ, Roses RE, Zimmitti G, Lozano RD, Hassan MM, Hassabo HM, Curley SA, Aloia TA, Abbruzzese JL, Vauthey JN. Modified cisplatin/interferon α-2b/doxorubicin/5-fluorouracil (PIAF) chemotherapy in patients with no hepatitis or cirrhosis is associated with improved response rate, resectability, and survival of initially unresectablehepatocellular carcinoma. Cancer 2013;119:3334-42.
29. Yeo W, Mok TS, Zee B, Leung TW, Lai PB, Lau WY, Koh J, Mo FK, Yu SC, Chan AT, Hui P, Ma B, Lam KC, Ho WM, Wong HT, Tang A, Johnson PJ. A randomized phase III study of doxorubicin versus cisplatin/interferon alpha-2b/doxorubicin/fluorouracil (PIAF) combination chemotherapy for unresectable hepatocellular carcinoma. J Natl Cancer Inst 2005;97:1532-8.
30. Sangro B, Gomez-Martin C, de la Mata M, I-arrairaegui M, Garralda E, Barrera P, Riezu-Boj JI, Larrea E, Alfaro C, Sarobe P, Lasarte JJ, Pérez-Gracia JL, Melero I, Prieto J. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol 2013;59:81-8.
31. El-Koueiry AB, Melero I, Crocenzi TS, Welling TH. Phase I/II safety and antitumor activity of nivolumab in patients with advanced hepatocellular carcinoma (HCC): CA209-040. In: Proceedings of the 51th Annual Meeting of the American Society of Clinical Oncology; 2015 May 30-June 2; Chicago. ASCO 2015. Abstract nr LBA 101.
32. El-Koueiry AB, Sangro B, Yau T, Crocenzi TS, Welling III TH, Yeo W, Chopra A, Anderson J, dela Cruz C, Lang L, Neely J, Melero I. Phase 1/2 safety and antitumor activity of Nivolumab in patients with advanced hepatocellular carcinoma: interim analysis of the Checkmate-040 dose escalation study. In: Proceedings of the 52th Annual Meeting of the American Society of Clinical Oncology; 2016 June 4-8; Chicago. ASCO 2016. Abstract nr 4012 .
33. El Ansary M, Mogawer S, Elhamind SA, Alwakil S, Aboelkasem F, Sabaawy HE, Abdelhalim O. Immunotherapy by autologous dendritic cell vaccine in patients with advanced HCC. J Cancer Res Clin Oncol 2013;139:39-48.
34. Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M, Cho M, Lim HY, Chung HC, Kim CW, Burke J, Lencioni R, Hickman T, Moon A, Lee YS, Kim MK, Daneshmand M, Dubois K, Longpre L, Ngo M, Rooney C, Bell JC, Rhee BG, Patt R, Hwang TH, Kirn DH. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med 2013;19:329-36.
35. Park BH, Hwang T, Liu TC, Sze DY, Kim JS, Kwon HC, Oh SY, Han SY, Yoon JH, Hong SH, Moon A, Speth K, Park C, Ahn YJ, Daneshmand M, Rhee BG, Pinedo HM, Bell JC, Kirn DH. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol 2008;9:533-42.
36. Breitbach CJ, Burke J, Jonker D, Stephenson J, Haas AR, Chow LQ, Nieva J, Hwang TH, Moon A, Patt R, Pelusio A, Le Boeuf F, Burns J, Evgin L, De Silva N, Cvancic S, Robertson T, Je JE, Lee YS, Parato K, Diallo JS, Fenster A, Daneshmand M, Bell JC, Kirn DH. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature 2011;477:99-102.
37. Nakamoto N, Kaplan DE, Coleclough J, Li Y, Kaminski M, Shaked A, Olthoff K, Gostick E, Price DA, Freeman GJ, Wherry EJ, Chang KM. Functional restoration of HCV-specific CD8 T cells by PD1 blockade is defined by PD1 expression and compartmentalization. Gastroenterology 2008;134:1927-37.
38. Nakamoto N, Cho H, Shaked A, Olthoff K, Valiga ME, Kaminski M, Gostick E, Price DA, Freeman GJ, Wherry EJ, Chang KM. Synergistic reversal of intrahepatic HCV-specific CD8 T cell exhaustion by combined PD-1/CTLA-4 blockade. PLos Pathog 2009;5:e1000313.
39. Liao J, Xiao J, Zhou Y, Liu Z, Wang C. Effect of transcatheter arterial chemoembolization on cellular immune function and regulatory T cells in patients with hepatocellular carcinoma. Mol Med Rep 2015;12:6065-71.
40. Guan H, Wang J, Yang M, Song L, Tong X, Zou YH. Changes in immunological function after treatment with transarterial chemoembolization plus radiofrequency ablation in hepatocellular carcinoma patients. Chin Med J (Engl) 2013;126:3651-5.
41. Duffy AG, Makarova-Rusher OV, Pratt D, Kleiner DE, Fioravanti S, Walker M, Carey S, Figg WD, Steinberg SM, Anderson V, Levy E, Krishnasamy V, Wood BJ, Greten TF. Tremelimumab, a monoclonal antibody against CTLA-4, in combination with subtotal ablation (trans-catheter arterial chemoembolization [TACE], radiofrequency ablation [RFA] or cryoablation) in patients with hepatocellular carcinoma (HCC) and biliary tract carcinoma (BTC). J Clin Oncol 2016;34 suppl 4:abstr 270.
42. Wang Q, Yu T, Yuan Y, Zhuang H, Wang Z, Liu X, Feng M. Sorafenib reduces hepatic infiltrated regulatory T cells in hepatocellular carcinoma patients by suppressing TGF-beta signal. J Surg Oncol 2013;107:422-7.
43. Hipp M, Hilf N, Walter S, Werth D, Brauer KM, Radsak MP, Weinschenk T, Singh-Jasuja H, Brossart P. Sorafenib, but not sunitinib, affects function of dendritic cells and induction of primary immune response. Blood 2008;111:5610-20.
44. Martin del Campo SE, Levine KM, Mundy-Bosse BL, Grignol VP, Fairchild ET, Campbell AR, Trikha P, Mace TA, Paul BK, Jaime-Ramirez AC, Markowitz J, Kondadasula SV, Guenterberg KD, McClory S, Karpa VI, Pan X, Olencki TE, Monk JP, Mortazavi A, Tridandapani S, Lesinski GB, Byrd JC, Caligiuri MA, Shah MH, Carson WE, 3rd. The Raf kinase inhibitor sorafenib inhibits JAK-STAT signal transduction in human immune cells. J Immunol 2015;195:1995-2005.
45. Chen ML, Yan BS, Lu WC, Chen MH, Yu SL, Yang PC, Cheng AL. Sorafenib relieves cell-intrinsic and cell-extrinsic inhibtions of effector T cells in tumor environment to augment antitumor immunity. Int J Cancer 2014;134:319-31.
46. Chen J, Ji T, Zhao J, Li G, Zhang J, Jin R, Liu J, Liu X, Liang X, Huang D, Xie A, Lin H, Cang Y, Cai X. Sorafenib-resistant hepatocellular carcinoma stratified by phosphorylated ERK activates PD-1 immune checkpoint. Oncotarget 2016;7:41274-84.
47. Calderaro J, Rousseau B, Amaddeo G, Mercey M, Charpy C, Costentin C, Luciani A, Zafrani ES, Laurent A, Azoulay D, Lafdil F, Pawlotsky JM. Programmed death ligand 1 expression in hepatocellular carcinoma: relationship with clinical and pathological features. Hepatology 2016;64:2038-46.
48. Capurro M, Wanless IR, Sherman M, Deboer G, Shi W, Miyoshi E, Filmus J. Glypican-3: a novel serum and histochemical marker for hepatocellular carcinoma. Gastroenterology 2003;125:89-97.
49. Filmus J, Capurro M. Glypican-3: a marker and a therapeutic target in hepatocellular carcinoma. FEBS J 2013;280:2471-6.
50. Capurro MI, Xiang YY, Lobe C, Filmus J. Glypican-3 promotes the growth of hepatocellular carcinoma by stimulating canonical Wnt signaling. Cancer Res 2005;65:6245-54.
51. Sawada Y, Yoshikawa T, Nobucka D, Shirakawa H, Kuronuma T, Motomura Y, Mizuno S, Ishii H, Nakachi K, Konishi M, Nakagohri T, Takahashi S, Gotohda N, Takayama T, Yamao K, Uesaka K, Furuse J, Kinoshita T, Nakatsura T. Phase I trial of a glypican-3-derived peptide vaccine for advanced hepatocellular carcinoma: immunologic evidence and potential for improving overall survival. Clin Cancer Res 2012;18:3686-96.
52. Zhu AX, Gold PJ, El-Koueiry AB, Abrams TA, Morikawa H, Ohtomo T, Philip PA. A phase I study of GC33, a recombinant humanized antibody against glypican-3, in patients with advanced hepatocellular carcinoma (HCC). J Clin Oncol 2011;29 suppl:abstr 4085.
53. Okusaka T, Ikeda M, Ohkawa S, Yamamoto S, Suzuki I, Furuse J. A phase I study of GC33 in Japanese patients with advanced hepatocellular carcinoma (HCC). J Clin Oncol 2013;31 suppl 4:abstr 235.
54. Abou-Alfa GK, Yen CJ, Carrasquillo JA, Hsu CH, Gansukh B, Ma J, Hollywood E, Wan PJ, Shao YY, Lin ZZ, Frenette C, O'Neil BH, Schwartz LH, Ohtomo T, Tanaka T, Chen YC, Ukrainskyj S, Saltz L, Lee RM, Cheng AL. Phase I b study of RO5137382/GC33 in combination with sorafenib in patients with advanced hepatocellular carcinoma (HCC) (NCT00976170). J Clin Oncol 2014;32 suppl:abstr 4100.
55. Hashimoto K, Perera A, Ogita Y, Nakamura M, Ishiguro T, Sano Y, Kinoshita Y, Sakurai MK, Frings W, Komatsu SI, Kaneko A, Ueda M, Kishishita S, Gianella-Borradori A. A phase I dose escalation and cohort expansion study of T-cell redirecting bispecific antibody against Glypican 3 in patients with advanced solid tumors. J Clin Oncol 2016;34 suppl:abstr TPS2592.
56. Donkor MK, Sarkar A, Li MO. TGF-β1 produced by activated CD4(+) T cells antagonized T cell surveillance of tumor development. Oncoimmunology 2012;1:162-71.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Feun LG, Li YY, Wangpaichitr M, Wu CJ, Savaraj N. Immunotherapy for hepatocellular carcinoma: the force awakens in HCC?. Hepatoma Res 2017;3:43-51. http://dx.doi.org/10.20517/2394-5079.2016.45
AMA Style
Feun LG, Li YY, Wangpaichitr M, Wu CJ, Savaraj N. Immunotherapy for hepatocellular carcinoma: the force awakens in HCC?. Hepatoma Research. 2017; 3: 43-51. http://dx.doi.org/10.20517/2394-5079.2016.45
Chicago/Turabian Style
Feun, Lynn G., Ying-Ying Li, Medhi Wangpaichitr, Chun-Jing Wu, Niramol Savaraj. 2017. "Immunotherapy for hepatocellular carcinoma: the force awakens in HCC?" Hepatoma Research. 3: 43-51. http://dx.doi.org/10.20517/2394-5079.2016.45
ACS Style
Feun, LG.; Li Y.Y.; Wangpaichitr M.; Wu C.J.; Savaraj N. Immunotherapy for hepatocellular carcinoma: the force awakens in HCC?. Hepatoma. Res. 2017, 3, 43-51. http://dx.doi.org/10.20517/2394-5079.2016.45
About This Article
Special Issue
Copyright

Author Biographies

Data & Comments
Data


 Cite This Article 1 clicks
Cite This Article 1 clicks

 Like This Article 1
likes
Like This Article 1
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.