
Epstein-Barr virus associated secondary hemophagocytic lymphohistiocytosis with an unusual presentation of abdominal compartment syndrome
 0
0Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a cytokine storm syndrome caused by an overactive but ineffective immune reaction. Without prompt diagnosis and treatment, HLH is life-threatening. However, presenting symptoms are often nonspecific, with fatigue and fever being the most common. A high index of suspicion is therefore critical for early diagnosis and timely management. A previously healthy, 65-year-old female who initially presented with fever and abdominal pain developed abdominal compartment syndrome (ACS) requiring decompressive laparotomy on hospital day 6. Intraoperative frozen sections of biopsied liver showed intense portal lymphohistiocytic infiltrates. Epstein-Barr virus DNA copy numbers escalated from 600 copies/ mL after admission to 134,000 copies/mL before death. The diagnostic criteria of HLH-2004 were met. Patient expired on hospital day 12. It is important to raise awareness of ACS being an unusual presentation of HLH. Recent changes in diagnostic criteria tailored to adult HLH cases are reviewed.
Keywords
Introduction
Hemophagocytic lymphohistiocytosis (HLH), as its name implies, is featured by the hallmark finding of hemophagocytosis in addition to uncontrolled lymphohistiocytic proliferation. The exact etiology remains unknown. A widely accepted explanation is cytokine storm due to an overactive but ineffective immune reaction.
HLH is classified into primary and secondary forms according to the World Health Organization classification. Primary HLH is typically seen in children, and caused by mutations inherited in an autosomal recessive pattern. In contrast, adults tend to have the secondary form, which is often triggered by malignancy, infection or autoimmune disorders, with T-cell lymphoma being the most common malignancy and Epstein-Barr virus (EBV) being the most common infection.[1] A genetic predisposition has been recognized in some but not all of the adult cases, even with targeted high-throughput sequencing.[2] The mutations in adult HLH, when present, are less likely to be bi-allelic.[3] From the genetic point of view, adult or secondary HLH cases are intrinsically different.[4] Because HLH-2004 diagnostic guidelines were established for pediatric cases, it has always been a question whether or not HLH-2004 can be readily applied to adult patients.
It is important yet challenging to recognize HLH in a timely manner because HLH can be quickly fatal without prompt diagnosis and treatment, but the presenting symptoms are often nonspecific. We herein present a fulminant fatal case in an elderly female with an unusual presentation of abdominal compartment syndrome (ACS), and review recent advances in diagnosing adult HLH.
Case Report
The patient was a previously healthy, 65-year-old female who presented with fever and chills for 4 days, and mild right upper quadrant abdominal pain for 1 day. Complete blood count (CBC) showed neutropenia (1.4 × 109/L) and thrombocytopenia (72 × 109/L), which progressed to pancytopenia with hemoglobin level of 7.2 g/dL in 3 days. EBV DNA copy numbers by quantitative real-time polymerase chain reaction (PCR) were 600 copies/mL on hospital day 2. Other viral tests were negative, including cytomegalovirus, herpes simplex virus, human immunodeficiency virus, and hepatitis B and C.
Ultrasonography at admission showed marked nonspecific gallbladder wall thickening in the setting of positive Murphy’s sign. Computed tomography (CT) next day suggested severe acute cholecystitis and hepatosplenomegaly, with the liver enlarged from 17.2 cm at admission to 22.3 cm within 21 h, and the spleen from 10.9 cm to 14.2 cm. Other minor findings include prominent portahepatic and periaortic lymph nodes measuring up to 1.0 cm in short axis, pyloric and duodenal wall edema, and the 12.3 cm uterus enlarged by a 9.5 cm fibroid. Subsequent endoscopic retrograde cholangiopancreatography showed gastric ulcers and large circumferential duodenal ulcers. Cholecystostomy was performed. Bacterial and fungal cultures of the biliary drainage were negative.
The patient progressively developed ACS, with abdominal pressures ranging from 15-26 mmHg. An emergent decompressive laparotomy was performed on hospital day 6. Because of worsening hepatic dysfunction and a diffusely enlarged firm liver, a liver biopsy was sent for intraoperative rapid frozen sections. The histologic sections showed large portal lymphohistiocytic infiltrates [Figure 1A], in a background of reactive hepatocytes and periportal karryorrhexis [Figure 1B]. No hemophagocytosis was identified. Hepatic parenchymal cells appeared to be uninvolved, with only mild limiting plate changes.
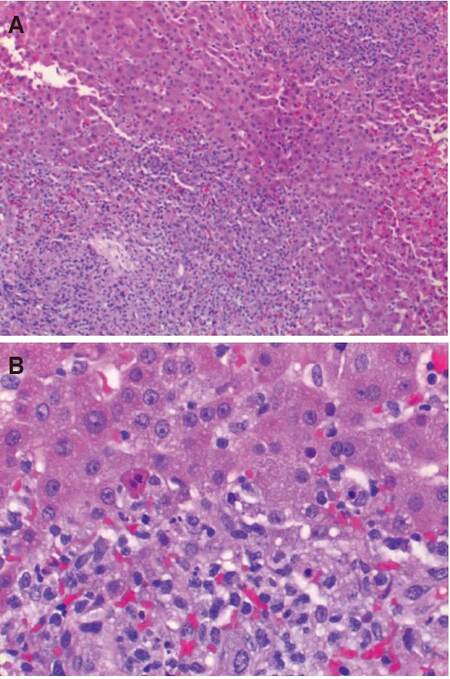
Figure 1. Low-power view (A) shows moderate-sized portal lymphohistiocytic infiltrates (HE, ×100); high-power view (B) shows lymphohistiocytic infiltrates, periportal karryorrhexis and background reactive hepatocytes (HE, ×400)
The portal lymphocytes were predominantly CD3 positive T cells [Figure 2A], with admixed rare CD20 positive B cells in the background [Figure 2B]. The T cells showed an inverted CD4: CD8 ratio of approximately 1:2 [Figure 2C and 2D], partial loss of CD7 [Figure 2E] and CD45 but appropriate expression of CD5 and CD43. Immunostaining for CD68 highlighted Kupffer cells as well as portal aggregates of histiocytes [Figure 2F].
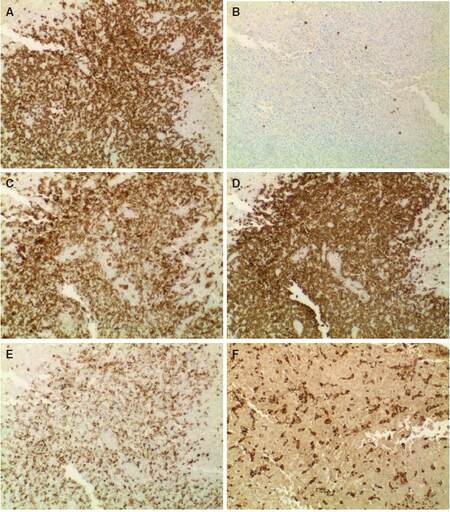
Figure 2. The lymphocytes were predominantly CD3 positive T cells (A), with admixed rare CD20 positive B cells in the background (B); the T cells showed an inverted CD4:CD8 ratio of approximately 1:2 (C, CD4 and D, CD8), partial loss of CD7 (E) and CD45; immunostaining for CD68 highlighted sinusoidal and portal aggregates of histiocytes (F). (IHC, ×100)
The paraffin block was sent to integrated oncology for Epstein-Barr virus-encoded small RNAs (EBER) by in situ hybridization and T-cell receptor (TCR) gene rearrangements analysis by multiplex PCR. The portal lymphohistiocytic infiltrate was negative for EBER, with adequate control. Clonalities were detected with primers targeting the conserved variable and joining regions in the TCR gamma and beta genes including TRG V1-8, 9 + J1/2, TRG alternate V + J1/2 and TRB V + J2.
Other relevant laboratory findings included hyperferritinemia (1,012 ng/mL), hypertriglyceridemia (662 mg/dL), hypofibrinogenemia (nadir < 50 mg/dL), hyperbilirubinemia (1.3 mg/dL), hyponatremia (126 mmol/L), elevated lactate dehydrogenase (LDH 711 U/L), elevated liver enzymes including aspartate aminotransferase (AST 189 U/L), alanine aminotransferase (143 U/L) and alkaline phosphatase (196 U/L), increased prothrombin time (14.7 s, international normalized ratio 1.5) and activated partial thromboplastin time (51.1 s). C-reactive protein (CRP) was within normal range (0.5 mg/dL). EBV DNA copy numbers escalated to 134,000 copies/mL on hospital day 11. A diagnosis of EBV associated HLH was made.
Patient’s clinical condition deteriorated rapidly, despite aggressive attempts at lowering intra-abdominal pressure via decompressive laparotomy, correcting severe acidosis, improving acute liver failure and acute renal failure, supporting acute respiratory failure with pressure control ventilation, and supporting cardiac failure with epinephrine and other vasopressors. A bone marrow biopsy and cytogenetic testing were therefore not performed. The patient expired on hospital day 12.
Discussion
Our patient initially presented with fever and mild right upper quadrant abdominal pain. Imaging at admission showed marked acalculous gallbladder wall thickening, which is most commonly seen in cholecystitis but can be encountered in a variety of conditions unrelated to intrinsic gallbladder disease. In a case series reviewing the diseases associated with gallbladder wall thickening, HLH accounted for 6.0%.[5] Though hepatosplenomegaly noted on CT next day is a common finding in HLH, rapid progression to ACS is unusual. To the best of the authors’ knowledge, this has never been reported in the PubMed listed literature.
With our single case report of ACS associated with HLH, it is difficult to ascertain the underlying mechanism(s) responsible for ACS in this clinical setting. However, the fulminant course of hepatosplenomegaly seen in our case would not allow compensative stretch of the abdominal wall, and is therefore expected to cause rapid elevation of intra-abdominal pressure and consequent ACS. In addition, prominent lymphadenopathy, gallbladder and duodenal wall edema, large uterine fibroid may also contribute more or less to the development of ACS in our patient. We postulate that, ACS is less likely to be encountered in HLH cases of relatively chronic clinical course; at the other extreme of HLH, ACS may not be fully developed or recognized promptly before the patients expire. Raising awareness of ACS as an unusual presentation of HLH would facilitate timely treatment and improve survival rate.
The HLH-2004 diagnostic guidelines proposed by the Histiocyte Society include a molecular diagnosis consistent with HLH or fulfillment of five out of the following eight criteria: fever, splenomegaly, cytopenia affecting two or more lineages (hemoglobin < 9 g/dL, platelets < 100 × 109/L and/or neutrophils < 1.0 × 109/L), hypertriglyceridemia (≥ 265 mg/dL) and/or hypofibrinogenemia (≤ 150 mg/dL), hemophagocytosis in bone marrow, spleen or lymph node, impaired natural killer (NK) cell function, hyperferritinemia (≥ 500 µg/L), and elevated soluble CD25/sIL-2R (≥ 2,400 U/mL).[6,7] Our patient met the HLH-2004 diagnostic criteria based on fever, splenomegaly, pancytopenia, hyperferritinemia, hypertriglyceridemia and hypofibrinogeneima. Other features that have been documented in adult HLH cases but not listed in the HLH-2004 guideline include hyponatremia, hyperbilirubinemia, elevated AST, LDH and CRP.[8] Except elevated CRP, all other ancillary features were observed in the present case.
Because HLH-2004 guidelines were established for primary HLH in pediatric patients, whether or not it can be readily applied to secondary HLH in adults has been questioned. For instance, significantly elevated ferritin is considered specific for HLH in the pediatric population but not in adults.[3,7] To define the diagnostic guidelines for secondary HLH, an international consensus survey was recently conducted.[9] Major revisions made to HLH-2004 for adult HLH are summarized as follows. First of all, unilineage cytopenia is emphasized as an absolutely required criterion, in contrast to bilineage involvement as a dispensable criterion in HLH-2004. Secondly, a known predisposing underlying disease is considered of major importance in diagnosing adult HLH, but not mentioned in HLH-2004. Thirdly, high LDH is included, which is not part of HLH-2004 either. Fourthly, NK cell activity and soluble CD25 are considered of limited use due to the poor availability of these tests. Molecular diagnosis, which is adequate by itself to diagnose primary HLH, is disregarded in the consensus survey for the adult HLH. Lastly, the value of hypertriglyceridemia and hypofibrinogeneima for diagnosing adult HLH fails to reach consensus among experts.
A scoring system, available online at http://saintantoine.aphp.fr/score/, has recently been proposed to estimate an individual's risk of having reactive HLH.[10] Additional differences reflected in this system include hepatomegaly and elevated AST. Degree of fever also contributes to the final score (HScore). An HScore ≥ 169 has been chosen as the cut-off value for confirming the diagnosis of HLH, with a reported sensitivity of 93%, specificity of 86% and correct classification rate of 90%.[10,11] Using the scoring system, our patient has an HScore of 203, and her probability of having HLH is estimated to be 90%.
Though hemophagocytosis documented in bone marrow, spleen or lymph nodes is one of the diagnostic criteria and a hallmark of HLH, it should be noted that hemophagocytosis per se is neither sensitive nor specific for HLH. The reported incidence of hemophagocytosis on bone marrow examination of patients with HLH ranges from 25% to 100%.[1,8,12] On the other hand, hemophagocytosis may be encountered in conditions other than HLH, including sepsis, post transfusion or cytotoxic therapies, and critically ill patients who fall short of diagnostic criteria of HLH.[8,13] Because hemophagocytosis is a systemic event, it can be observed in many other organs, such as liver and brain.[14,15] However, on liver biopsy, hemophagocytic histiocytes are present in variable numbers, and therefore not always seen.[16] A more common but less specific finding is portal, periportal and intrasinusoidal infiltrates of T lymphocytes and histiocytes,[17-19] as seen in the present case. Interestingly, destruction of interlobular bile ducts has been described as an important feature of hepatic involvement by primary HLH,[18] but not in cases of secondary HLH for reasons that are poorly understood.
The trigger of HLH in our patient is most likely EBV infection, as evidenced by the dramatic increase of EBV DNA copies to 1.3 × 105 copies/mL. Teramura et al.[20] reported that the median EBV genome copy number at diagnosis was 3.0 × 103 (range: undetectable to 5.5 × 107) copies/mL in EBV associated HLH, in contrast to 6.6 × 101 (range: undetectable to 1.0 × 103) copies/mL in infectious mononucleosis. EBER negativity may be explained by the relatively early stage of the clinical course when the liver biopsy was performed, or simply technical difficulties encountered in some cases.[21] In addition, the liver biopsy specimen of our patient had previously been frozen for intraoperative consultation. The freeze-and-thaw process may have damaged the EBV RNA to cause a falsely negative test.
Since our patient had no prior history of immunodeficiency, it is intriguing what initiated the defect of her cellular immunity. According to two independent large cohort studies,[8,22] concomitant hematologic malignancy and active infection were found in 2.9-3.7% of adult HLH cases. Given the predominant T-lymphocytic infiltrate on the liver biopsy, a T-cell lymphoma is high in our differential diagnoses. However, the possibility of an underlying T-cell lymphoma is difficult to confirm or exclude in our case due to the fulminant clinical course. Flow cytometry and cytogenetics might have aided in the diagnosis if the patient had been able to tolerate additional biopsies, particularly a bone marrow study. A sIL-2R/ferritin ratio of ≥ 2.0 has been proposed as a useful marker for lymphoma associated HLH.[23,24] Serum beta2 microglobulin level was also reported to be significantly higher in lymphoma associated HLH than benign disease-associated HLH.[25] Unfortunately, serum sIL-2R and beta2 microglobulin were not evaluated in a timely fashion in this case.
Monoclonal TCR gene rearrangements support the diagnosis of T-cell lymphoma in the proper clinical scenario. However, TCR clonality is not uncommon in EBV associated HLH.[26-28] The clonality is likely due to monoclonal proliferation of EBV-infected T cells,[29] and can become polyclonal after eradication of EBV-infected T cells using immunochemotherapy.[28] Though some of these patients may eventually progress to lymphoma should they survive, clonality assay does not help identify patients with underlying lymphoma in the context of EBV associated HLH. Indeed, EBV associated HLH and systemic EBV-positive lymphoproliferative disease may represent a biologic continuum rather than discrete entities.[21] A clear-cut distinction is not always possible.
Despite the progress in the management of HLH, one-month mortality rate is 20-44% for secondary HLH, much worse than primary HLH.[1,8,30] Among all the clinical features and laboratory findings, underlying malignancy, particularly T-cell lymphoma, is most consistently associated with worse prognosis,[1,8,31] followed by older age[11,22,32] and abnormal karyotype.[21,26] Other factors found to correlate with poor prognosis include high EBV viral load (≥ 1,000 copies/mL),[27] organ failure at admission,[11] hyperferritinemia,[31] hypoalbuminemia,[1] male, splenomegaly and thrombocytopenia.[22] TCR clonality does not appear to be of prognostic value.[26,27]
Financial support and sponsorship
None.
Conflicts of interest
There are no conflicts of interest.
Patient consent
A Notice of Privacy Practices signed by the patient for approval of use of patient information for research purposes is available for review upon request.
Ethics approval
Case report is automatically waived and does not require further approval by Institutional Review Board at Loma Linda University Medical Center.
REFERENCES
1. Parikh SA, Kapoor P, Letendre L, Kumar S, Wolanskyj AP. Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis. Mayo Clin Proc 2014;89:484-92.
2. Tesi B, Lagerstedt-Robinson K, Chiang SC, Bdira EB, Abboud M, Belen B, Devecioglu O, Fadoo Z, Yeoh AE, Erichsen HC, Möttönen M, Akar HH, Hästbacka J, Kaya Z, Nunes S, Patiroglu T, Sabel M, Saribeyoglu ET, Tvedt TH, Unal E, Unal S, Unuvar A, Meeths M, Henter JI, Nordenskjöld M, Bryceson YT. Targeted high-throughput sequencing for genetic diagnostics of hemophagocytic lymphohistiocytosis. Genome Med 2015;7:130.
3. Nikiforow S, Berliner N. The unique aspects of presentation and diagnosis of hemophagocytic lymphohistiocytosis in adults. Hematology Am Soc Hematol Educ Program 2015;2015:183-9.
4. Brisse E, Imbrechts M, Put K, Avau A, Mitera T, Berghmans N, Rutgeerts O, Waer M, Ninivaggi M, Kelchtermans H, Boon L, Snoeck R, Wouters CH, Andrei G, Matthys P. Mouse cytomegalovirus infection in BALB/c mice resembles virus-associated secondary hemophagocytic lymphohistiocytosis and shows a pathogenesis distinct from primary hemophagocytic lymphohistiocytosis. J Immunol 2016;196:3124-34.
5. Lee JH, No YE, Lee YJ, Hwang JY, Lee JW, Park JH. Acalculous diffuse gallbladder wall thickening in children. Pediatr Gastroenterol Hepatol Nutr 2014;17:98-103.
6. Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski J, Janka G. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48:124-31.
7. Schram AM, Berliner N. How I treat hemophagocytic lymphohistiocytosis in the adult patient. Blood 2015;125:2908-14.
8. Rivière S, Galicier L, Coppo P, Marzac C, Aumont C, Lambotte O, Fardet L. Reactive hemophagocytic syndrome in adults: a retrospective analysis of 162 patients. Am J Med 2014;127:1118-25.
9. Hejblum G, Lambotte O, Galicier L, Coppo P, Marzac C, Aumont C, Fardet L. A web-based delphi study for eliciting helpful criteria in the positive diagnosis of hemophagocytic syndrome in adult patients. PLoS One 2014;9:e94024.
10. Fardet L, Galicier L, Lambotte O, Marzac C, Aumont C, Chahwan D, Coppo P, Hejblum G. Development and validation of the HScore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol 2014;66:2613-20.
11. Barba T, Maucort-Boulch D, Iwaz J, Bohé J, Ninet J, Hot A, Lega JC, Guérin C, Argaud L, Broussolle C, Jamilloux Y, Richard JC, Sève P. Hemophagocytic lymphohistiocytosis in intensive care unit: a 71-case strobe-compliant retrospective study. Medicine (Baltimore) 2015;94:e2318.
12. Gupta A, Tyrrell P, Valani R, Benseler S, Weitzman S, Abdelhaleem M. The role of the initial bone marrow aspirate in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2008;51:402-4.
13. Strauss R, Neureiter D, Westenburger B, Wehler M, Kirchner T, Hahn EG. Multifactorial risk analysis of bone marrow histiocytic hyperplasia with hemophagocytosis in critically ill medical patients--a postmortem clinicopathologic analysis. Crit Care Med 2004;32:1316-21.
14. Jung SY. Hemophagocytic syndrome diagnosed by liver biopsy in a female patient with systemic lupus erythematosus. J Clin Rheumatol 2013;19:449-51.
15. Ju HY, Hong CR, Kim SJ, Lee JW, Kim H, Kang HJ, Park KD, Shin HY, Chae JH, Phi JH, Cheon JE, Park SH, Ahn HS. Hemophagocytic lymphohistiocytosis diagnosed by brain biopsy. Korean J Pediatr 2015;58:358-61.
16. Ferry JA. Hematolymphoid tumors of the gastrointestinal tract, hepatobiliary tract, and pancreas Odze and Goldblum's Surgical Pathology of the GI Tract, Liver, Biliary Tract, and Pancreas. 3rd ed. Amsterdam, The Netherlands: Elsevier; 2015. p. 879.
17. Favara BE. Histopathology of the liver in histiocytosis syndromes. Pediatr Pathol Lab Med 1996;16:413-33.
18. Kapelari K, Fruehwirth M, Heitger A, Königsrainer A, Margreiter R, Simma B, Offner FA. Loss of intrahepatic bile ducts: an important feature of familial hemophagocytic lymphohistiocytosis. Virchows Arch 2005;446:619-25.
19. Billiau AD, Roskams T, Van Damme-Lombaerts R, Matthys P, Wouters C. Macrophage activation syndrome: characteristic findings on liver biopsy illustrating the key role of activated, IFN-gamma-producing lymphocytes and IL-6- and TNF-alpha-producing macrophages. Blood 2005;105:1648-51.
20. Teramura T, Tabata Y, Yagi T, Morimoto A, Hibi S, Imashuku S. Quantitative analysis of cell-free Epstein-Barr virus genome copy number in patients with EBV-associated hemophagocytic lymphohistiocytosis. Leuk Lymphoma 2002;43:173-9.
21. Smith MC, Cohen DN, Greig B, Yenamandra A, Vnencak-Jones C, Thompson MA, Kim AS. The ambiguous boundary between EBV-related hemophagocytic lymphohistiocytosis and systemic EBV-driven T cell lymphoproliferative disorder. Int J Clin Exp Pathol 2014;7:5738-49.
22. Li J, Wang Q, Zheng W, Ma J, Zhang W, Wang W, Tian X. Hemophagocytic lymphohistiocytosis: clinical analysis of 103 adult patients. Medicine (Baltimore) 2014;93:100-5.
23. Tsuji T, Hirano T, Yamasaki H, Tsuji M, Tsuda H. A high sIL-2R/ferritin ratio is a useful marker for the diagnosis of lymphoma-associated hemophagocytic syndrome. Ann Hematol 2014;93:821-6.
24. Tabata C, Tabata R. Possible prediction of underlying lymphoma by high sIL-2R/ferritin ratio in hemophagocytic syndrome. Ann Hematol 2012;91:63-71.
25. Jiang T, Ding X, Lu W. The prognostic significance of beta2 microglobulin in patients with hemophagocytic lymphohistiocytosis. Dis Markers 2016;2016:1523959.
26. Imashuku S, Hibi S, Tabata Y, Itoh E, Hashida T, Tsunamoto K, Ishimoto K, Kawano F. Outcome of clonal hemophagocytic lymphohistiocytosis: analysis of 32 cases. Leuk Lymphoma 2000;37:577-84.
27. Ahn JS, Rew SY, Shin MG, Kim HR, Yang DH, Cho D, Kim SH, Bae SY, Lee SR, Kim YK, Kim HJ, Lee JJ. Clinical significance of clonality and Epstein-Barr virus infection in adult patients with hemophagocytic lymphohistiocytosis. Am J Hematol 2010;85:719-22.
28. Matsuda K, Nakazawa Y, Yanagisawa R, Honda T, Ishii E, Koike K. Detection of T-cell receptor gene rearrangement in children with Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis using the BIOMED-2 multiplex polymerase chain reaction combined with GeneScan analysis. Clin Chim Acta 2011;412:1554-8.
29. Kawaguchi H, Miyashita T, Herbst H, Niedobitek G, Asada M, Tsuchida M, Hanada R, Kinoshita A, Sakurai M, Kobayashi N. Epstein-Barr virus-infected T lymphocytes in Epstein-Barr virus-associated hemophagocytic syndrome. J Clin Invest 1993;92:1444-50.
30. Trottestam H, Horne A, Aricò M, Egeler RM, Filipovich AH, Gadner H, Imashuku S, Ladisch S, Webb D, Janka G, Henter JI, Histiocyte Society. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood 2011;118:4577-84.
31. Otrock ZK, Eby CS. Clinical characteristics, prognostic factors, and outcomes of adult patients with hemophagocytic lymphohistiocytosis. Am J Hematol 2015;90:220-4.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Lei L, Cobb CJ, Cao J, Raza AS. Epstein-Barr virus associated secondary hemophagocytic lymphohistiocytosis with an unusual presentation of abdominal compartment syndrome. Hepatoma Res 2016;2:287-92. http://dx.doi.org/10.20517/2394-5079.2016.18
AMA Style
Lei L, Cobb CJ, Cao J, Raza AS. Epstein-Barr virus associated secondary hemophagocytic lymphohistiocytosis with an unusual presentation of abdominal compartment syndrome. Hepatoma Research. 2016; 2: 287-92. http://dx.doi.org/10.20517/2394-5079.2016.18
Chicago/Turabian Style
Lei, Li, Camilla J. Cobb, Jeffrey Cao, Anwar S. Raza. 2016. "Epstein-Barr virus associated secondary hemophagocytic lymphohistiocytosis with an unusual presentation of abdominal compartment syndrome" Hepatoma Research. 2: 287-92. http://dx.doi.org/10.20517/2394-5079.2016.18
ACS Style
Lei, L.; Cobb CJ.; Cao J.; Raza AS. Epstein-Barr virus associated secondary hemophagocytic lymphohistiocytosis with an unusual presentation of abdominal compartment syndrome. Hepatoma. Res. 2016, 2, 287-92. http://dx.doi.org/10.20517/2394-5079.2016.18
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 3 clicks
Cite This Article 3 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.