
Hepatitis B virus molecular biology and pathogenesis


Abstract
As obligate intracellular parasites, viruses need a host cell to provide a milieu favorable to viral replication. Consequently, viruses often adopt mechanisms to subvert host cellular signaling processes. While beneficial for the viral replication cycle, virus-induced deregulation of host cellular signaling processes can be detrimental to host cell physiology and can lead to virus-associated pathogenesis, including, for oncogenic viruses, cell transformation and cancer progression. Included among these oncogenic viruses is the hepatitis B virus (HBV). Despite the availability of an HBV vaccine, 350-500 million people worldwide are chronically infected with HBV, and a significant number of these chronically infected individuals will develop hepatocellular carcinoma (HCC). Epidemiological studies indicate that chronic infection with HBV is the leading risk factor for the development of HCC. Globally, HCC is the second highest cause of cancer-associated deaths, underscoring the need for understanding mechanisms that regulate HBV replication and the development of HBV-associated HCC. HBV is the prototype member of the Hepadnaviridae family; members of this family of viruses have a narrow host range and predominately infect hepatocytes in their respective hosts. The extremely small and compact hepadnaviral genome, the unique arrangement of open reading frames, and a replication strategy utilizing reverse transcription of an RNA intermediate to generate the DNA genome are distinguishing features of the Hepadnaviridae. In this review, the authors provide a comprehensive description of HBV biology, summarize the model systems used for studying HBV infections, and highlight potential mechanisms that link a chronic HBV-infection to the development of HCC. For example, the HBV X protein (HBx), a key regulatory HBV protein that is important for HBV replication, is thought to play a cofactor role in the development of HBV-induced HCC, and the authors highlight the functions of HBx that may contribute to the development of HBV-associated HCC.
Keywords
Introduction
The discovery by Baruch Blumberg and colleagues of the Australia antigen, which would later be identified as the hepatitis B virus (HBV) surface antigen, was a major breakthrough towards improving global health.[1,2] For decades prior to Blumberg’s discovery, an unknown virus in blood and plasma samples had been the suspected cause of post-transfusion hepatitis.[3] Recognition that the Australia antigen was a marker of viral hepatitis facilitated the generation of a blood-screening protocol that led to a two- to three-fold reduction in the incidence of post-transfusion hepatitis,[4] with the remaining cases likely caused by hepatitis C virus (HCV), which would not be identified for another 23 years.[5] Retrospective studies also associated the presence of the Australia antigen with chronic liver diseases such as cirrhosis and hepatocellular carcinoma (HCC).[6,7] Finally, the discovery of the Australia antigen also facilitated the eventual development of a vaccine that has greatly reduced the global burden of HBV infection,[8-10] and Baruch Blumberg was awarded the 1976 Nobel Prize in Medicine for his discovery of HBV.
Despite the decades of work between the discovery of HBV and our current understanding of the virus, many aspects of the HBV life cycle and pathogenesis remain unclear. The fact that it is estimated that as much as a third of the world’s population has been infected with HBV at some point, that roughly 5% of the population (350-500 million people) are chronically infected with the virus, and that about 800,000 people die annually from acute or chronic HBV-related consequences underscores the importance of a more complete understanding of HBV biology and pathogenesis.[11,12]
Hepadnaviridae
While the discovery of human HBV, hereafter denoted as HBV, occurred in the 1960s, recent research has shown that hepatitis B viruses have actually been present since the time of the dinosaurs. In fact, the earliest known hepatitis B virus is approximately 82 million years old and was identified from the DNA of infected birds from the Mesozoic period.[13] Although multiple theories of the origins of HBV exist, it appears that the infection of mammals is a much more recent event. The jump into humans, in particular, may have been only about 40,000 years ago.[14] Despite the evolutionary timeline, modern HBV is remarkably similar to these ancient hepatitis B viruses.[13]
The present day Hepadnaviridae family is a group of small, hepatotropic, DNA viruses that are divided into two distinct genera based on their divergent genomic sequences and narrow host range of infection. The avihepadnaviruses, such as duck HBV (DHBV) and heron HBV, infect birds. In contrast, the orthohepadnaviruses infect mammals and include HBV and woodchuck hepatitis virus (WHV), among others. Each member of the Hepadnaviridae family is primarily species specific. For example, the only non-human hosts of HBV are chimpanzee and treeshrew, each of which can be experimentally infected.[15,16] Additionally, a primate virus similar to HBV, called woolly monkey HBV, has been identified in woolly monkeys and designated as the prototype of a new species of hepatitis B-like viruses. A maximum of 40% sequence divergence exists between orthohepadnaviruses, while only 20% sequence divergence exists among avihepadnaviruses; however, little to no homology exists between the two genera. All mammalian HBV encode an X protein, which has been shown to be required for viral replication and has oncogenic properties (discussed below). This X protein is either lacking or highly divergent in avian viruses, and the acquisition of this X protein could have been an essential factor for the evolution of hepadnaviruses from avian into mammalian hosts.[13] Genomic diversity between species of hepadnaviruses is reviewed in detail in the literature.[11,17]
While significant genomic diversity exists between viral species and particularly between the hepadnaviridae genera, all hepadnaviruses share a large number of common features. Among these, all members have an extremely small (3.0-3.3 kb) and compact DNA genome that encodes overlapping open reading frames (ORFs). Additionally, all hepadnaviruses use a genome replication strategy in which the virus replicates its DNA genome by reverse transcription of an RNA intermediate using the reverse-transcriptase activity of the viral polymerase. Hepadnaviruses are also distinguished from nearly all other viruses utilizing reverse transcription for viral replication by a number of unique features, including envelopment of a DNA genome, rather than RNA, and the fact that integration of the hepadnavirus DNA genome into the host-cell genome is not required for viral replication. These features, common to all members of the hepadnavirus family, contributed to the designation of Hepadnaviridae as a distinct family of viruses.[11]
HBV
Studies have identified a minimum of eight HBV genotypes, designated A-H, with genetic differences greater than 8%, but less than 17% between each genotype.[11,17,18] Two additional potential genotypes have been described. Genotype I has genetic divergence around 8% with a strong homology to genotype C,[19] making its classification as a distinct genotype more controversial than that of the more well-accepted genotypes.[20] A potential 10th genotype, genotype J, has also been described recently and is likely the result of recombination of genotype C and gibbon HBV.[18]
There is a distinct distribution of HBV genotypes within specific populations and geographic locations. Similarly, there is an association between genotype and disease severity and outcome. In the United States, where chronic HBV infection is relatively uncommon, each genotype is present, though not at equal levels. Within the United States population, genotypes A and D are most prevalent overall, and the distribution of genotypes can be further divided based on ethnicity.[21,22] For example, genotype C is most common in Asian Americans, which correlates with the prevalence of this genotype in much of Asia. This is significant because genotype C has been associated with a more severe disease and a lower response rate to interferon therapy.[23,24]
HBV genome organization
HBV has a small (3.2 kb), partially double-stranded, relaxed-circular DNA genome that encodes four overlapping ORFs [Figure 1a]. The largest ORF encodes the viral polymerase, which also has reverse transcriptase (RT) activity that generates the first strand of the DNA genome from an RNA intermediate. The second largest ORF encodes the three viral envelope proteins: large (L-), middle (M-), and small (S-) surface antigen (HBsAg). Another ORF encodes precore, also referred to as HBV E antigen (HBeAg), and the core protein, which makes up the viral capsid. Finally, the smallest ORF encodes the HBV X protein (HBx), a small regulatory protein that has been shown to be required for HBV replication both in vitro and in vivo.[25-29] The viral ORFs are encoded in distinct capped and polyadenylated RNAs that can be divided into genomic and subgenomic transcripts. The subgenomic transcripts act only as templates for HBV proteins and consist of the 0.7 kb transcript, which encodes HBx, and the 2.1 kb and 2.4 kb HBsAg transcripts encoding M- and S-HBsAg, and L-HBsAg, respectively. The genomic transcripts act as mRNAs for precore, core, and polymerase. The genomic transcript that encodes both core and polymerase is multifunctional and referred to as pregenomic RNA (pgRNA). The pgRNA is the template for HBV replication and is reverse transcribed to generate the HBV DNA genome. As the viral genome is only 3.2 kb and the pgRNA is 3.5 kb, the pgRNA is a greater than unit length, terminally redundant copy of the viral genome. All HBV RNA transcripts share the same polyadenylation site, and each of the smaller transcripts makes up the 3’ end of each of the larger transcripts. This means that the sequence of the HBx transcript is contained at the 3’ end of all HBV mRNA transcripts, while the largest transcript is the only viral transcript to contain sequence that is not shared with the other transcripts.[11,30,31]
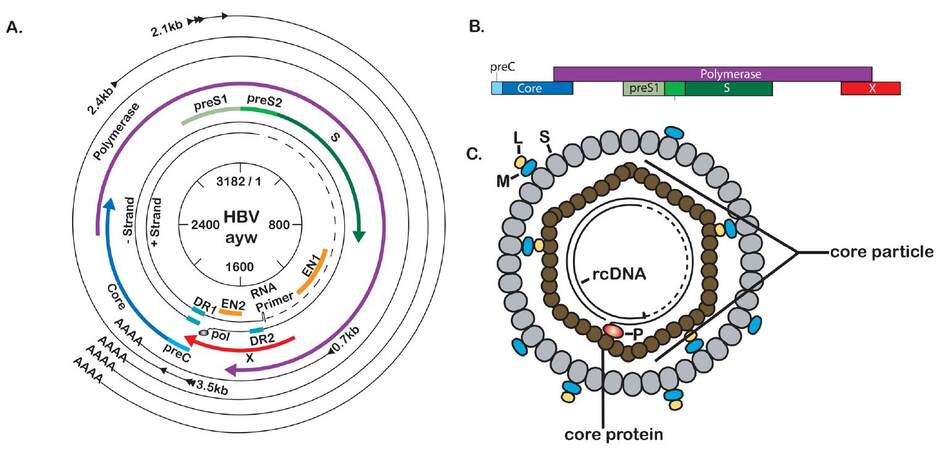
Figure 1. Molecular biology of hepatitis B virus (HBV). (A) Scaled depiction of the HBV (genotype ayw) genome. Internal circle shows genomic position relative to EcoRI site at position 1. Partially double-stranded genome is depicted with attached RNA primer and polymerase protein. Open reading frames (ORFs) are indicated by the thicker, colored lines. The outermost black circles represent the viral transcripts with the shared polyadenylation site; (B) schematic representation of the overlapping nature of the HBV ORFs; (C) the mature HBV virion (Dane particle) consists of two main parts: a nucleocapsid (or core particle) consisting of a partially double-stranded DNA genome bound to polymerase (P) and encapsidated by dimers of core protein, and a viral envelope consisting primarily of S-HBsAg (S), with an intermediate amount of M-HBsAg (M) and lower levels of L-HBsAg (L)
Transcription of HBV RNAs is driven from specific promoter sequences within the viral genome. At least some of the hepatotropic restriction of HBV can be attributed to transcriptional activation by hepatocyte-specific transcription factors. For example, activation of the Enhancer I/HBx promoter is a required first step in viral transcription, as this is believed to enhance transcription from downstream promoters. A number of the transcription factors that have been mapped to the EN1/HBx promoter are liver specific, including hepatocyte nuclear factor (HNF) 1, HNF3, and HNF4. Many of the transcription factor binding sites that have been identified within the 4 promoter regions of HBV are for transcription factors that are activated by HBV proteins, oftentimes HBx, implying a specific cascade of transcription.[32] Transcription factor-mediated regulation of HBV transcription has been reviewed in more detail elsewhere.[11,33]
HBV proteins
The HBV genome encodes seven proteins: HBx, core, polymerase, L-, M-, and S-HBsAg, and precore/HBeAg [Figure 1a]. Of these proteins, HBx is a non-structural regulatory protein, HBeAg is not incorporated into virions and is independently secreted from the cells, the polymerase is responsible for genome replication, and the core and HBsAg proteins form the structural aspects of the virion.[11] Each of these will be discussed in further detail below.
E antigen
HBeAg is the final product of post-translational processing of the translated precore ORF [Figure 1a and b]. As one of the proteins encoded by a genomic transcript, the genomic promoter drives its expression. The HBeAg ORF encodes an endoplasmic reticulum (ER) targeting sequence that co-translationally traffics the peptide to the ER, where the protein is processed to the final 15 kD HBeAg that is secreted from HBV-infected cells.[34]
The function of HBeAg remains incompletely defined. Multiple groups have hypothesized that HBeAg can facilitate HBV immune evasion, and studies with HBc/HBeAg-transgenic (tg) mice crossed with T cell receptor (TCR)-Tg mice expressing receptors for the HBc/HBeAgs specifically suggest that a function of HBeAg is to suppress the immune response to the HBV core protein.[35,36] The secretion of a viral marker that is not present in the HBV infectious virion may help to dampen the neutralizing immune response by diverting this response away from infectious viral particles.[11] From a diagnostic perspective, HBeAg is an important marker of HBV replication, and the levels of serum HBeAg are generally considered to correlate with viral titer. In fact, HBeAg seroconversion is considered an important aspect of the transition to the inactive carrier state of infection (described below).[37]
Surface antigens
HBV encodes three envelope proteins, or surface antigens, that make up the viral envelope: large (L), middle (M), and small (S) surface antigen [Figure 1a and c]. The smallest envelope protein, or S (24 kD), is 226 amino acids (aa) in length and makes up a shared C-terminal region of the two longer envelope proteins. The M protein (31 kD) contains the S sequence with a 55aa N-terminal extension known as preS2. Expression of the M- and S-encoding mRNA is driven by the S promoter, with translation initiating from an upstream (M) or downstream (S) AUG. The L protein (39 kD), the largest of the envelope proteins, contains S, preS2, and an additional 108aa or 119aa (depending on the genotype) N-terminal extension known as preS1. L-HBsAg is encoded by its own mRNA transcript that is controlled by the preS1 promoter.
The envelope proteins are synthesized at the ER, where they attain their transmembrane configuration. Because all three proteins contain an identical C-terminal sequence, the transmembrane topology of this region is the same across all three proteins. Specifically, an N-terminal signal sequence initiates insertion of S into the ER membrane, followed by another signal that pushes the downstream peptide sequence into the ER lumen. The sequence upstream of this signal remains in the cytosol, with the signal domain itself acting as a transmembrane anchor domain. This orientation forms two loops; one loop, between aa 23-79, remains on the cytosolic side, while the other loop, between aa 99-169, remains in the ER lumen.[38] Importantly, the luminal loop contains the major conformational epitope of HBsAg and is glycosylated in nearly half of all S-protein moieties.[39] Once budding of the membrane occurs, these epitopes are on the outer surface of the viral particles. The topology of M is identical to that of S, except for the presence of preS2 within the ER lumen.[40]
A major characteristic of L is that it exists in two conformations that vary in the localization of the N-terminal domain. In the first conformation of L, the preS1 and preS2 domains are present in the cytosol. This conformation of L is essential for binding of capsids and for the assembly of HBV virions. In the second conformation of L, the N-terminus is located in the ER lumen and, as a result, exposed on the surface of viral particles. Thus, this conformation of L plays a role in the infection of hepatocytes. The conformational change is facilitated by interactions of molecular chaperones Hsc70/Hsp40 and BiP with L; however, the exact details of the mechanism underlying this step are not yet understood.[11] The preS1 domain contains the receptor-binding region for HBV,[41,42] thus it needs to be exposed out of the cell. A myristylated peptide containing a portion of the N-terminal preS1 region is sufficient to inhibit infection[41] and is currently being developed as a therapeutic.[43]
The main function of the surface antigen proteins is to form the HBV envelope. Three different forms of viral particles are secreted from an HBV-infected cell as a result of the unequal expression of each of the three surface antigens. S-HBsAg is the highest expressed of the three envelope proteins and makes up the majority of the viral envelope. Intact, infectious HBV virions, called Dane particles, also include M-HBsAg and L-HBsAg. In addition, an HBV-infected cell produces non-infectious subviral particles (SVP) made primarily of S-HBsAg containing varying (but much lower) amounts of M-HBsAg and little to no L-HBsAg. These SVPs can reach a concentration 10,000-fold higher than infectious HBV particles in the serum of an infected individual.[44,45] SVPs are produced in two forms: 25 nm spheres, which are almost exclusively made up of S-HBsAg, and 22 nm filaments, which are made up primarily of S-HBsAg, with some M-HBsAg and potentially small amounts of L-HBsAg. It is currently unknown why HBV produces SVPs in such excess compared to the level of infectious virions, but multiple hypotheses have been proposed. For example, it has been suggested that the excess SVPs act to divert neutralizing antibodies away from infectious particles and that SVPs play a role in inducing the immune tolerance required to sustain a long-term chronic infection. A study of DHBV SVPs showed that the SVP-to-infectious-particle ratio plays a role in determining the efficiency of hepatocyte infection, with SVPs acting to either enhance or inhibit infection based on the ratio of SVP-to-infectious-particles.[46]
Core protein
The 21 kD HBV core protein, or HBcAg, is the organizing framework for the virion [Figure 1c]. When expressed in cells, core mainly exists as soluble dimers, or in T = 3 or T = 4 icosahedral capsids. About 95% of mature nucleocapsids isolated from Dane particles contain T = 4 capsids made up of 120 core dimers, with the remaining 5% being the smaller T = 3 with 90 dimers.[47] Core is translated from the pgRNA and the first 149aa of core form the assembly domain, which is sufficient for in vitro formation of capsids that are indistinguishable from capsids isolated from Dane particles.[48] The remaining 34-36aa makes up the arginine-rich C-terminal domain (CTD); phosphorylation of various aa in the CTD regulates multiple stages of the HBV life cycle.[49-52]
While the best-described role for core protein is assembling the nucleocapsid, the results of recent studies also suggest that the core protein does more than simply act as an inert container for the HBV genome. In fact, core protein binds to HBV covalently closed circular DNA (cccDNA), potentially to regulate spacing of nucleosomes on cccDNA; cccDNA is a nuclear-localized replication intermediate of hepadnaviruses that forms a minichromosome (described in more detail below).[53] In addition, the CTD is required for pgRNA packaging,[54] and core protein also plays an active role in initiating reverse transcription[55-57] and in mature nucleocapsid envelopment.[58] The many potential roles of core protein in the HBV life cycle were recently reviewed, along with a detailed description of the mechanism of capsid assembly.[59]
Polymerase/reverse transcriptase
Not long after the identification of an HBV-like virus in ducks,[60] the DHBV model was used to demonstrate that DHBV genome replication utilizes an RNA intermediate, implying that hepadnaviruses replicate via reverse transcription.[61] While reverse transcription is a mechanism employed by many viruses, hepadnaviruses approach genome replication with a number of unique features. The 90 kD, 838aa polymerase protein of HBV (reverse transcriptase/RT/Pol/P) is made up of 3 functional domains and a variable spacer region. At the N-terminus is the terminal protein (TP) domain, which is important for multiple facets of the initiation of genome replication. This region, despite its important role in P binding to the pgRNA, RNA packaging, and protein-priming,[62-64] is a unique domain that is not shared by any non-hepadnavirus RTs. A variable spacer separates the TP domain from the RT domain, and studies have shown that nearly all aa within the variable spacer region can be mutated without altering P function.[65] In fact, only 3 cysteine residues within the C-terminal end of the spacer region, along with a fourth in the N-terminal side of the RT domain, are thought to be important for RT/pol function.[66]
The RT domain is responsible for genome replication by reverse transcribing the pgRNA to form the (-)-strand of the DNA genome and subsequent use of the (-)-strand as a template for synthesis of the (+)-strand of the DNA. This domain shares significant homology to the RT of other retroviruses.[67] The RT domain is the only current anti-HBV therapeutic target,[12] which is based on the efficacy of nucleoside analogs to inhibit the human immunodeficiency virus (HIV) RT.[68] In fact, portions of HBV RT can be replaced by homologous portions of HIV RT; this can generate an active RT that can function to produce mature HBV virions.[69]
The final domain, P, is the RNase H domain. This domain is responsible for degrading the pgRNA template during synthesis of the (-)-strand of the DNA genome. Coordination of metal ion binding, which is important for RNase H activity, is achieved through 4 conserved carboxylates.[70] Studies of the RNase H domain have also shown that purified recombinant RNase H domain is functional in vitro and that the RNase H domain of P is important for pgRNA packaging.[71] Further information on the HBV RT/pol, including a detailed description of the RT domain active sites and binding motifs, can be found in the literature.[68,72]
X protein
HBx is the only regulatory protein encoded by HBV. It is a 154aa, 17 kD protein that is encoded by the smallest HBV ORF. Various studies have provided considerable evidence that HBx plays an essential role during HBV replication. Specifically, studies have shown that HBx is bound to cccDNA,[73] that HBx is required for transcription from cccDNA,[28,74] and that downstream HBx-mediated effects are required for HBV replication. Importantly, studies of other mammalian hepadnaviruses have also supported the role of their respective X proteins in viral replication. For example, two different studies demonstrated that the WHV X protein is required for WHV replication in vivo,[25,27] although another study did show a low level of viral replication from a WHx-deficient WHV mutant in infected woodchucks.[75] Similarly, viral replication was detected from tg mice expressing either wild-type or an HBx-null HBV mutant; however, when the HBx-null mice were crossed with HBx-tg mice, levels of HBV replication surpassed those seen in wild-type HBV-tg mice.[76] A similar experiment using hydrodynamic tail vein injection of a plasmid encoding either the wild type HBV genome or an HBx-deficient mutant HBV showed a significant decrease in the levels of HBV replication in the absence of HBx, which could be restored through co-injection of the HBx-deficient mutant HBV and a plasmid encoding HBx.[26] This indicates that while HBx may not be absolutely required for HBV replication in these systems, it undoubtedly enhances the levels of replication. Moreover, studies of direct HBV infection of mice with humanized livers demonstrated that only infection with wild type HBV, and not HBx-deficient virus, could result in HBV replication.[29,77] A similar requirement for HBx in HBV replication has been shown in human HepG2 hepatoblastoma cells[78-82] and in primary rat hepatocytes.[83-86] Importantly, the requirement for HBx was also confirmed in primary human hepatocytes directly infected with either wild-type or HBx-deficient HBV.[28]
The lack of a single accepted model for studies of HBV and HBx has created some confusion about the overall consequences of HBx expression for HBV replication and hepatocyte physiology. HBx-related studies have often been performed in transformed or immortalized cell lines and with different levels of HBx expression, leaving the impact of HBx on a normal hepatocyte incompletely understood.[87] While HBx is generally considered to have oncogenic potential, it is yet to be determined if it is directly oncogenic or simply acts as a co-factor in HCC development, as both effects have been demonstrated in different HBx-tg mouse models.[88-91] It is important to recognize that a strongly oncogenic HBx would not be consistent with the biology of HBV-associated HCC, which involves decades of a chronic HBV infection, and it is more likely that HBx plays a cofactor role in the development of HBV-induced liver cancer. The hypothesis that HBx-induced subtle changes in hepatocyte physiology sensitize cells to other oncogenic signals, while facilitating HBV replication, is more consistent with the biology of HBV-associated HCC.[92] Peripheral evidence for the oncogenic potential of HBx comes from the fact that hepadnavirus-associated HCC seems to be restricted to mammalian hepadnaviruses, which each express a form of the X protein. Avian hepadnaviruses, which do develop a chronic infection but do not cause HCC, either do not express an X protein or express a highly divergent form.[78,93]
HBx is a multifunctional protein that can modulate many hepatocyte signaling cascades and factors that have also been linked to mechanisms that underlie cellular transformation. For example, HBx can modulate calcium,[84,85] apoptosis,[83,86] and proliferation signals, among other pathways, and can activate numerous transcription factors, including activator proteins 1[94] and 2[95] (AP-1 and AP-2), nuclear factor of activated T cells (NFAT),[96] and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB).[97-99] HBx can also regulate cellular signaling factors, such as Wnt/β-catenin,[100] p53,[101] and Akt,[86,102] that have been implicated in HCC. Recently, modulation of miRNA expression has also been included in the functions of HBx. It is possible that the many functions attributed to HBx could actually be the result of a few fundamental upstream HBx functions that can affect multiple downstream cellular signal-transduction pathways in a context-dependent manner. Interestingly, while HBV replication in established HBV-associated HCCs is typically absent, a number of groups have shown that these tumors can still express HBx from fragments of the HBV genome that have integrated into the host genome. The presence of HBx in these cells could mean that HBx might be active in these HCC cells, even in the absence of replicating HBV, and potentially contribute to HCC development or maintenance.
HBV life cycle
Studies have shown that the species specificity and hepatotropic nature of HBV are due to at least two different layers of cellular factors. The first is the hepatocyte-specific expression of the recently described HBV receptor, human sodium taurocholate cotransporting peptide (hNTCP/SLC10A1) [Figure 2]. hNTCP is only expressed on human hepatocytes, and mouse NTCP cannot be bound by HBV, which correlates with the inability of HBV to directly infect mouse hepatocytes.[42] The second level of cell-specificity of an HBV infection is controlled by hepatocyte-specific transcription factors such as HNF1α and HNF4α; these control post-entry, downstream stages of the HBV life cycle. Evidence for the additional role of intracellular factors for controlling the cell-specificity of an HBV infection comes from the observation that humanized-mouse NTCP, in which the binding residues from mouse NTCP are replaced by hNTCP, allows binding of HBV to the receptor but does not result in a productive HBV infection when expressed in mouse cells.[103] Studies using hepatitis D virus (HDV), which is a satellite virus requiring HBV envelope proteins for entry into a cell, demonstrated that the 75 aa at the N-terminal portion of the PreS1 domain of L-HBsAg are required residues responsible for binding to the viral receptor.[104] In addition, it was shown that N-myristylation of the PreS1 domain is required for infectivity, but not HBV virion assembly.[105] In fact, a myristylated peptide consisting of only the first 47 aa of the preS1 domain is able to bind to hNTCP and inhibit the binding of HBV.[41] Additional studies have suggested a role for heparin sulfate proteoglycans in the initial stages of HBV binding to hepatocytes,[106] including the recent identification, using an RNAi-based screen in Huh7 cells stably expressing hNTCP, of glypican 5 as an HBV and HDV entry factor.[107]
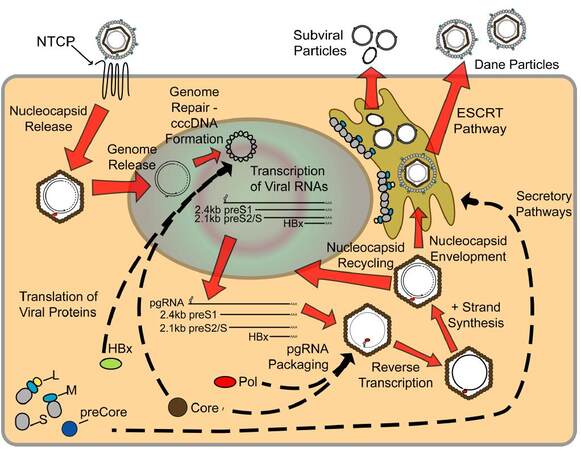
Figure 2. Life cycle of hepatitis B virus (HBV). Mature HBV virions enter hepatocytes through the sodium taurocholate cotransporting polypeptide receptor on the cell membrane. After release from the viral envelope, the nucleocapsid is then transported to the nucleus where the genome is repaired to form covalently-closed circular DNA (cccDNA). Using cccDNA as the template, viral RNAs are transcribed and exported into the cytoplasm where they are translated to form the viral proteins. Additionally, pregenomic RNA (pgRNA) is packaged by core protein, along with the polymerase protein, and the viral genome is replicated through reverse transcription of the pgRNA to form the - strand, followed by partial synthesis of the + strand. Mature nucleocapsids can then either be recycled back to the nucleus to maintain a pool of cccDNA, or enveloped and secreted through the ESCRT pathway. See text for a more detailed description of viral life cycle
Although amino acid sequences of both preS1 and hNTCP that affect binding of HBV to hNTCP are known, the lack of an effective model system that mimics a robust natural infection has hampered a complete understanding of aspects of the HBV life cycle immediately following receptor-binding. The observation that preS1 binds to clathrin heavy chain and the adapter protein AP-2 in immortalized primary human hepatocytes, and that knockdown of these proteins inhibits infection, suggests that the HBV-hNTCP complex may enter the cell through clathrin-mediated endocytosis.[108] Once in the cell, the HBV DNA is delivered into the nucleus by mechanisms that remain unclear. One potential mechanism is the active transport of the nucleocapsid through nuclear pores.[109] Another potential mechanism involves CTD phosphorylation of the core protein, which is thought to expose nuclear localization signals,[49] leading to nucleocapsid disintegration at the nuclear pore and transfer of the polymerase-bound, relaxed circular (rc) form of the HBV DNA into the nucleus.[110,111] The single-stranded gaps in the rcDNA are repaired either through (+)-strand extension by the HBV polymerase or through repair activity of host proteins, and cccDNA is formed as a nucleosome-bound minichromosome in the nucleus. The observations that some HBV-tg mice do not produce cccDNA,[112] and that nucleoside analogues that inhibit the RT function of polymerase do not prevent cccDNA formation,[113] suggest that the production of cccDNA likely involves specific host factors. In addition to studies suggesting a role for cellular histones in cccDNA formation, evidence also exists showing that cccDNA is bound to both core protein[53] and HBx[73] and that this influences the structural arrangement of the cccDNA episome and the epigenetic regulation of cccDNA. Although multiple studies have suggested that HBx is not required for cccDNA formation, transcription of viral RNA from cccDNA is lost in the absence of HBx,[28,114] and HBx has been suggested to regulate levels of cccDNA histone acetylation and methylation.[115] Host RNA polymerase II uses cell-specific transcription factors and cccDNA, which serves as the template for all viral transcripts, to produce 5’-capped and 3’-polyadenylated RNA transcripts. Translation of the viral transcripts occurs in the cytoplasm following nuclear export.
While a portion of the pgRNA is translated, forming the pool of core and polymerase proteins, pgRNA also serves as the template for reverse transcription [Figure 2]. This requires encapsidation of pgRNA by 120 dimers of core protein to form the nucleocapsid. This occurs through a complex cascade of events involving multiple viral and host proteins. Specifically, the 5’ end of the pgRNA contains an encapsidation signal, termed ε, which is recognized and bound by polymerase. Studies have also shown that the 5’ cap structure is required for packaging of the pgRNA;[116] however, polyadenylation is not required.[117] In addition, interaction of pgRNA-bound polymerase with the 5’ cap and host eIF4E leads to encapsidation of this entire RNP complex,[118] resulting in cellular eIF4E within the viral nucleocapsid. Cellular heat shock proteins have also been suggested to play a similar role in stabilizing the binding of polymerase to ε.[119]
Once packaged, reverse transcription is initiated through priming by the polymerase from a specific tyrosine residue within the N-terminal, TP domain of the polymerase[63,64][Figure 1a]. A bulge region within ε supplies the template for the first 3-4nt of the (-)-DNA strand before translocation to a matching acceptor motif in the 3’ direct repeat 1* (DR1*).[120] This strand is then extended until completion, resulting in a unit length (-)-DNA strand copy of the pgRNA that contains an additional 10nt terminal redundancy (r). The majority of pgRNA is degraded during DNA synthesis by the RNase H activity of polymerase, with the remaining bases serving as the 5’ primer for synthesis of the (+)-strand.[121] Direct extension of this primer from its 5’ position results in a double-stranded linear form of the genome that is replication incompetent.[122] This double-stranded linear form does, however, seem to play a role as the main form of HBV DNA that can be integrated into the host genome.[123] Instead of direct extension of the RNA primer from its 5’ location, successful rcDNA formation can occur only after the RNA primer is translocated to the 3’ DR2 sequence. Once on DR2, the RNA primer is extended towards the 5’ end of the (-)-DNA strand. Because r on the other end has the same sequence, exchange of the two ends allows (+)-strand synthesis to continue. As with the previous translocations, additional cis-acting elements are likely playing a role in long-range base-pairing, which allows the close juxtaposition of these donor and acceptor sites that can otherwise be separated by kilobases of sequence.[124,125] In addition, recent evidence has suggested a role for core protein in regulating DNA synthesis, as mutations in core protein inhibit the synthesis of the (+)-strand of DNA.[126] The complex process of reverse transcription has been reviewed in more detail elsewhere.[30,31,68]
Replication occurs in core particles in the cytosol of an HBV-infected hepatocyte, and the final product of DNA synthesis is the encapsidated, partially double-stranded rcDNA with the polymerase still bound to the 5’ end of the (-)-DNA strand [Figure 2]. This nucleocapsid can then proceed in one of two directions. The first is shuttling of the nucleocapsid back to the nucleus to amplify and maintain a stable pool of cccDNA.[127,128] The levels of envelope proteins influence this recycling, with decreased amounts of HBsAg promoting shuttling of the nucleocapsid to the nucleus.[129] In particular, levels of L-HBsAg directly influence shuttling back to the nucleus,[130] and these findings correlate well with the early establishment of a cccDNA pool, followed by identification of secreted infectious HBV.[127] The result is a pool of cccDNA that contains a fluctuating number of copies (typically less than 10) of cccDNA per cell,[131-133] which can be maintained in the cell for years. Additionally, it has been suggested the half-life of a single cccDNA molecule is between 33 and 57 days,[132,134] underscoring the role of cccDNA in maintaining HBV persistence.
The second potential HBV nucleocapsid-associated process is envelopment by HBV envelope glycoproteins residing in the ER membrane [Figure 2]. Interestingly, mechanisms exist that may limit envelopment of capsids containing immature HBV genome; however, these mechanisms remain incompletely understood. For example, it has been suggested that only mature rcDNA-containing nucleocapsids are enveloped, while ssDNA or RNA containing nucleocapsids are not secreted from the cell.[135] Studies utilizing an RNase H-deficient polymerase, which renders the virus unable to initiate (+)-DNA strand synthesis, have suggested that only completion of the (-)-DNA strand is required for envelopment,[136] and specific mutations in core protein can allow envelopment of immature nucleocapsids.[137] The mechanisms associated with this selectivity are unknown, although the phosphorylation state of core protein, likely influenced by the nucleic acid species inside the capsid, could be playing a role. Specifically, studies have shown that core protein isolated from DNA-containing capsids is dephosphorylated (after the prior phosphorylation required for pgRNA packaging and reverse transcription) in a specific C-terminal region, while immature nucleocapsids remain phosphorylated at at least 6 different sites.[50] The overall secretion of infectious HBV Dane particles has been hypothesized to be as little as 1-10 virions per cell per day,[138] which, because of the large number of cells in the liver, can account for high in vivo HBV titers, but can hinder in vitro research requiring isolation of large amounts of infectious virus. Secretion of Dane particles was originally thought to follow the same secretory pathway as the much more abundant SVP, with the envelope proteins residing within the ER-golgi intermediate compartment from where they could bind the DNA-containing capsid, enter the lumen, and be secreted from the cell. Recent evidence has suggested, however, that mature HBV virions are secreted from the cell using a pathway that is dependent on proteins involved in the endosomal sorting complex required for transport (ESCRT) pathway, which forms multivesicular bodies.[139] One characteristic that needs to be considered regardless of the pathway of HBV secretion is the seemingly contradictory conformations of L-HBsAg, with the domains required for both interaction with the nucleocapsid and hNTCP being needed on opposite sides of the membrane. This is addressed by the fact that nearly half of L-HBsAg changes transmembrane conformation after translation, to expose the preS domains within the ER lumen.[140]
Model systems used in the study of HBV
Each member of the hepadnavirus family has a narrow host range that is thought to be defined primarily by the interaction between the virus and a specific cell-surface receptor that is present on host hepatocytes.[11,90] Available cell culture systems for studying the life cycle of the Hepadnaviridae are limited. Typically, members of a hepadnavirus family can only directly infect hepatocytes within the liver of their respective avian or mammalian hosts or cultured, well-differentiated primary hepatocytes that are derived from these hosts. This has hampered the capabilities of researchers to study a natural HBV infection. An overview of the in vivo model systems that exist for studying HBV biology is provided below.
Due to the limited host range of HBV, few suitable animal models exist for studying an in vivo HBV infection. Closely related viruses, such as DHBV[60] and WHV,[141] have been used in their respective host animals as surrogate models for understanding overall hepadnavirus biology. These studies have been instrumental in establishing our understanding of the viral life cycle, including the identification of DNA replication through an RNA intermediate,[61] the establishment of a pool of cccDNA as a mechanism for maintaining chronic infection,[142,143] and the course of both acute[144-146] and chronic[147-149] infection.
The treeshrew, Tupaia belangeri, is a small animal model and is one of the very few animals that can be experimentally infected with HBV.[150] Genomic analysis has placed the treeshrew phylogenetically between humans and rodents,[151,152] and this similarity to primates has spurred its use as a model for a broad range of studies, including as a model for viral hepatitis.[16,153] Specifically, Tupaia belangeri has been used as a model to study the immediate effects of HBV infection on gene expression in the liver[16] and to identify genes potentially contributing to the development of HBV-associated HCC.[154] In fact, freshly isolated primary treeshrew hepatocytes were recently used in multiple studies in which hNTCP was identified as the HBV receptor.[42,155] Recent studies also suggest that neonatal exposure of treeshews to HBV can lead to a disease progression similar to what is seen in humans, with development of a chronic infection leading to the eventual development of HCC.[156] Unfortunately, a relatively low HBV infection efficiency and lack of genetically uniform tree shrew strains has limited their use.[157]
The chimpanzee is the only non-human primate model for HBV infection and, along with the tree shrew, represents one of the only animal models that can be directly infected with HBV. HBV can establish both acute[158] and chronic infections[159] in chimpanzees, and this model has been used most often for modeling the immune response to HBV and the interaction between the virus and host.[160-164] Studies in chimpanzees have helped to establish the relationship between the innate and adaptive immune response to HBV infection, demonstrating minimal early activation of innate immune mediators[160] and a reliance on CD8+ T cells for viral clearance through interferon γ- and tumor necrosis factor α-dependent mechanisms,[161] in agreement with previous work in HBV-tg mice.[165,166] Another important use of the chimpanzee model has been as a surrogate model for preclinical drug and vaccine testing.[167-170] The ethical issues and high costs associated with non-human primate use, however, have limited the use of this model and the recent reevaluation in the United States (one of only two countries to allow chimpanzee research) of the need for chimpanzees in preclinical research[171] will likely diminish their future use even further.
A number of mouse models exist for the study of HBV, and have been reviewed more extensively in the literature.[157,172] Typically these models can be separated into two categories: HBV-/HBx-tg mice, which constitutively express HBV or HBx, respectively, and mice that are delivered the HBV genome or an HBx-expressing plasmid by hydrodynamic tail-vein injection. Although mouse hepatocytes cannot be directly infected with HBV, the use of tail-vein-delivered DNA or HBV-tg mice allows studies of the impact of HBV replication on hepatocyte physiology; HBx-tg mice similarly aid in the study of HBx-mediated effects. While these mouse models are valuable tools, they do have their drawbacks. For example, there is no inflammatory response against HBV in an HBV-tg mouse, which could alter the cellular pathways activated in these models compared to a natural HBV infection. Additionally, because some HBV-tg mice do not produce HBV cccDNA, there is some concern over whether this system accurately mimics all aspect of HBV replication in humans.[112] Despite this, mouse models have been instrumental in determining a number of important aspects of the HBV life cycle, including the requirement for HBx in HBV replication[26] and the oncogenic potential of HBV[173] and HBx.[89,91]
More recently, two additional mouse models have been described that may greatly enhance our understanding of the HBV life cycle and HBV-associated disease. The first of these systems is the humanized mouse model, in which the majority of the mouse liver is repopulated with either primary human hepatocytes or human induced pluripotent stem cells. The use of these animals represents a significant advancement, as they support direct infection with HBV and can develop a chronic HBV infection,[174,175] thereby allowing studies of the impact of an HBV infection on the liver in a cellular context more similar to that seen in the human liver. Some of these chimeric mouse models include both a humanized liver and humanized immune cells, offering the unique opportunity to study the human immune response in a small animal model. Although different techniques for the development of the humanized mouse model exist, a number of groups have adapted their use for the study of HBV. These studies cover a broad range of aspects of HBV biology, including studies of the HBV-mediated immune response,[175,176] investigation of potential HBV therapeutics,[177,178] and aspects of the HBV life cycle such as particle formation,[179] receptor binding,[180] and cccDNA regulation.[181]
Another recently described mouse model expresses hNTCP to allow receptor binding by HBV. Currently these mice are limited in their utility for studying HBV, as multiple groups have shown that while HBV can bind to hNTCP expressed in mouse cells, the HBV life cycle does not appear to proceed beyond receptor binding.[103,182,183] Conversely, HDV utilizes HBV envelope proteins for its envelopment, and hNTCP-expressing mice have been used for the study of HDV infection.[184] Further work with hNTCP-expressing systems may help to determine species-specificity factors that could ultimately lead to the development of an hNTCP-expressing mouse model useful for the study of HBV infection. Together with the humanized-liver model, these mouse models could greatly contribute to our understanding of the early stages of an HBV infection, including entry, HBV genome transport to the nucleus, and genome repair.
The paucity of in vivo models for studying direct HBV infections, and the limited availability of cultured primary human hepatocytes, has lead many researchers to study HBV replication and the activities of HBV-encoded proteins in immortalized or transformed liver cell lines and in cultured primary hepatocytes derived from small-animal models such as rats or mice.[11,92] Use of these systems necessitates bypassing the initial receptor-mediated infection of the cell by direct transfection of the HBV DNA genome. Although primary hepatocytes derived from small-animal models, namely rat and mouse, cannot be directly infected with HBV, they can support HBV replication and serve as a surrogate model system for studying the effects of HBV replication and HBV proteins on cellular physiology.[86,185,186] Here we provide a summary of the available cell culture model systems that are used to study HBV biology, each one possessing its own benefits and limitations.
Most hepatocyte cell lines that are used in HBV-related studies are tumor-derived and thus are transformed. Since cellular signaling pathways are significantly altered in cancer, cell lines derived from tumors do not recapitulate the physiology of normal hepatocytes. While the results obtained by using transformed cell lines may be valid in a specific cellular context, caution should be exercised in the interpretation of such results because they may not necessarily represent the effects of HBV on cellular physiology in normal, untransformed hepatocytes, the authentic site of an HBV infection. In addition to tumor-derived cell lines, some cell lines have been specifically derived from HBV-positive tumors. Examples of cell lines isolated from HBV-positive tumors include the PLC/PRF/5 cell line and the Hep3B cell line, which are human HCC-derived cell lines containing multiple copies of HBV DNA integrated into the host DNA. While the PLC/PRF/5[187] and the Hep3B[188] cells are active in HBsAg production, they do not produce HBV virions and display no indicators of HBV replication,[187,189-192] so results of studies using these cell lines require careful interpretation.
In an attempt to establish a system to study the biology of HBV, and specifically HBV replication, the results of numerous studies demonstrated that HBV DNA could be transfected into many different cell lines, including the hepatoblastoma-derived cell line HepG2[15,193] and the hepatoma-derived cell line Huh7[188,194,195] and that HBV could replicate efficiently in these cells. Consequently, HepG2 and Huh7 cells expressing HBV DNA are frequently used in the study of HBV biology. While HepG2 cells and Huh7 cells support HBV replication, similar to nearly all existing human liver cell lines, they cannot be directly infected with HBV, partly due to the low expression levels of hNTCP, the functional cellular receptor for HBV.[42] In order to analyze differences between cells with and without replicating HBV, HepG2.2.15 cells have sometimes been compared to HepG2 cells. HepG2.215 cells were originally derived from HepG2 cells and stably express HBV from two integrated head-to-tail dimers of the HBV genome.[196] Results obtained by comparing HepG2.215 cells to the parental HepG2 cells, however, need to be interpreted with caution; because of the continuous passaging of HepG2.215 cells since their development in 1987, dissimilarities beyond the expression of HBV may exist between HepG2.2.15 cells and the parental HepG2 cells. Due to these dissimilarities, phenotypic differences that are observed between HepG2.215 and HepG2 cells might not be an exclusive consequence of replicating HBV.
Together, the use of exogenously delivered HBV DNA into established cell lines, such as HepG2 and Huh7, and the use of cell lines stably expressing HBV, such as the HepG2-derived cell lines HepG2.2.15 and HepAD38,[196,197] make up the majority of the studies that have been conducted to understand HBV biology. While these cell culture models have proven extremely valuable to study HBV DNA replication, viral assembly, and virion secretion, they have limitations that prevent them from recapitulating all the aspects of an authentic human HBV infection.[198]
Some recent developments have lead to increased optimism for the development of an effective in vitro system to study the complete HBV life cycle. For example, fusion of primary human hepatocytes with hypoxanthine-guanine phosphoribosyltransferase null [HGPRT(-)] HepG2 cells led to the establishment of the immortal cell line, HepCHLine-4/-7, that may provide a model system for HBV infection. This cell line supports HBV replication and is susceptible to HBV infection when incubated with serum from HBV-positive patients.[199,200] However, an uncertain genetic stability during maintenance hampers the use of this system.[198] In addition, the HepaRG cell line, a human hepatoma cell line, can also be directly infected with HBV and supports HBV replication. While this cell line is often used in studies of HBV infection, its use is limited by a low HBV infection efficiency of only about 10-20% and the need to induce differentiation prior to infection, which involves the maintenance of cells in 2% DMSO for 2 weeks before the induction of differentiation.[198,201]
The recent discovery of hNTCP as the functional HBV receptor has important implications for basic HBV research and antiviral development. In particular, identification of hNTCP has opened new avenues for the establishment of novel cell culture model systems that can be utilized to understand the effects of natural HBV infection. HepG2 cells expressing hNTCP (HepG2-hNTCP), theoretically, provide a convenient in vitro system for HBV infection. The exogenous expression of hNTCP in HepG2 cells does render them susceptible to HBV infection; however, low levels of infection, typically around 10%, and a requirement for large viral inoculums limit their use. Infection-based studies are hampered even further by the low levels of virus released by HBV-infected cells, believed to be around 1 virion per day, making it difficult to produce the large quantities of virus needed for these types of studies. Despite these issues, HepG2-hNTCP cells provide a valuable in vitro model system for elucidating the effects of natural HBV infection, investigating the complete HBV life cycle including the early steps of an HBV infection, and identifying novel therapeutic options.[42,103,172,198,202-204]
As the natural target of an HBV infection, primary human hepatocytes would be the ideal in vitro system for studies of HBV. Unfortunately, cultured primary human hepatocytes lose susceptibility to HBV infection within days of isolation and culture, potentially because hNTCP expression rapidly decreases over time in culture.[42,198,205] Interestingly, Rice and colleagues recently reported that induced pluripotent stem cell-derived hepatocytes(iPSC-derived iHeps) can support HBV infection, opening potential new avenues to study HBV biology and virus-host interaction and to test antiviral candidates. However, a long induction process involving differentiation of the iPSCs is required prior to HBV infection, and viral markers of infection can only be detected more than a week after inoculation.[198,205,206]
Although studies in immortalized or transformed cells have served as powerful models for studying various aspects of HBV replication and the functions of HBV-encoded proteins, these studies have also demonstrated that HBV-mediated activities, particularly those associated with HBV proteins such as HBx, may vary in different cellular contexts.[92,207] Alternatively, studies in cultured primary hepatocytes have begun to clarify HBV replication strategies and the function of HBV proteins in a more relevant context.[92] Recently, cultured primary rat hepatocytes have been used to study HBV replication and functions of the HBx protein;[83-86,208] HBx activities in cultured primary rat hepatocytes were similar to HBx activities in cultured primary human hepatocytes, supporting the use of cultured primary rat hepatocytes as a model system for studying the impact of HBV on hepatocyte physiology.[86,185,186]
HBV natural history
HBV infection can lead to high viral titers in the blood of HBV-infected individuals, with levels of HBV virions reaching as high as 1010 particles/mL.[209] Because of these high titers of HBV in blood, the main mechanism for the transmission of the virus is through the blood. In particular, exposure during childbirth from an HBV-infected mother is the leading global cause of HBV infections, with the potential of vertical transmission being as high as 90% in some parts of Asia. Additional routes of exposure to bodily fluids from infected individuals, such as sexual contact or sharing of needles, are also common routes of transmission.[11]
The natural history of HBV has been divided into two types of infection [Figure 3]. For about 90-95% of HBV infections in adults, the result is “acute hepatitis” where the infected individual resolves infection to the point of undetectable viral DNA and the presence of antibodies against HBsAg. Symptomatic HBV-infected individuals present with inflammation of the liver, which is known as hepatitis, and associated nausea, jaundice, abdominal pain, and vomiting. For many cases of HBV infection, the infected person is asymptomatic, and acute infections are generally cleared within 6 months. In models of acute infection in WHV-infected woodchucks and HBV-infected chimpanzees, the first several weeks of infection are typically characterized by minimal innate[160,210] or adaptive[211] immune activation, with viral spread throughout the entire hepatocyte population.[145,211] Eventually, the activation of an effective antiviral response, including activation of cytotoxic T lymphocytes (CTLs), results in inflammation in the liver and killing of the majority of HBV-infected hepatocytes over the length of a few weeks. Interestingly, studies of integrated WHV DNA in woodchucks treated with clevudine, a viral polymerase inhibitor, demonstrated that repopulation of the liver seems to occur from the population of infected hepatocytes and not from a smaller population of uninfected hepatocytes.[212] Clearance appears to be mostly mediated by antiviral cytokines, with CTLs directly killing HBV-infected hepatocytes once the viral load has dropped below specific levels.[213,214]
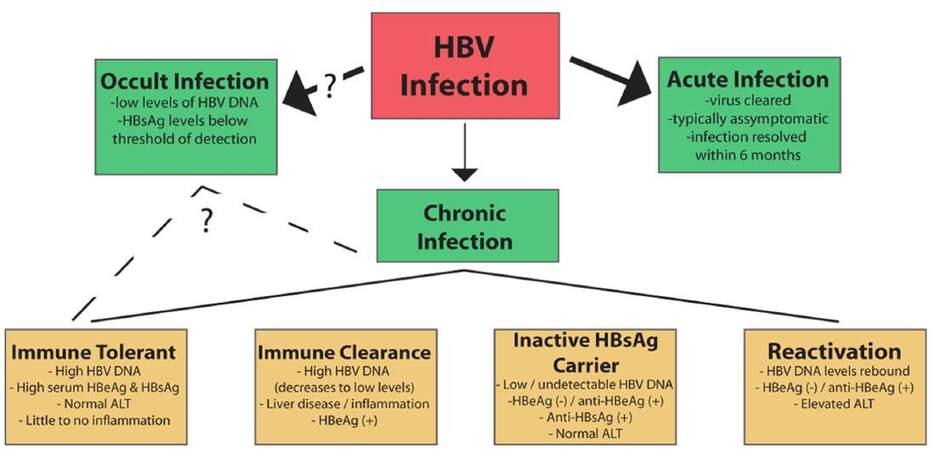
Figure 3. The natural history of an hepatitis B virus (HBV) infection. Infection with HBV can result in an acute, self-clearing, or chronic HBV infection; the development of a chronic HBV infection positively correlates with younger age. A chronic infection usually follows a long-term course in which the virus replicates at high levels, followed by immune-mediated control of viral replication associated with liver inflammation. Seroconversion and maintenance of undetectable or low levels of viral replication are markers of a favorable prognosis, but long-term disease can lead to the development of cirrhosis and hepatocellular carcinoma. (See text for additional details)
Approximately 5-10% of cases of HBV-infected adults, and a significantly higher percentage of HBV-infected infants and children, develop a chronic HBV infection[215] as indicated by continued, detectable expression of HBsAg for at least 6 months after the initial infection. More recently, the application of more sensitive detection techniques, such as polymerase chain reaction (PCR)-based methods that can detect < 250 HBV virions/mL, has also shown that many individuals who were believed to be HBV-free following purported HBV clearance (indicated by the absence of detectable levels of HBsAg expression) actually have low levels of detectable serum and liver HBV DNA. In fact, low levels of HBV DNA can be detected in up to 30% of patients with liver disease of previously unknown etiology.[215-217] This result has led to the recognition of occult HBV infections, in which the level of virus is persistently low and below the level of detection by traditional HBsAg detection techniques. Because of the relatively recent identification of this group of HBV-infected individuals, the risk factors associated with an occult HBV infection remain incompletely understood, although some evidence does suggest that occult infections retain much of the same risk factors as chronic HBV infection.[218]
Clinically, a chronic HBV infection can be divided into multiple phases,[11,209,215,219] though not all patients progress through each stage. The “immune tolerant” phase is characterized by high titers of HBV DNA (> 100,000 copies/mL), the presence of HBeAg, and little liver disease. This phase can last decades, especially in perinatally infected patients, but is typically short or absent in childhood- and adult-acquired HBV.
The “immune clearance” phase also has high levels of HBV, though usually less than is present in the immune tolerant phase, as well as HBeAg expression, but is also characterized by more advanced liver disease with increased inflammation and progression of fibrosis. In addition, this phase is associated with spikes in levels of aminotransferases, which are believed to be a result of an HBV-specific cytotoxic T-cell-mediated immune response and destruction of HBV-infected hepatocytes.[220] This is important, as a longer duration of this phase and higher frequency/severity of the HBV flares are associated with the development of cirrhosis and HCC.[221] Typically this phase can last from several weeks to years and likely represents immunological attempts to control HBV levels. Seroconversion from HBeAg to anti-HBe is considered an important clinical outcome of the immune clearance phase, with immune control of the virus leading to very low or undetectable levels of serum HBV along with normal aminotransferase levels. Importantly, HBeAg seroconversion is associated with a favorable long-term outcome and with decreased risk of developing cirrhosis or HCC.[37]
The “inactive HBsAg carrier” phase is characterized by multiple changes to the disease state. Specifically, there is a loss of HBeAg expression corresponding to an increased presence of anti-HBe. Spontaneous seroconversion from HBsAg to anti-HBs and low to undetectable levels of serum HBV DNA are also hallmarks of this phase. Additionally, aminotransferase levels remain consistently normal; low to mild hepatitis and fibrosis may be observed based on the length of the immune clearance phase. The inactive carrier phase could potentially be maintained indefinitely and is associated with a favorable clinical outcome;[215,219] however, some individuals in the inactive HBV carrier phase enter a “reactivation/HBeAg-negative chronic hepatitis B” phase during which HBV replication rebounds either spontaneously or as a result of immune suppression. These patients are HBeAg negative/anti-HBe positive and have elevated liver enzymes with increased necroinflammation. Serum HBV DNA levels can reach as high as 108-109 copies/mL, though levels are typically lower than in HBeAg-positive patients.[44,209,215,219]
Ultimately, for many patients the end result of a chronic HBV infection is the development of HBV-associated HCC. While seven therapeutics are currently approved for the treatment of chronic HBV, none has proven successful at achieving an “absolute cure” or a complete loss of HBV DNA and a lifetime risk of development of HCC equal to natural clearing of the infection. Five of these therapeutics are nucleoside analogs, designed to directly inhibit the RT. The other two, standard and pegylated interferon-α, function as antiviral cytokines, signaling through the interferon receptor to activate the JAK-STAT pathway.[11,219] While generally effective at lowering viral load, the fact that none of these anti-HBV therapies is curative means these therapies must be life-long treatments, which eventually leads to the development of HBV mutants that are resistant to these therapies. Because of this, specific guidelines have been developed for when to use antiviral therapy and which therapeutic to use.[219]
HBV and HCC
HCC, which accounts for 80-90% of all liver cancers, is one of the most common and most deadly cancers worldwide. Globally, liver cancer is the sixth most common and second deadliest cancer, with an incidence to mortality rate near 1.[222] Epidemiological studies have identified chronic HBV infection of the liver as the leading risk factor for HCC development.[223,224] Despite the availability of a vaccine, 350-500 million people worldwide are chronically infected with HBV and, depending on age and route of infection, as many as 25% of these individuals could go on to develop HBV-associated HCC.[224,225] The number of cases of HCC that are attributed to HBV will likely increase as occult infections become both better reported and better understood.
The molecular mechanisms that link a chronic HBV infection to HCC development are incompletely understood but are likely subtle considering that HBV-associated HCC usually occurs in the context of a decades-long chronic HBV infection. Studies have focused on three main factors that may be involved in the development of HBV-associated HCC: chronic inflammation accompanied by destruction and regeneration of hepatocytes, consequences of HBV DNA integration into host genome, and the potential effects of HBV proteins such as HBx.[88,92,93,225,226] Some potential mechanisms that might link an HBV infection to HCC development have already been described above. Here we summarize additional mechanisms that have been suggested to link a chronic HBV infection to the development of HCC.
One particularly important intermediate aspect of a decades-long chronic HBV infection includes the development of HBV-associated cirrhosis prior to HCC development.[219] It is generally accepted that the majority, potentially as much as 70-90%, of all HCC occurs in the context of decompensated cirrhosis,[224] and a strong relationship exists between chronic HBV infection and cirrhosis. In fact, a recent cohort study demonstrated that the cumulative lifetime risk of developing HBV-associated cirrhosis is 41.5% for chronically infected patients, with a cumulative risk of developing HCC of 21.7%.[209] Therefore, establishing a clearer understanding of the cellular mechanisms associated with the intermediate stages of chronic disease, particularly development of cirrhosis, could enhance the overall understanding of causes of HBV-associated HCC.
Numerous aspects of an HBV infection could be playing a role in the development of HCC. It is logical to assume that hepatotropic viruses such as HBV, which alter hepatocyte physiology as part of, or a consequence of their replication, may disrupt normal hepatocyte and overall liver functions. Many of these disruptions and alterations, either through viral replication or activities of viral proteins such as HBx, could be playing a role in the development of downstream HBV-associated HCC and have been extensively reviewed elsewhere in the literature. For example, HBV has been shown to disrupt cell cycle regulation,[227,228] alter apoptotic pathways,[229] alter hepatocyte metabolism,[33] and alter miRNA expression and miRNA-mediated regulation.[230] Many of these studies have focused on multiple cellular signal transduction pathways, including those involving Ras/mitogen-associated protein kinases (MAPK),[231] mechanistic target of rapamycin (mTOR),[232] PI3K/Akt,[86,233] and NFκB,[229] among many others. Each of these pathways and factors, while also important for HBV replication, are main mediators of hepatocyte functions. As such, disruption can have a major impact on hepatocyte physiology, which has generated a considerable amount of interest in their potential role as mechanisms for the development of HBV-associated HCC. The results of some of these studies are summarized here.
HBV and the cell cycle
As with many viruses, HBV must optimize the cellular environment for viral replication. In the case of HBV, this involves inducing hepatocytes to exit quiescence and enter into an active cell cycle, and the status of cell proliferation pathways can have a significant impact on HBV replication.[91] For example, in primary rat hepatocytes, HBV moved cells from G0 into and through G1, but stalled progression before the hepatocytes were able to reach S phase, and this regulation of the cell cycle is required for HBV replication in primary rat hepatocytes.[85] Studies in cell lines suggest a similar HBV-mediated regulation of cell cycle progression, with HBV stalling progression of the cell cycle before entry into S phase in both Huh7 cells expressing HBV and the HBV-expressing HepG2.2.15 cells.[234] Interestingly, studies have also shown decreased proliferation of HepG2.2.15 cells, in comparison to HepG2 cells, along with HBV-mediated alteration of cell cycle regulators leading to a G1 phase arrest.[235] Another study, however, in Huh7 cells and primary marmoset hepatocytes, demonstrated an HBV-mediated stall in the G2 phase of the cell cycle.[236] While somewhat contradictory, these results together correlate well with the results of other studies showing that HBV replication is increased when the cell cycle is arrested in either G1 or G2, but HBV replication is decreased during S phase, when cellular DNA synthesis would be higher, potentially depleting the pool of nucleotides that would be available for HBV replication.[237,238]
Much of the HBV-mediated regulation of the cell cycle appears to be through the activity of the HBx protein. Multiple studies in primary hepatocytes have demonstrated that HBx alters cell cycle regulators, including decreasing p15 and p16 expression, decreasing DNA synthesis, and increasing p21, p27, cyclin D1, and cyclin E expression.[85,185,239] Together these results suggest that HBx drives hepatocytes into the cell cycle but increases expression of inhibitors that prevent progression beyond G1. This HBx-mediated regulation of the cell cycle could have a long-term impact on hepatocyte physiology, altering hepatocyte proliferation pathways and contributing to the development of HBV-associated disease and HCC.
HBV and metabolism
Because of the primary role of the liver as a metabolic organ, a growing body of research has begun to investigate the impact of HBV infection on metabolic pathways in HBV-infected cells. In fact, HBV has been referred to as a “metabolovirus” due to the perceived intersection between HBV gene expression and control of cellular metabolism.[206,240] Specifically, a number of groups have examined the role of HBV in lipid metabolism, especially considering the well-established link between hepatocyte lipids and various stages of the HCV life cycle[241] and the recent identification of a bile salt transporter as a functional receptor for HBV.
The influence of HBV infection on hepatocyte metabolism was recently brought to the forefront with the identification of hNTCP, the primary bile salt transporter in hepatocytes, as a functional HBV receptor. Interestingly, the binding of HBV, specifically the preS1 domain of L-HBsAg, to hNTCP directly interferes with the normal function of hNTCP suggesting competition for binding motifs within the receptor. Furthermore, point mutations in hNTCP that abolished binding of preS1 also blocked the ability of the receptor to bind taurocholate,[242] suggesting that by binding to hNTCP, HBV could dramatically alter hepatic bile acid uptake.
Recently, HBV-mediated inhibition of normal hNTCP function was extended further using a biochemical profiling approach in which human liver chimeric mice were infected with HBV, and the impact on cholesterol metabolism was determined. Indeed, this study was able to demonstrate overall modest HBV-mediated changes in lipid metabolism, but specific factors involved in both cholesterol and bile acid metabolism were significantly altered. Interestingly, similar results were seen in a comparison of HBV-infected humanized mice, mice treated with the HBV entry inhibitor Myrcludex-B, which mimics the preS1 domain and binds to hNTCP to block HBV infection, and liver biopsy samples from chronically HBV-infected individuals.[180] Together these results indicate that the binding of HBV to hNTCP inhibits bile acid uptake, which stimulates bile acid synthesis pathways. One interesting caveat to these studies is the relatively novel use of a direct infection system, which drastically alters the question being asked in the experiment. For example, by also using the preS1 mimic Myrcludex-B, the studies are specifically addressing the impact of receptor binding by HBV, and not the cellular impact of HBV replication. This is in contrast to some previous work, which has typically been done using systems that bypass the infection step. An example of the importance of this distinction is that while the study using HBV-infected human-liver chimeric mice demonstrated that by binding to hNTCP, HBV alters the levels of nuclear farnesoid X receptor (FXR) and small heterodimer partner (SHP), previous work in an HBV-tg mouse model (which bypasses the infection step, among other differences) showed that depletion of FXR and SHP signaling did not diminish viral replication or transcription.[243] This suggests that although HBV binding to its receptor alters the expression of these transcription factors, this alteration might not affect HBV replication. Further research would be needed to determine the relevance of similar contrasting results within different model systems.
In addition to the functional inhibition of a major bile salt transporter, evidence from other studies has suggested that HBV replication may be intimately associated with central metabolic pathways. For example, multiple transcription factors associated with hepatic metabolic processes, including HNFs,[244,245] peroxisome proliferator-activated receptors (PPARs),[245-247] and FXR[248-250] can all be recruited to the HBV genome.[33] Moreover, studies in vitro have shown that exogenous addition of bile acids to HBV-expressing cells can increase HBV replication.[250,251]
Induction of gluconeogenesis enhances HBV replication,[252] and HBx has been shown to increase expression of multiple gluconeogenic genes,[253] potentially contributing to the central role of HBx in HBV replication. Recent RNA-seq analyses of HBV-expressing Huh7 cells[254] and primary rat hepatocytes[186] also detected decreased expression of GLUT2, the main hepatic glucose transporter. Investigation of the effect of fasting glucose levels on HBV replication revealed a link, albeit minor, between the metabolic state of the cell and the level of HBV replication,[245] and both gluconeogenesis and lipogenesis are under the same transcription factor control as HBV replication.[255] Studies have linked metabolic changes to effects of HBV proteins. For example, some of the earliest functions attributed to HBx involved its regulation of metabolic pathways, such as HBx-mediated activation of the Ras-Raf-MAPK pathway,[256,257] a central pathway involved in the response to nearly all changes that affect metabolism.[258] Protein kinase C (PI3K) is also activated by HBx,[259,260] which correlates with recent results suggesting HBV and HBx activate the PI3K/Akt pathway, reducing HBV replication.[86,233] In addition, studies have shown that mutant L-HBsAg can activate mTOR signaling,[261-263] ultimately causing increased lipogenesis.[262]
When considered in combination, these studies support the characterization of HBV as a “metabolovirus”, and HBV responds to and causes significant metabolic changes in the cell. While the clinical impact of this altered metabolic regulation remains unknown, some studies have suggested that HBV can “help” reduce fatty liver disease.[264] The overall link between HBV and metabolic syndrome remains less clear; however, studies that consider the impact of direct infection through a major metabolic receptor may help to enhance our understanding of the link between HBV and metabolic pathways, and how this relationship may impact the metabolic state of the liver and the development of HBV-associated disease and HCC.
HBV and apoptosis
Despite the many studies that have investigated the effect of an HBV infection and expression of HBV proteins on hepatocyte pro- and anti-apoptotic signaling pathways, the interplay of an HBV infection and hepatocyte apoptotic signaling pathways remains incompletely understood. Because an HBV infection is non-cytopathic, it would be expected that HBV either inhibits or has little effect on apoptotic pathways. Evidence has suggested, however, an HBV-mediated effect on cellular apoptosis that is cell-type- or cell-context-dependent. Some of these differing effects can be attributed to HBx activities, which often have divergent functions depending on context. In the case of apoptosis, some studies have shown that HBx can inhibit apoptosis[86,265-269] or have no effect on apoptosis,[99,270,271] while other studies have shown that HBx can activate apoptotic pathways[272-276] or sensitize cells to pro-apoptotic stimuli.[277-279] The context-dependent apoptotic effects of HBx were clearly shown by studies demonstrating that HBx sensitized dedifferentiated hepatocytes to apoptosis, while HBx-expressing hepatocytes that remained differentiated were resistant to apoptotic stimuli.[279] This underscores the importance of using relevant cell systems for studying the cellular impact of HBV replication and protein expression on cell physiology. HBx was also shown to have divergent apoptotic functions in the context of HBV replication. Studies in primary hepatocytes demonstrated that HBx can have both a pro- and anti-apoptotic effect, depending on the cellular context of HBx expression. Specifically, inhibition of apoptosis was linked to HBx-mediated activation of NFκB; however, when activation of NFκB was blocked, HBx induced apoptosis through pathways involving the mitochondria permeability transition pore (MPTP), a critical pore that spans the inner mitochondrial and outer mitochondrial membranes and affects numerous mitochondrial functions, including mitochondrial control of apoptosis.[83] Whether HBV, through functions of HBx, regulates apoptosis as a mechanism for regulating viral replication or enhancing viral spread is currently unknown. Although activation of apoptosis may impact both viral spread,[280,281] and immune evasion,[93] recent evidence suggests that alteration of apoptosis during HBV infection is unlikely to impact viral spread.[282] Due to the regenerative nature of hepatocytes, it is also possible that the impact of HBV on apoptosis may fluctuate during the course of infection, as regenerating hepatocytes have different active signaling pathways than quiescent cells, and these could have differing influences on apoptotic stimuli.[280,283,284] Interestingly, both the activation and the inactivation of apoptosis could be playing a role in the long-term development of HBV-associated HCC: enhanced regeneration associated with HBV-mediated activation of apoptosis could lead to selection of apoptosis-resistant cells,[285] while inhibition of apoptosis could lead to unchecked proliferation and the accumulation of transforming mutations.[93] Although the exact mechanisms that underlie HBV and HBx regulation of apoptosis remain incompletely understood, the cellular impact of altered apoptotic signaling could significantly contribute to the downstream development of HBV-associated disease and HCC.
HBV and microRNAs
Potentially spurred by the discovery of the required role of miR-122 in successful HCV replication,[286,287] multiple groups have begun to investigate how cellular miRNAs may impact various aspects of HBV biology, and alternatively, how HBV may impact the expression of cellular miRNAs. These effects have been reviewed in more detail elsewhere.[230,288]
While a wide range of cellular miRNAs has been investigated for their role in regulating or being regulated by HBV, none has been studied as extensively as miR-122. This miRNA, which makes up 50-70% of the total miRNA pool in hepatocytes[289,290] has been shown to have many different roles in the context of HBV replication. Although conflicting reports do exist, it seems that miR-122 functions as an antiviral miRNA, potentially through multiple mechanisms. These mechanisms include direct targeting of viral RNAs through miR-122 recognition sites in the HBV genome[291,292] and altered miR-122-mediated regulation of cellular factors involved in regulation of HBV replication, such as heme oxygenase-1,[293] cyclin G1,[294,295] and pituitary tumor-transforming gene 1 binding factor (PBF).[292] Importantly, multiple studies have also shown an HBV-mediated decrease in the levels of functional miR-122,[291,292,296-299] although the mechanism for this reduction remains unclear. One potential mechanism is a sponge effect, where the HBV transcripts act as a sponge to divert miR-122 away from endogenous targets,[291,292] although it is unclear whether the levels of HBV transcripts in the cell reach the high levels of target required for this sponge effect to have a biological impact.[300] Interestingly, primary tree shrew hepatocytes, which can be directly infected with human HBV, showed an increase in the levels of miR-122 in response to HBV infection.[301] Further research will be needed to determine if this effect is the result of using a more biologically relevant system, with direct infection, or is an inherent difference between tree shrew and human hepatocytes.
Other miRNA families have also been assessed for their role in HBV replication, including miRNAs with well-established roles as either oncomirs or tumor suppressors. For example, the let-7 family, which function as tumor suppressor miRNAs and are downregulated in multiple cancers including HCC,[302] are decreased in the context of HBV replication, HBx expression, and HBV-associated HCC.[303-307] The miR-15 family,[305,307-312] miR-125 family,[303,305,310,313,314] miR-17/92 cluster,[289,303,307,310,315] and miR-199a-3p[289] are all HCC-related miRNAs that multiple groups have studied in the context of HBV. Although the field is still developing and contradictory reports exist, when taken together these reports support the overall impact of HBV on cellular miRNAs. How these miRNAs impact HBV replication, and ultimately HBV-associated disease and development of HBV-associated HCC, remains incompletely understood.
Conclusion
Hepatocytes are the main target of an HBV infection, and a chronic HBV infection is the major global cause for the development of HCC.[92,207] While the association between chronic HBV infections and HCC is well established, there are still gaps in our understanding of how a chronic HBV infection can lead to HCC development. The high worldwide prevalence of chronic HBV infections, the limited therapeutic options currently available for the treatment of chronic HBV infections, the increased global incidence of HCC, the high mortality rate of individuals with HCC, and the close correlation between chronic HBV infections and HCC development have generated considerable interest in understanding HBV biology and elucidating the molecular mechanisms that underlie the development of HBV-associated HCC. In this article, we provided a review of HBV biology and highlighted the potential mechanisms that could underlie the development of HBV-associated HCC. These mechanisms are thought to involve a combination of continuous immune-mediated destruction of HBV-infected hepatocytes and concomitant hepatocyte regeneration, the activities of certain HBV proteins such as the HBx, and potential consequences of HBV genome integration into the host genome.[92,207]
Although there are treatments for a chronic HBV infection, resistance to currently available anti-HBV drugs, which develops due to the emergence of HBV mutants, is one major drawback of continuing nucleoside analog therapy. Moreover, existing antiviral treatments can control but not entirely eliminate HBV because of the persistence of HBV nuclear-localized cccDNA, and the persistence of cccDNA remains a major obstacle for the treatment and cure of chronic HBV infections.[316-320] While there has been substantial progress in identifying mechanisms that underlie HBV infection, replication and clearance, there are still gaps in our understanding of the HBV lifecycle. The paucity of cell culture model systems that can recapitulate all the aspects of a human HBV infection and the scarcity of in vivo models for studying direct HBV infections has impeded our understanding of HBV biology. The recent discovery of hNTCP as the functional HBV receptor has provided new opportunities for the creation of novel cell culture model systems that can be used to understand the outcomes of a natural HBV infection. It would also be interesting to utilize tg mice expressing a hNTCP to study HBV biology and examine the activities of HBV-encoded proteins. However, currently, hNTCP tg mice do not permit the establishment of a productive HBV infection and it is likely that identification of additional species-specific determinants of HBV infection will be required before small rodent models of HBV infection and pathogenesis can be fully utilized.[103,182,183] Although mice with humanized-livers and immune systems provide another promising model for studying HBV infection and pathogenesis, the complexity of generating these models have limited their use for studying HBV biology.[175] Overall, studies aimed at enhancing our current understanding of the HBV life cycle and identifying central factors involved in the development of HBV-associated HCC are still needed and remain critical for the generation of novel therapeutics to inhibit HBV replication and the development of HBV-associated HCC.
Financial support and sponsorship
The work was partially supported by an NIH predoctoral fellowship to RJL; the grant number is F31CA171712.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
2. Blumberg BS, Gerstley BJ, Hungerford DA, London WT, Sutnick AI. A serum antigen (Australia antigen) in Down's syndrome, leukemia, and hepatitis. Ann Intern Med 1967;66:924-31.
3. Maccallum FO. Homologous serum hepatitis. Proc R Soc Med 1946;39:655-7.
4. Alter HJ, Holland PV, Purcell RH, Lander JJ, Feinstone SM, Morrow AG, Schmidt PJ. Posttransfusion hepatitis after exclusion of commercial and hepatitis-B antigen-positive donors. Ann Intern Med 1972;77:691-9.
5. Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 1989;244:359-62.
6. Blumberg BS, Larouzé B, London WT, Werner B, Hesser JE, Millman I, Saimot G, Payet M. The relation of infection with the hepatitis B agent to primary hepatic carcinoma. Am J Pathol 1975;81:669-82.
7. Prince AM, Szmuness W, Michon J, Demaille J, Diebolt G, Linhard J, Quenum C, Sankale M. A case/control study of the association between primary liver cancer and hepatitis B infection in Senegal. Int J Cancer 1975;16:376-83.
8. Blumberg BS, Millman I, inventor; Cancer Res Inst, assignee. Vaccine against viral hepatitis and process. United States patent US3636191 A. 1972 Jan 18 .
9. Buynak EB, Roehm RR, Tytell AA, Bertland AU 2nd, Lampson GP, Hilleman MR. Vaccine against human hepatitis B. JAMA 1976;235:2832-4.
10. Purcell RH, Gerin JL. Hepatitis B subunit vaccine: a preliminary report of safety and efficacy tests in chimpanzees. Am J Med Sci 1975;270:395-9.
11. Seeger C, Zoulim F, Mason WS. Hepadnaviruses. In: Knipe DM, Howley PM, editors. Field's Virology. 6th ed. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins Health; 2014. pp. 3376-436.
12. Scaglione SJ, Lok AS. Effectiveness of hep atitis B treatment in clinical practice. Gastroenterology 2012;142:1360-8.
13. Suh A, Brosius J, Schmitz J, Kriegs JO. The genome of a Mesozoic paleovirus reveals the evolution of hepatitis B viruses. Nat Commun 2013;4:1791.
14. Paraskevis D, Magiorkinis G, Magiorkinis E, Ho SY, Belshaw R, Allain JP, Hatzakis A. Dating the origin and dispersal of hepatitis B virus infection in humans and primates. Hepatology 2013;57:908-16.
15. Sureau C, Eichberg JW, Hubbard GB, Romet-Lemonne JL, Essex M. A molecularly cloned hepatitis B virus produced in vitro is infectious in a chimpanzee. J Virol 1988;62:3064-7.
16. Yan RQ, Su JJ, Huang DR, Gan YC, Yang C, Huang GH. Human hepatitis B virus and hepatocellular carcinoma. I. Experimental infection of tree shrews with hepatitis B virus. J Cancer Res Clin Oncol 1996;122:283-8.
17. Locarnini S, Littlejohn M, Aziz MN, Yuen L. Possible origins and evolution of the hepatitis B virus (HBV). Semin Cancer Biol 2013;23:561-75.
18. Tatematsu K, Tanaka Y, Kurbanov F, Sugauchi F, Mano S, Maeshiro T, Nakayoshi T, Wakuta M, Miyakawa Y, Mizokami M. A genetic variant of hepatitis B virus divergent from known human and ape genotypes isolated from a Japanese patient and provisionally assigned to new genotype J. J Virol 2009;83:10538-47.
19. Tran TT, Trinh TN, Abe K. New complex recombinant genotype of hepatitis B virus identified in Vietnam. J Virol 2008;82:5657-63.
20. Kurbanov F, Tanaka Y, Kramvis A, Simmonds P, Mizokami M. When should "I" consider a new hepatitis B virus genotype? J Virol 2008;82:8241-2.
21. Chu CJ, Keeffe EB, Han SH, Perrillo RP, Min AD, Soldevila-Pico C, Carey W, Brown RS Jr, Luketic VA, Terrault N, Lok AS. Hepatitis B virus genotypes in the United States: results of a nationwide study. Gastroenterology 2003;125:444-51.
22. Shi W, Zhang Z, Ling C, Zheng W, Zhu C, Carr MJ, Higgins DG. Hepatitis B virus subgenotyping: history, effects of recombination, misclassifications, and corrections. Infect Genet Evol 2013;16:355-61.
23. Kao JH, Chen PJ, Lai MY, Chen DS. Hepatitis B genotypes correlate with clinical outcomes in patients with chronic hepatitis B. Gastroenterology 2000;118:554-9.
24. Seo Y, Yano Y. Short- and long-term outcome of interferon therapy for chronic hepatitis B infection. World J Gastroenterol 2014;20:13284-92.
25. Chen HS, Kaneko S, Girones R, Anderson RW, Hornbuckle WE, Tennant BC, Cote PJ, Gerin JL, Purcell RH, Miller RH. The woodchuck hepatitis virus X gene is important for establishment of virus infection in woodchucks. J Virol 1993;67:1218-26.
26. Keasler VV, Hodgson AJ, Madden CR, Slagle BL. Enhancement of hepatitis B virus replication by the regulatory X protein in vitro and in vivo. J Virol 2007;81:2656-62.
27. Zoulim F, Saputelli J, Seeger C. Woodchuck hepatitis virus X protein is required for viral infection in vivo. J Virol 1994;68:2026-30.
28. Lucifora J, Arzberger S, Durantel D, Belloni L, Strubin M, Levrero M, Zoulim F, Hantz O, Protzer U. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J Hepatol 2011;55:996-1003.
29. Tsuge M, Hiraga N, Akiyama R, Tanaka S, Matsushita M, Mitsui F, Abe H, Kitamura S, Hatakeyama T, Kimura T, Miki D, Mori N, Imamura M, Takahashi S, Hayes CN, Chayama K. HBx protein is indispensable for development of viraemia in human hepatocyte chimeric mice. J Gen Virol 2010;91:1854-64.
30. Nassal M. HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015;64:1972-84.
31. Seeger C, Mason WS. Molecular biology of hepatitis B virus infection. Virology 2015;479-480C:672-86.
32. Doitsh G, Shaul Y. Enhancer I predominance in hepatitis B virus gene expression. Mol Cell Biol 2004;24:1799-808.
33. Bar-Yishay I, Shaul Y, Shlomai A. Hepatocyte metabolic signalling pathways and regulation of hepatitis B virus expression. Liver Int 2011;31:282-90.
34. Wang J, Lee AS, Ou JH. Proteolytic conversion of hepatitis B virus e antigen precursor to end product occurs in a postendoplasmic reticulum compartment. J Virol 1991;65:5080-3.
35. Chen MT, Billaud JN, Sällberg M, Guidotti LG, Chisari FV, Jones J, Hughes J, Milich DR. A function of the hepatitis B virus precore protein is to regulate the immune response to the core antigen. Proc Natl Acad Sci U S A 2004;101:14913-8.
36. Chen M, Sällberg M, Hughes J, Jones J, Guidotti LG, Chisari FV, Billaud JN, Milich DR. Immune tolerance split between hepatitis B virus precore and core proteins. J Virol 2005;79:3016-27.
37. Liaw YF. HBeAg seroconversion as an important end point in the treatment of chronic hepatitis B. Hepatol Int 2009;3:425-33.
38. Eble BE, MacRae DR, Lingappa VR, Ganem D. Multiple topogenic sequences determine the transmembrane orientation of the hepatitis B surface antigen. Mol Cell Biol 1987;7:3591-601.
39. Peterson DL, Nath N, Gavilanes F. Structure of hepatitis B surface antigen. Correlation of subtype with amino acid sequence and location of the carbohydrate moiety. J Biol Chem 1982;257:10414-20.
40. Eble BE, Lingappa VR, Ganem D. The N-terminal (pre-S2) domain of a hepatitis B virus surface glycoprotein is translocated across membranes by downstream signal sequences. J Virol 1990;64:1414-9.
41. Gripon P, Cannie I, Urban S. Efficient inhibition of hepatitis B virus infection by acylated peptides derived from the large viral surface protein. J Virol 2005;79:1613-22.
42. Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, Huang Y, Qi Y, Peng B, Wang H, Fu L, Song M, Chen P, Gao W, Ren B, Sun Y, Cai T, Feng X, Sui J, Li W. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012;1:e00049.
43. Urban S, Bartenschlager R, Kubitz R, Zoulim F. Strategies to inhibit entry of HBV and HDV into hepatocytes. Gastroenterology 2014;147:48-64.
44. Ganem D, Prince AM. Hepatitis B virus infection--natural history and clinical consequences. N Engl J Med 2004;350:1118-29.
46. Bruns M, Miska S, Chassot S, Will H. Enhancement of hepatitis B virus infection by noninfectious subviral particles. J Virol 1998;72:1462-8.
47. Stannard LM, Hodgkiss M. Morphological irregularities in Dane particle cores. J Gen Virol 1979;45:509-14.
48. Birnbaum F, Nassal M. Hepatitis B virus nucleocapsid assembly: primary structure requirements in the core protein. J Virol 1990;64:3319-30.
49. Li HC, Huang EY, Su PY, Wu SY, Yang CC, Lin YS, Chang WC, Shih C. Nuclear export and import of human hepatitis B virus capsid protein and particles. PLoS Pathog 2010;6:e1001162.
50. Perlman DH, Berg EA, O'connor PB, Costello CE, Hu J. Reverse transcription-associated dephosphorylation of hepadnavirus nucleocapsids. Proc Natl Acad Sci U S A 2005;102:9020-5.
51. Ludgate L, Ning X, Nguyen DH, Adams C, Mentzer L, Hu J. Cyclin-dependent kinase 2 phosphorylates s/t-p sites in the hepadnavirus core protein C-terminal domain and is incorporated into viral capsids. J Virol 2012;86:12237-50.
52. Melegari M, Wolf SK, Schneider RJ. Hepatitis B virus DNA replication is coordinated by core protein serine phosphorylation and HBx expression. J Virol 2005;79:9810-20.
53. Bock CT, Schwinn S, Locarnini S, Fyfe J, Manns MP, Trautwein C, Zentgraf H. Structural organization of the hepatitis B virus minichromosome. J Mol Biol 2001;307:183-96.
54. Nassal M. The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J Virol 1992;66:4107-16.
55. Le Pogam S, Chua PK, Newman M, Shih C. Exposure of RNA templates and encapsidation of spliced viral RNA are influenced by the arginine-rich domain of human hepatitis B virus core antigen (HBcAg 165-173). J Virol 2005;79:1871-87.
56. Chu TH, Liou AT, Su PY, Wu HN, Shih C. Nucleic acid chaperone activity associated with the arginine-rich domain of human hepatitis B virus core protein. J Virol 2014;88:2530-43.
57. Lewellyn EB, Loeb DD. The arginine clusters of the carboxy-terminal domain of the core protein of hepatitis B virus make pleiotropic contributions to genome replication. J Virol 2011;85:1298-309.
58. Le Pogam S, Shih C. Influence of a putative intermolecular interaction between core and the pre-S1 domain of the large envelope protein on hepatitis B virus secretion. J Virol 2002;76:6510-7.
59. Zlotnick A, Venkatakrishnan B, Tan Z, Lewellyn E, Turner W, Francis S. Core protein: a pleiotropic keystone in the HBV lifecycle. Antiviral Res 2015;121:82-93.
60. Mason WS, Seal G, Summers J. Virus of Pekin ducks with structural and biological relatedness to human hepatitis B virus. J Virol 1980;36:829-36.
61. Summers J, Mason WS. Replication of the genome of a hepatitis B--like virus by reverse transcription of an RNA intermediate. Cell 1982;29:403-15.
62. Bartenschlager R, Schaller H. The amino-terminal domain of the hepadnaviral P-gene encodes the terminal protein (genome-linked protein) believed to prime reverse transcription. EMBO J 1988;7:4185-92.
63. Zoulim F, Seeger C. Reverse transcription in hepatitis B viruses is primed by a tyrosine residue of the polymerase. J Virol 1994;68:6-13.
64. Weber M, Bronsema V, Bartos H, Bosserhoff A, Bartenschlager R, Schaller H. Hepadnavirus P protein utilizes a tyrosine residue in the TP domain to prime reverse transcription. J Virol 1994;68:2994-9.
65. Radziwill G, Tucker W, Schaller H. Mutational analysis of the hepatitis B virus P gene product: domain structure and RNase H activity. J Virol 1990;64:613-20.
66. Kim S, Lee J, Ryu WS. Four conserved cysteine residues of the hepatitis B virus polymerase are critical for RNA pregenome encapsidation. J Virol 2009;83:8032-40.
67. Xiong Y, Eickbush TH. Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J 1990;9:3353-62.
68. Nassal M. Hepatitis B viruses: reverse transcription a different way. Virus Res 2008;134:235-49.
69. Wang YX, Luo C, Zhao D, Beck J, Nassal M. Extensive mutagenesis of the conserved box E motif in duck hepatitis B virus P protein reveals multiple functions in replication and a common structure with the primer grip in HIV-1 reverse transcriptase. J Virol 2012;86:6394-407.
70. Tavis JE, Cheng X, Hu Y, Totten M, Cao F, Michailidis E, Aurora R, Meyers MJ, Jacobsen EJ, Parniak MA, Sarafianos SG. The hepatitis B virus ribonuclease H is sensitive to inhibitors of the human immunodeficiency virus ribonuclease H and integrase enzymes. PLoS Pathog 2013;9:e1003125.
71. Bartenschlager R, Junker-Niepmann M, Schaller H. The P gene product of hepatitis B virus is required as a structural component for genomic RNA encapsidation. J Virol 1990;64:5324-32.
72. Jones SA, Hu J. Hepatitis B virus reverse transcriptase: diverse functions as classical and emerging targets for antiviral intervention. Emerg Microbes Infect 2013;2:e56.
73. Belloni L, Pollicino T, De Nicola F, Guerrieri F, Raffa G, Fanciulli M, Raimondo G, Levrero M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc Natl Acad Sci U S A 2009;106:19975-9.
74. Seeger C, Sohn JA. Targeting hepatitis B virus with CRISPR/Cas9. Mol Ther Nucleic Acids 2014;3:e216.
75. Zhang Z, Torii N, Hu Z, Jacob J, Liang TJ. X-deficient woodchuck hepatitis virus mutants behave like attenuated viruses and induce protective immunity in vivo. J Clin Invest 2001;108:1523-31.
76. Xu Z, Yen TS, Wu L, Madden CR, Tan W, Slagle BL, Ou JH. Enhancement of hepatitis B virus replication by its X protein in transgenic mice. J Virol 2002;76:2579-84.
77. Tsuge M, Hiraga N, Takaishi H, Noguchi C, Oga H, Imamura M, Takahashi S, Iwao E, Fujimoto Y, Ochi H, Chayama K, Tateno C, Yoshizato K. Infection of human hepatocyte chimeric mouse with genetically engineered hepatitis B virus. Hepatology 2005;42:1046-54.
79. Bouchard MJ, Wang LH, Schneider RJ. Calcium signaling by HBx protein in hepatitis B virus DNA replication. Science 2001;294:2376-8.
80. Melegari M, Scaglioni PP, Wands JR. Hepatitis B virus mutants associated with 3TC and famciclovir administration are replication defective. Hepatology 1998;27:628-33.
81. Tang H, Delgermaa L, Huang F, Oishi N, Liu L, He F, Zhao L, Murakami S. The transcriptional transactivation function of HBx protein is important for its augmentation role in hepatitis B virus replication. J Virol 2005;79:5548-56.
82. Leupin O, Bontron S, Schaeffer C, Strubin M. Hepatitis B virus X protein stimulates viral genome replication via a DDB1-dependent pathway distinct from that leading to cell death. J Virol 2005;79:4238-45.
83. Clippinger AJ, Gearhart TL, Bouchard MJ. Hepatitis B virus X protein modulates apoptosis in primary rat hepatocytes by regulating both NF-kappaB and the mitochondrial permeability transition pore. J Virol 2009;83:4718-31.
84. Gearhart TL, Bouchard MJ. Replication of the hepatitis B virus requires a calcium-dependent HBx-induced G1 phase arrest of hepatocytes. Virology 2010;407:14-25.
85. Gearhart TL, Bouchard MJ. The hepatitis B virus X protein modulates hepatocyte proliferation pathways to stimulate viral replication. J Virol 2010;84:2675-86.
86. Rawat S, Bouchard M. The hepatitis B virus (HBV) HBx protein activates AKT to simultaneously regulate HBV replication and hepatocyte survival. J Virol 2015;89:999-1012.
87. Slagle BL, Andrisani OM, Bouchard MJ, Lee CG, Ou JH, Siddiqui A. Technical standards for hepatitis B virus X protein (HBx) research. Hepatology 2015;61:1416-24.
88. Keng VW, Tschida BR, Bell JB, Largaespada DA. Modeling hepatitis B virus X-induced hepatocellular carcinoma in mice with the sleeping beauty transposon system. Hepatology 2011;53:781-90.
89. Kim CM, Koike K, Saito I, Miyamura T, Jay G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 1991;351:317-20.
90. Koike K. Hepatitis B virus X gene is implicated in liver carcinogenesis. Cancer Lett 2009;286:60-8.
91. Madden CR, Finegold MJ, Slagle BL. Hepatitis B virus X protein acts as a tumor promoter in development of diethylnitrosamine-induced preneoplastic lesions. J Virol 2001;75:3851-8.
92. Bouchard MJ, Navas-Martin S. Hepatitis B and C virus hepatocarcinogenesis: lessons learned and future challenges. Cancer Lett 2011;305:123-43.
93. Arbuthnot P, Capovilla A, Kew M. Putative role of hepatitis B virus X protein in hepatocarcinogenesis: effects on apoptosis, DNA repair, mitogen-activated protein kinase and JAK/STAT pathways. J Gastroenterol Hepatol 2000;15:357-68.
94. Tanaka Y, Kanai F, Ichimura T, Tateishi K, Asaoka Y, Guleng B, Jazag A, Ohta M, Imamura J, Ikenoue T, Ijichi H, Kawabe T, Isobe T, Omata M. The hepatitis B virus X protein enhances AP-1 activation through interaction with Jab1. Oncogene 2006;25:633-42.
95. Kim JH, Rho HM. Activation of the human transforming growth factor alpha (TGF-alpha) gene by the hepatitis B viral X protein (HBx) through AP-2 sites. Mol Cell Biochem 2002;231:155-61.
96. Lara-Pezzi E, Armesilla AL, Majano PL, Redondo JM, López-Cabrera M. The hepatitis B virus X protein activates nuclear factor of activated T cells (NF-AT) by a cyclosporin A-sensitive pathway. EMBO J 1998;17:7066-77.
97. Lucito R, Schneider RJ. Hepatitis B virus X protein activates transcription factor NF-kappa B without a requirement for protein kinase C. J Virol 1992;66:983-91.
98. Um HR, Lim WC, Chae SY, Park S, Park JH, Cho H. Raf-1 and protein kinase B regulate cell survival through the activation of NF-kappaB in hepatitis B virus X-expressing cells. Virus Res 2007;125:1-8.
99. Yun C, Um HR, Jin YH, Wang JH, Lee MO, Park S, Lee JH, Cho H. NF-kappaB activation by hepatitis B virus X (HBx) protein shifts the cellular fate toward survival. Cancer Lett 2002;184:97-104.
100. Hsieh A, Kim HS, Lim SO, Yu DY, Jung G. Hepatitis B viral X protein interacts with tumor suppressor adenomatous polyposis coli to activate Wnt/β-catenin signaling. Cancer Lett 2011;300:162-72.
101. Iyer S, Groopman JD. Interaction of mutant hepatitis B X protein with p53 tumor suppressor protein affects both transcription and cell survival. Mol Carcinog 2011;50:972-80.
102. Chung TW, Lee YC, Kim CH. Hepatitis B viral HBx induces matrix metalloproteinase-9 gene expression through activation of ERK and PI-3K/AKT pathways: involvement of invasive potential. FASEB J 2004;18:1123-5.
103. Yan H, Peng B, He W, Zhong G, Qi Y, Ren B, Gao Z, Jing Z, Song M, Xu G, Sui J, Li W. Molecular determinants of hepatitis B and D virus entry restriction in mouse sodium taurocholate cotransporting polypeptide. J Virol 2013;87:7977-91.
104. Blanchet M, Sureau C. Infectivity determinants of the hepatitis B virus pre-S domain are confined to the N-terminal 75 amino acid residues. J Virol 2007;81:5841-9.
105. Gripon P, Le Seyec J, Rumin S, Guguen-Guillouzo C. Myristylation of the hepatitis B virus large surface protein is essential for viral infectivity. Virology 1995;213:292-9.
106. Schulze A, Gripon P, Urban S. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology 2007;46:1759-68.
107. Verrier ER, Colpitts CC, Bach C, Heydmann L, Weiss A, Renaud M, Durand SC, Habersetzer F, Durantel D, Abou-Jaoudé G, López Ledesma MM, Felmlee DJ, Soumillon M, Croonenborghs T, Pochet N, Nassal M, Schuster C, Brino L, Sureau C, Zeisel MB. A targeted functional RNA interference screen uncovers glypican 5 as an entry factor for hepatitis B and D viruses. Hepatology 2016;63:35-48.
108. Huang HC, Chen CC, Chang WC, Tao MH, Huang C. Entry of hepatitis B virus into immortalized human primary hepatocytes by clathrin-dependent endocytosis. J Virol 2012;86:9443-53.
109. Rabe B, Delaleau M, Bischof A, Foss M, Sominskaya I, Pumpens P, Cazenave C, Castroviejo M, Kann M. Nuclear entry of hepatitis B virus capsids involves disintegration to protein dimers followed by nuclear reassociation to capsids. PLoS Pathog 2009;5:e1000563.
110. Schmitz A, Schwarz A, Foss M, Zhou L, Rabe B, Hoellenriegel J, Stoeber M, Panté N, Kann M. Nucleoporin 153 arrests the nuclear import of hepatitis B virus capsids in the nuclear basket. PLoS Pathog 2010;6:e1000741.
111. Guidotti LG, Martinez V, Loh YT, Rogler CE, Chisari FV. Hepatitis B virus nucleocapsid particles do not cross the hepatocyte nuclear membrane in transgenic mice. J Virol 1994;68:5469-75.
112. Guidotti LG, Matzke B, Schaller H, Chisari FV. High-level hepatitis B virus replication in transgenic mice. J Virol 1995;69:6158-69.
113. Fourel I, Saputelli J, Schaffer P, Mason WS. The carbocyclic analog of 2'-deoxyguanosine induces a prolonged inhibition of duck hepatitis B virus DNA synthesis in primary hepatocyte cultures and in the liver. J Virol 1994;68:1059-65.
114. Luo L, Chen S, Gong Q, Luo N, Lei Y, Guo J, He S. Hepatitis B virus X protein modulates remodelling of minichromosomes related to hepatitis B virus replication in HepG2 cells. Int J Mol Med 2013;31:197-204.
115. Rivière L, Gerossier L, Ducroux A, Dion S, Deng Q, Michel ML, Buendia MA, Hantz O, Neuveut C. HBx relieves chromatin-mediated transcriptional repression of hepatitis B viral cccDNA involving SETDB1 histone methyltransferase. J Hepatol 2015;63:1093-102.
116. Jeong JK, Yoon GS, Ryu WS. Evidence that the 5'-end cap structure is essential for encapsidation of hepatitis B virus pregenomic RNA. J Virol 2000;74:5502-8.
117. Lee HJ, Lee J, Shin MK, Ryu WS. Polyadenylation is dispensable for encapsidation and reverse transcription of hepatitis B viral pregenomic RNA. Mol Cells 2008;25:545-52.
118. Kim S, Wang H, Ryu WS. Incorporation of eukaryotic translation initiation factor eIF4E into viral nucleocapsids via interaction with hepatitis B virus polymerase. J Virol 2010;84:52-8.
119. Hu J, Toft DO, Seeger C. Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. EMBO J 1997;16:59-68.
120. Nassal M, Rieger A. A bulged region of the hepatitis B virus RNA encapsidation signal contains the replication origin for discontinuous first-strand DNA synthesis. J Virol 1996;70:2764-73.
121. Loeb DD, Hirsch RC, Ganem D. Sequence-independent RNA cleavages generate the primers for plus strand DNA synthesis in hepatitis B viruses: implications for other reverse transcribing elements. EMBO J 1991;10:3533-40.
122. Staprans S, Loeb DD, Ganem D. Mutations affecting hepadnavirus plus-strand DNA synthesis dissociate primer cleavage from translocation and reveal the origin of linear viral DNA. J Virol 1991;65:1255-62.
123. Bill CA, Summers J. Genomic DNA double-strand breaks are targets for hepadnaviral DNA integration. Proc Natl Acad Sci U S A 2004;101:11135-40.
124. Maguire ML, Loeb DD. cis-Acting sequences that contribute to synthesis of minus-strand DNA are not conserved between hepadnaviruses. J Virol 2010;84:12824-31.
125. Lewellyn EB, Loeb DD. Base pairing between cis-acting sequences contributes to template switching during plus-strand DNA synthesis in human hepatitis B virus. J Virol 2007;81:6207-15.
126. Tan Z, Pionek K, Unchwaniwala N, Maguire ML, Loeb DD, Zlotnick A. The interface between hepatitis B virus capsid proteins affects self-assembly, pregenomic RNA packaging, and reverse transcription. J Virol 2015;89:3275-84.
127. Tuttleman JS, Pourcel C, Summers J. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell 1986;47:451-60.
128. Wu TT, Coates L, Aldrich CE, Summers J, Mason WS. In hepatocytes infected with duck hepatitis B virus, the template for viral RNA synthesis is amplified by an intracellular pathway. Virology 1990;175:255-61.
129. Summers J, Smith PM, Horwich AL. Hepadnavirus envelope proteins regulate covalently closed circular DNA amplification. J Virol 1990;64:2819-24.
130. Lentz TB, Loeb DD. Roles of the envelope proteins in the amplification of covalently closed circular DNA and completion of synthesis of the plus-strand DNA in hepatitis B virus. J Virol 2011;85:11916-27.
131. Zhang YY, Zhang BH, Theele D, Litwin S, Toll E, Summers J. Single-cell analysis of covalently closed circular DNA copy numbers in a hepadnavirus-infected liver. Proc Natl Acad Sci U S A 2003;100:12372-7.
132. Laras A, Koskinas J, Dimou E, Kostamena A, Hadziyannis SJ. Intrahepatic levels and replicative activity of covalently closed circular hepatitis B virus DNA in chronically infected patients. Hepatology 2006;44:694-702.
133. Mason WS, Litwin S, Xu C, Jilbert AR. Hepatocyte turnover in transient and chronic hepadnavirus infections. J Viral Hepat 2007;14:22-8.
134. Addison WR, Walters KA, Wong WW, Wilson JS, Madej D, Jewell LD, Tyrrell DL. Half-life of the duck hepatitis B virus covalently closed circular DNA pool in vivo following inhibition of viral replication. J Virol 2002;76:6356-63.
135. Ning X, Nguyen D, Mentzer L, Adams C, Lee H, Ashley R, Hafenstein S, Hu J. Secretion of genome-free hepatitis B virus--single strand blocking model for virion morphogenesis of para-retrovirus. PLoS Pathog 2011;7:e1002255.
136. Gerelsaikhan T, Tavis JE, Bruss V. Hepatitis B virus nucleocapsid envelopment does not occur without genomic DNA synthesis. J Virol 1996;70:4269-74.
137. Yuan TT, Sahu GK, Whitehead WE, Greenberg R, Shih C. The mechanism of an immature secretion phenotype of a highly frequent naturally occurring missense mutation at codon 97 of human hepatitis B virus core antigen. J Virol 1999;73:5731-40.
138. Nowak MA, Bonhoeffer S, Hill AM, Boehme R, Thomas HC, McDade H. Viral dynamics in hepatitis B virus infection. Proc Natl Acad Sci U S A 1996;93:4398-402.
139. Watanabe T, Sorensen EM, Naito A, Schott M, Kim S, Ahlquist P. Involvement of host cellular multivesicular body functions in hepatitis B virus budding. Proc Natl Acad Sci U S A 2007;104:10205-10.
141. Summers J, Smolec JM, Snyder R. A virus similar to human hepatitis B virus associated with hepatitis and hepatoma in woodchucks. Proc Natl Acad Sci U S A 1978;75:4533-7.
142. Lenhoff RJ, Luscombe CA, Summers J. Acute liver injury following infection with a cytopathic strain of duck hepatitis B virus. Hepatology 1999;29:563-71.
143. Newbold JE, Xin H, Tencza M, Sherman G, Dean J, Bowden S, Locarnini S. The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J Virol 1995;69:3350-7.
144. Guo JT, Zhou H, Liu C, Aldrich C, Saputelli J, Whitaker T, Barrasa MI, Mason WS, Seeger C. Apoptosis and regeneration of hepatocytes during recovery from transient hepadnavirus infections. J Virol 2000;74:1495-505.
145. Kajino K, Jilbert AR, Saputelli J, Aldrich CE, Cullen J, Mason WS. Woodchuck hepatitis virus infections: very rapid recovery after a prolonged viremia and infection of virtually every hepatocyte. J Virol 1994;68:5792-803.
146. Vickery K, Cossart Y. DHBV manipulation and prediction of the outcome of infection. J Virol 1996;25:504-9.
147. Cote PJ, Toshkov I, Bellezza C, Ascenzi M, Roneker C, Ann Graham L, Baldwin BH, Gaye K, Nakamura I, Korba BE, Tennant BC, Gerin JL. Temporal pathogenesis of experimental neonatal woodchuck hepatitis virus infection: increased initial viral load and decreased severity of acute hepatitis during the development of chronic viral infection. Hepatology 2000;32:807-17.
148. Snyder RL, Tyler G, Summers J. Chronic hepatitis and hepatocellular carcinoma associated with woodchuck hepatitis virus. Am J Pathol 1982;107:422-5.
149. Marion PL, Knight SS, Ho BK, Guo YY, Robinson WS, Popper H. Liver disease associated with duck hepatitis B virus infection of domestic ducks. Proc Natl Acad Sci U S A 1984;81:898-902.
150. Kock J, Nassal M, MacNelly S, Baumert TF, Blum HE, von Weizsäcker F. Efficient infection of primary tupaia hepatocytes with purified human and woolly monkey hepatitis B virus. J Virol 2001;75:5084-9.
151. Tsukiyama-Kohara K, Kohara M. Tupaia belangeri as an experimental animal model for viral infection. Exp Anim 2014;63:367-74.
152. Yang EB, Cao J, Su JJ, Chow P. The tree shrews: useful animal models for the viral hepatitis and hepatocellular carcinoma. Hepatogastroenterology 2005;52:613-6.
153. Zhao X, Tang ZY Klumpp B, Wolff-Vorbeck G, Barth H, Levy S, von Weizsäcker F, Blum HE, Baumert TF. Primary hepatocytes of tupaia belangeri as a potential model for hepatitis C virus infection. J Clin Invest 2002;109:221-32.
154. Li Y, Wan D, Wei W, Su J, Cao J, Qiu X, Ou C, Ban K, Yang C, Yue H. Candidate genes responsible for human hepatocellular carcinoma identified from differentially expressed genes in hepatocarcinogenesis of the tree shrew (tupaia belangeri chinesis). Hepatol Res 2008;38:85-95.
155. Meier A, Mehrle S, Weiss TS, Mier W, Urban S. Myristoylated PreS1-domain of the hepatitis B virus L-protein mediates specific binding to differentiated hepatocytes. Hepatology 2013;58:31-42.
156. Yang C, Ruan P, Ou C, Su J, Cao J, Luo C, Tang Y, Wang Q, Qin H, Sun W, Li Y. Chronic hepatitis B virus infection and occurrence of hepatocellular carcinoma in tree shrews (tupaia belangeri chinensis). Virol J 2015;12:26.
157. Cheng L, Li F, Bility MT, Murphy CM, Su L. Modeling hepatitis B virus infection, immunopathology and therapy in mice. Antiviral Res 2015;121:1-8.
158. Barker LF, Chisari FV, McGrath PP, Dalgard DW, Kirschstein RL, Almeida JD, Edington TS, Sharp DG, Peterson MR. Transmission of type B viral hepatitis to chimpanzees. J Infect Dis 1973;127:648-62.
159. Shouval D, Chakraborty PR, Ruiz-Opazo N, Baum S, Spigland I, Muchmore E, Gerber MA, Thung SN, Popper H, Shafritz DA. Chronic hepatitis in chimpanzee carriers of hepatitis B virus: morphologic, immunologic, and viral DNA studies. Proc Natl Acad Sci U S A 1980;77:6147-51.
160. Wieland S, Thimme R, Purcell RH, Chisari FV. Genomic analysis of the host response to hepatitis B virus infection. Proc Natl Acad Sci U S A 2004;101:6669-74.
161. Thimme R, Wieland S, Steiger C, Ghrayeb J, Reimann KA, Purcell RH, Chisari FV. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J Virol 2003;77:68-76.
162. Wieland SF, Asabe S, Engle RE, Purcell RH, Chisari FV. Limited hepatitis B virus replication space in the chronically hepatitis C virus-infected liver. J Virol 2014;88:5184-8.
163. Asabe S, Wieland SF, Chattopadhyay PK, Roederer M, Engle RE, Purcell RH, Chisari FV. The size of the viral inoculum contributes to the outcome of hepatitis B virus infection. J Virol 2009;83:9652-62.
164. Murray JM, Purcell RH, Wieland SF. The half-life of hepatitis B virions. Hepatology 2006;44:1117-21.
165. Guidotti LG, Ishikawa T, Hobbs MV, Matzke B, Schreiber R, Chisari FV. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity 1996;4:25-36.
166. Heise T, Guidotti LG, Cavanaugh VJ, Chisari FV. Hepatitis B virus RNA-binding proteins associated with cytokine-induced clearance of viral RNA from the liver of transgenic mice. J Virol 1999;73:474-81.
167. Lanford RE, Guerra B, Chavez D, Giavedoni L, Hodara VL, Brasky KM, Fosdick A, Frey CR, Zheng J, Wolfgang G, Halcomb RL, Tumas DB. GS-9620, an oral agonist of toll-like receptor-7, induces prolonged suppression of hepatitis B virus in chronically infected chimpanzees. Gastroenterology 2013;144:1508-17.
168. Kamili S, Sozzi V, Thompson G, Campbell K, Walker CM, Locarnini S, Krawczynski K. Efficacy of hepatitis B vaccine against antiviral drug-resistant hepatitis B virus mutants in the chimpanzee model. Hepatology 2009;49:1483-91.
169. Payette PJ, Ma X, Weeratna RD, McCluskie MJ, Shapiro M, Engle RE, Davis HL, Purcell RH. Testing of CpG-optimized protein and DNA vaccines against the hepatitis B virus in chimpanzees for immunogenicity and protection from challenge. Intervirology 2006;49:144-51.
170. Prince AM, Brotman B, Purcell RH, Gerin JL. A final report on safety and immunogenicity of a bivalent aqueous subunit HBV vaccine. J Med Virol 1985;15:399-419.
172. Dandri M, Lutgehetmann M. Mouse models of hepatitis B and delta virus infection. J Immunol Methods 2014;410:39-49.
173. Na B, Huang Z, Wang Q, Qi Z, Tian Y, Lu CC, Yu J, Hanes MA, Kakar S, Huang EJ, Ou JH, Liu L, Yen TS. Transgenic expression of entire hepatitis B virus in mice induces hepatocarcinogenesis independent of chronic liver injury. PloS One 2011;6:e26240.
174. Lütgehetmann M, Mancke LV, Volz T, Helbig M, Allweiss L, Bornscheuer T, Pollok JM, Lohse AW, Petersen J, Urban S, Dandri M. Humanized chimeric uPA mouse model for the study of hepatitis B and D virus interactions and preclinical drug evaluation. Hepatology 2012;55:685-94.
175. Bility MT, Cheng L, Zhang Z, Luan Y, Li F, Chi L, Zhang L, Tu Z, Gao Y, Fu Y, Niu J, Wang F, Su L. Hepatitis B virus infection and immunopathogenesis in a humanized mouse model: induction of human-specific liver fibrosis and M2-like macrophages. PLoS Pathog 2014;10:e1004032.
176. Giersch K, Allweiss L, Volz T, Helbig M, Bierwolf J, Lohse AW, Pollok JM, Petersen J, Dandri M, Lütgehetmann M. Hepatitis Delta co-infection in humanized mice leads to pronounced induction of innate immune responses in comparison to HBV mono-infection. J Hepatol 2015;63:346-53.
177. Volz T, Allweiss L, Ben MBarek M, Warlich M, Lohse AW, Pollok JM, Alexandrov A, Urban S, Petersen J, Lütgehetmann M, Dandri M. The entry inhibitor Myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus. J Hepatol 2013;58:861-7.
178. Brezillon N, Brunelle MN, Massinet H, Giang E, Lamant C, DaSilva L, Berissi S, Belghiti J, Hannoun L, Puerstinger G, Wimmer E, Neyts J, Hantz O, Soussan P, Morosan S, Kremsdorf D. Antiviral activity of Bay 41-4109 on hepatitis B virus in humanized Alb-uPA/SCID mice. PloS One 2011;6:e25096.
179. Redelsperger F, Lekbaby B, Mandouri Y, Giang E, Duriez M, Desire N, Roque Afonso AM, Brichler S, Dubreuil P, Dobrin A, Perlemuter G, Prevot S, Bacon N, Grange JD, Zatla F, Le Pendeven C, Pol S, Strick-Marchand H, Di Santo J, Kremsdorf D, Soussan P. Production of hepatitis B defective particles is dependent on liver status. Virology 2012;431:21-8.
180. Oehler N, Volz T, Bhadra OD, Kah J, Allweiss L, Giersch K, Bierwolf J, Riecken K, Pollok JM, Lohse AW, Fehse B, Petersen J, Urban S, Lütgehetmann M, Heeren J, Dandri M. Binding of hepatitis B virus to its cellular receptor alters the expression profile of genes of bile acid metabolism. Hepatology 2014;60:1483-93.
181. Belloni L, Allweiss L, Guerrieri F, Pediconi N, Volz T, Pollicino T, Petersen J, Raimondo G, Dandri M, Levrero M. IFN-α inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J Clin Invest 2012;122:529-37.
182. Ni Y, Lempp FA, Mehrle S, Nkongolo S, Kaufman C, Fälth M, Stindt J, Königer C, Nassal M, Kubitz R, Sültmann H, Urban S. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 2014;146:1070-83.
183. Li H, Zhuang Q, Wang Y, Zhang T, Zhao J, Zhang Y, Zhang J, Lin Y, Yuan Q, Xia N, Han J. HBV life cycle is restricted in mouse hepatocytes expressing human NTCP. Cell Mol Immunol 2014;11:175-83.
184. He W, Ren B, Mao F, Jing Z, Li Y, Liu Y, Peng B, Yan H, Qi Y, Sun Y, Guo JT, Sui J, Wang F, Li W. Hepatitis D virus infection of mice expressing human sodium taurocholate co-transporting polypeptide. PLoS Pathog 2015;11:e1004840.
185. Gearhart TL, Bouchard MJ. The hepatitis B virus HBx protein modulates cell cycle regulatory proteins in cultured primary human hepatocytes. Virus Res 2011;155:363-7.
186. Lamontagne J, Mell JC, Bouchard MJ. Transcriptome-wide analysis of hepatitis B virus-mediated changes to normal hepatocyte gene expression. PLoS Pathog 2016;12:e1005438.
187. Alexander JJ, Bey EM, Geddes EW, Lecatsas G. Establishment of a continuously growing cell line from primary carcinoma of the liver. S Afr Med J 1976;50:2124-8.
188. Knowles BB, Howe CC, Aden DP. Human hepatocellular carcinoma cell lines secrete the major plasma proteins and hepatitis B surface antigen. Science 1980;209:497-9.
189. Aden DP, Fogel A, Plotkin S, Damjanov I, Knowles BB. Controlled synthesis of HBsAg in a differentiated human liver carcinoma-derived cell line. Nature 1979;282:615-6.
190. Shaul Y, Ziemer M, Garcia PD, Crawford R, Hsu H, Valenzuela P, Rutter WJ. Cloning and analysis of integrated hepatitis virus sequences from a human hepatoma cell line. J Virol 1984;51:776-87.
191. Edman JC, Gray P, Valenzuela P, Rall LB, Rutter WJ. Integration of hepatitis B virus sequences and their expression in a human hepatoma cell. Nature 1980;286:535-8.
192. Twist EM, Clark HF, Aden DP, Knowles BB, Plotkin SA. Integration pattern of hepatitis B virus DNA sequences in human hepatoma cell lines. J Virol 1981;37:239-43.
193. Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res 1982;42:3858-63.
194. Chang CM, Jeng KS, Hu CP, Lo SJ, Su TS, Ting LP, Chou CK, Han SH, Pfaff E, Salfeld J. Production of hepatitis B virus in vitro by transient expression of cloned HBV DNA in a hepatoma cell line. EMBO J 1987;6:675-80.
195. Yaginuma K, Shirakata Y, Kobayashi M, Koike K. Hepatitis B virus (HBV) particles are produced in a cell culture system by transient expression of transfected HBV DNA. Proc Natl Acad Sci U S A 1987;84:2678-82.
196. Sells MA, Chen ML, Acs G. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc Natl Acad Sci U S A 1987;84:1005-9.
197. Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, Seeger C, King RW. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother 1997;41:1715-20.
198. Chen JL, Wu M, Liu K, Zhang W, Li Y, Zhou XH, Bai L, Yuan ZH. New insights into Hepatitis B virus biology and implications for novel antiviral strategies. Natl Sci Rev 2015;2:296-313.
199. Jiang Y, Wang AH, Shao LH, Wang G, Yao YY, Sai LT, Chen FZ, Zheng F, Li Y, Ma LX. A new cell culture system for infection with hepatitis B virus that fuses HepG2 cells with primary human hepatocytes. J Int Med Res 2009;37:650-61.
200. Sai LT, Yao YY, Guan YY, Shao LH, Ma RP, Ma LX. Hepatitis B virus infection and replication in a new cell culture system established by fusing HepG2 cells with primary human hepatocytes. J Microbiol Immunol Infect 2014; doi: 10.1016/j.jmii.2014.08.008.
201. Gripon P, Rumin S, Urban S, Le Seyec J, Glaise D, Cannie I, Guyomard C, Lucas J, Trepo C, Guguen-Guillouzo C. Infection of a human hepatoma cell line by hepatitis B virus. Proc Natl Acad Sci U S A 2002;99:15655-60.
202. Yan R, Zhang Y, Cai D, Liu Y, Cuconati A, Guo H. Spinoculation enhances HBV infection in NTCP-reconstituted hepatocytes. PloS One 2015;10:e0129889.
203. Zeisel MB, Lucifora J, Mason WS, Sureau C, Beck J, Levrero M, Kann M, Knolle PA, Benkirane M, Durantel D, Michel ML, Autran B, Cosset FL, Strick-Marchand H, Trépo C, Kao JH, Carrat F, Lacombe K, Schinazi RF, Barre-Sinoussi F, Delfraissy JF, Zoulim F. Towards an HBV cure: state-of-the-art and unresolved questions--report of the ANRS workshop on HBV cure. Gut 2015;64:1314-26.
204. Nkongolo S, Ni Y, Lempp FA, Kaufman C, Lindner T, Esser-Nobis K, Lohmann V, Mier W, Mehrle S, Urban S. Cyclosporin A inhibits hepatitis B and hepatitis D virus entry by cyclophilin-independent interference with the NTCP receptor. J Hepatol 2014;60:723-31.
205. Yan H, Liu Y, Sui J, Li W. NTCP opens the door for hepatitis B virus infection. Antiviral Res 2015;121:24-30.
206. Shlomai A, Schwartz RE, Ramanan V, Bhatta A, de Jong YP, Bhatia SN, Rice CM. Modeling host interactions with hepatitis B virus using primary and induced pluripotent stem cell-derived hepatocellular systems. Proc Natl Acad Sci U S A 2014;111:12193-8.
207. Neuveut C, Wei Y, Buendia MA. Mechanisms of HBV-related hepatocarcinogenesis. J Hepatol 2010;52:594-604.
208. Clippinger AJ, Bouchard MJ. Hepatitis B virus HBx protein localizes to mitochondria in primary rat hepatocytes and modulates mitochondrial membrane potential. J Virol 2008;82:6798-811.
209. Chen CJ, Yang HI. Natural history of chronic hepatitis B revealed. J Gastroenterol Hepatol 2011;26:628-38.
210. Guy CS, Mulrooney-Cousins PM, Churchill ND, Michalak TI. Intrahepatic expression of genes affiliated with innate and adaptive immune responses immediately after invasion and during acute infection with woodchuck hepadnavirus. J Virol 2008;82:8579-91.
211. Wieland SF, Spangenberg HC, Thimme R, Purcell RH, Chisari FV. Expansion and contraction of the hepatitis B virus transcriptional template in infected chimpanzees. Proc Natl Acad Sci U S A 2004;101:2129-34.
212. Summers J, Mason WS. Residual integrated viral DNA after hepadnavirus clearance by nucleoside analog therapy. Proc Natl Acad Sci U S A 2004;101:638-40.
213. Schultz U, Summers J, Staeheli P, Chisari FV. Elimination of duck hepatitis B virus RNA-containing capsids in duck interferon-alpha-treated hepatocytes. J Virol 1999;73:5459-65.
214. Guo JT, Pryce M, Wang X, Barrasa MI, Hu J, Seeger C. Conditional replication of duck hepatitis B virus in hepatoma cells. J Virol 2003;77:1885-93.
215. Yim HJ, Lok AS. Natural history of chronic hepatitis B virus infection: what we knew in 1981 and what we know in 2005. Hepatology 2006;43:S173-81.
216. Blackberg J, Kidd-Ljunggren K. Occult hepatitis B virus after acute self-limited infection persisting for 30 years without sequence variation. J Hepatol 2000;33:992-7.
217. Chazouillères O, Mamish D, Kim M, Carey K, Ferrell L, Roberts JP, Ascher NL, Wright TL. "Occult" hepatitis B virus as source of infection in liver transplant recipients. Lancet 1994;343:142-6.
218. Pollicino T, Squadrito G, Cerenzia G, Cacciola I, Raffa G, Craxi A, Farinati F, Missale G, Smedile A, Tiribelli C, Villa E, Raimondo G. Hepatitis B virus maintains its pro-oncogenic properties in the case of occult HBV infection. Gastroenterology 2004;126:102-10.
219. Gish RG, Given BD, Lai CL, Locarnini SA, Lau JY, Lewis DL, Schluep T. Chronic hepatitis B: virology, natural history, current management and a glimpse at future opportunities. Antiviral Res 2015;121:47-58.
220. Chang ML, Liaw YF. Hepatitis B flares in chronic hepatitis B: pathogenesis, natural course, and management. J Hepatol 2014;61:1407-17.
221. Liaw YF, Tai DI, Chu CM, Chen TJ. The development of cirrhosis in patients with chronic type B hepatitis: a prospective study. Hepatology 1988;8:493-6.
222. Globocan 2012 v1.0 [Internet]. IARC CancerBase No. 11. 2013. Available from: http://globocan.iarc.fr.
223. El-Serag HB. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012;142:1264-73.
224. El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 2007;132:2557-76.
225. Block TM, Mehta AS, Fimmel CJ, Jordan R. Molecular viral oncology of hepatocellular carcinoma. Oncogene 2003;22:5093-107.
226. McClune AC, Tong MJ. Chronic hepatitis B and hepatocellular carcinoma. Clin Liver Dis 2010;14:461-76.
227. Bagga S, Bouchard MJ. Cell cycle regulation during viral infection. Methods Mol Biol 2014;1170:165-227.
228. Casciano JC, Bagga S, Yang B, Bouchard MJ. Modulation of cell proliferation pathways by the hepatitis B virus X protein: a potential contributor to the development of hepatocellular carcinoma. In: Lau WY, editor. Hepatocellular carcinoma - basic research. Shanghai, China: InTech; 2011:103-53.
229. Rawat S, Clippinger AJ, Bouchard MJ. Modulation of apoptotic signaling by the hepatitis B virus X protein. Viruses 2012;4:2945-72.
230. Lamontagne J, Steel LF, Bouchard MJ. Hepatitis B virus and microRNAs: complex interactions affecting hepatitis B virus replication and hepatitis B virus-associated diseases. World J Gastroenterol 2015;21:7375-99.
231. Panteva M, Korkaya H, Jameel S. Hepatitis viruses and the MAPK pathway: is this a survival strategy? Virus Res 2003;92:131-40.
232. Wang Z, Jin W, Jin H, Wang X. mTOR in viral hepatitis and hepatocellular carcinoma: function and treatment. Biomed Res Int 2014;2014:735672.
233. Guo H, Zhou T, Jiang D, Cuconati A, Xiao GH, Block TM, Guo JT. Regulation of hepatitis B virus replication by the phosphatidylinositol 3-kinase-akt signal transduction pathway. J Virol 2007;81:10072-80.
234. Friedrich B, Wollersheim M, Brandenburg B, Foerste R, Will H, Hildt E. Induction of anti-proliferative mechanisms in hepatitis B virus producing cells. J Hepatol 2005;43:696-703.
235. Wang CM, Wang Y, Fan CG, Xu FF, Sun WS, Liu YG, Jia JH. miR-29c targets TNFAIP3, inhibits cell proliferation and induces apoptosis in hepatitis B virus-related hepatocellular carcinoma. Biochem Biophys Res Commun 2011;411:586-92.
236. Chin R, Earnest-Silveira L, Koeberlein B, Franz S, Zentgraf H, Dong X, Gowans E, Bock CT, Torresi J. Modulation of MAPK pathways and cell cycle by replicating hepatitis B virus: factors contributing to hepatocarcinogenesis. J Hepatol 2007;47:325-37.
237. Huang YQ, Wang LW, Yan SN, Gong ZJ. Effects of cell cycle on telomerase activity and on hepatitis B virus replication in HepG2 2.2.15 cells. Hepatobiliary Pancreat Dis Int 2004;3:543-7.
238. Ozer A, Khaoustov VI, Mearns M, Lewis DE, Genta RM, Darlington GJ, Yoffe B. Effect of hepatocyte proliferation and cellular DNA synthesis on hepatitis B virus replication. Gastroenterology 1996;110:1519-28.
239. Qiao L, Leach K, McKinstry R, Gilfor D, Yacoub A, Park JS, Grant S, Hylemon PB, Fisher PB, Dent P. Hepatitis B virus X protein increases expression of p21(Cip-1/WAF1/MDA6) and p27(Kip-1) in primary mouse hepatocytes, leading to reduced cell cycle progression. Hepatology 2001;34:906-17.
240. Shlomai A, Shaul Y. The "metabolovirus" model of hepatitis B virus suggests nutritional therapy as an effective anti-viral weapon. Med Hypotheses 2008;71:53-7.
241. Popescu CI, Riva L, Vlaicu O, Farhat R, Rouille Y, Dubuisson J. Hepatitis C virus life cycle and lipid metabolism. Biology (Basel) 2014;3:892-921.
242. Yan H, Peng B, Liu Y, Xu G, He W, Ren B, Jing Z, Sui J, Li W. Viral entry of hepatitis B and D viruses and bile salts transportation share common molecular determinants on sodium taurocholate cotransporting polypeptide. J Virol 2014;88:3273-84.
243. Reese VC, Moore DD, McLachlan A. Limited effects of bile acids and small heterodimer partner on hepatitis B virus biosynthesis in vivo. J Virol 2012;86:2760-8.
244. Waris G, Siddiqui A. Interaction between STAT-3 and HNF-3 leads to the activation of liver-specific hepatitis B virus enhancer 1 function. J Virol 2002;76:2721-9.
245. Li L, Oropeza CE, Kaestner KH, McLachlan A. Limited effects of fasting on hepatitis B virus (HBV) biosynthesis in HBV transgenic mice. J Virol 2009;83:1682-8.
246. Ondracek CR, Reese VC, Rushing CN, Oropeza CE, McLachlan A. Distinct regulation of hepatitis B virus biosynthesis by peroxisome proliferator-activated receptor gamma coactivator 1alpha and small heterodimer partner in human hepatoma cell lines. J Virol 2009;83:12545-51.
247. Reese V, Ondracek C, Rushing C, Li L, Oropeza CE, McLachlan A. Multiple nuclear receptors may regulate hepatitis B virus biosynthesis during development. Int J Biochem Cell Biol 2011;43:230-7.
248. Curtil C, Enache LS, Radreau P, Dron AG, Scholtès C, Deloire A, Roche D, Lotteau V, Andre P, Ramière C. The metabolic sensors FXRα, PGC-1α, and SIRT1 cooperatively regulate hepatitis B virus transcription. FASEB J 2014;28:1454-63.
249. Reese VC, Oropeza CE, McLachlan A. Independent activation of hepatitis B virus biosynthesis by retinoids, peroxisome proliferators, and bile acids. J Virol 2013;87:991-7.
250. Ramière C, Scholtès C, Diaz O, Icard V, Perrin-Cocon L, Trabaud MA, Lotteau V, André P. Transactivation of the hepatitis B virus core promoter by the nuclear receptor FXRalpha. J Virol 2008;82:10832-40.
251. Kim HY, Cho HK, Choi YH, Lee KS, Cheong J. Bile acids increase hepatitis B virus gene expression and inhibit interferon-alpha activity. FEBS J 2010;277:2791-802.
252. Shlomai A, Paran N, Shaul Y. PGC-1alpha controls hepatitis B virus through nutritional signals. Proc Natl Acad Sci U S A 2006;103:16003-8.
253. Shin HJ, Park YH, Kim SU, Moon HB, Park do S, Han YH, Lee CH, Lee DS, Song IS, Lee DH, Kim M, Kim NS, Kim DG, Kim JM, Kim SK, Kim YN, Kim SS, Choi CS, Kim YB, Yu DY. Hepatitis B virus X protein regulates hepatic glucose homeostasis via activation of inducible nitric oxide synthase. J Biol Chem 2011;286:29872-81.
254. Jagya N, Varma SP, Thakral D, Joshi P, Durgapal H, Panda SK. RNA-seq based transcriptome analysis of hepatitis E virus (HEV) and hepatitis B virus (HBV) replicon transfected Huh-7 cells. PLoS One 2014;9:e87835.
255. Jhuang HJ, Hsu WH, Lin KT, Hsu SL, Wang FS, Chou CK, Lee KH, Tsou AP, Lai JM, Yeh SF, Huang CY. Gluconeogenesis, lipogenesis, and HBV replication are commonly regulated by PGC-1α-dependent pathway. Oncotarget 2015;6:7788-803.
256. Benn J, Schneider RJ. Hepatitis B virus HBx protein activates Ras-GTP complex formation and establishes a Ras, Raf, MAP kinase signaling cascade. Proc Natl Acad Sci U S A 1994;91:10350-4.
257. Doria M, Klein N, Lucito R, Schneider RJ. The hepatitis B virus HBx protein is a dual specificity cytoplasmic activator of Ras and nuclear activator of transcription factors. EMBO J 1995;14:4747-57.
258. Gehart H, Kumpf S, Ittner A, Ricci R. MAPK signalling in cellular metabolism: stress or wellness? EMBO Rep 2010;11:834-40.
259. Luber B, Lauer U, Weiss L, Hohne M, Hofschneider PH, Kekulé AS. The hepatitis B virus transactivator HBx causes elevation of diacylglycerol and activation of protein kinase C. Res Virol 1993;144:311-21.
260. Zhu M, Guo J, Li W, Lu Y, Fu S, Xie X, Xia H, Dong X, Chen Y, Quan M, Zheng S, Xie K, Li M. Hepatitis B virus X protein induces expression of alpha-fetoprotein and activates PI3K/mTOR signaling pathway in liver cells. Oncotarget 2015;6:12196-208.
261. Teng CF, Hsieh WC, Wu HC, Lin YJ, Tsai HW, Huang W, Su IJ. Hepatitis B virus Pre-S2 mutant induces aerobic glycolysis through mammalian target of rapamycin signal cascade. PLoS One 2015;10:e0122373.
262. Teng CF, Wu HC, Hsieh WC, Tsai HW, Su IJ. Activation of ATP citrate lyase by mTOR signal induces disturbed lipid metabolism in hepatitis B virus pre-S2 mutant tumorigenesis. J Virol 2015;89:605-14.
263. Teng CF, Wu HC, Tsai HW, Shiah HS, Huang W, Su IJ. Novel feedback inhibition of surface antigen synthesis by mammalian target of rapamycin (mTOR) signal and its implication for hepatitis B virus tumorigenesis and therapy. Hepatology 2011;54:1199-207.
264. Wang CC, Tseng TC, Kao JH. Hepatitis B virus infection and metabolic syndrome: fact or fiction? J Gastroenterol Hepatol 2015;30:14-20.
265. Diao J, Khine AA, Sarangi F, Hsu E, Iorio C, Tibbles LA, Woodgett JR, Penninger J, Richardson CD. X protein of hepatitis B virus inhibits Fas-mediated apoptosis and is associated with up-regulation of the SAPK/JNK pathway. J Biol Chem 2001;276:8328-40.
266. Elmore LW, Hancock AR, Chang SF, Wang XW, Chang S, Callahan CP, Geller DA, Will H, Harris CC. Hepatitis B virus X protein and p53 tumor suppressor interactions in the modulation of apoptosis. Proc Natl Acad Sci U S A 1997;94:14707-12.
267. Gottlob K, Fulco M, Levrero M, Graessmann A. The hepatitis B virus HBx protein inhibits caspase 3 activity. J Biol Chem 1998;273:33347-53.
268. Lee YI, Kang-Park S, Do SI, Lee YI. The hepatitis B virus-X protein activates a phosphatidylinositol 3-kinase-dependent survival signaling cascade. J Biol Chem 2001;276:16969-77.
269. Pan J, Duan LX, Sun BS, Feitelson MA. Hepatitis B virus X protein protects against anti-Fas-mediated apoptosis in human liver cells by inducing NF-kappa B. J Gen Virol 2001;82:171-82.
270. Klein A, Guhl E, Tzeng YJ, Fuhrhop J, Levrero M, Graessmann M, Graessmann A. HBX causes cyclin D1 overexpression and development of breast cancer in transgenic animals that are heterozygous for p53. Oncogene 2003;22:2910-9.
271. Madden CR, Finegold MJ, Slagle BL. Expression of hepatitis B virus X protein does not alter the accumulation of spontaneous mutations in transgenic mice. J Virol 2000;74:5266-72.
272. Bergametti F, Prigent S, Luber B, Benoit A, Tiollais P, Sarasin A, Transy C. The proapoptotic effect of hepatitis B virus HBx protein correlates with its transactivation activity in stably transfected cell lines. Oncogene 1999;18:2860-71.
273. Chirillo P, Pagano S, Natoli G, Puri PL, Burgio VL, Balsano C, Levrero M. The hepatitis B virus X gene induces p53-mediated programmed cell death. Proc Natl Acad Sci U S A 1997;94:8162-7.
274. Kim H, Lee H, Yun Y. X-gene product of hepatitis B virus induces apoptosis in liver cells. J Biol Chem 1998;273:381-5.
275. Su F, Theodosis CN, Schneider RJ. Role of NF-kappaB and myc proteins in apoptosis induced by hepatitis B virus HBx protein. J Virol 2001;75:215-25.
276. Terradillos O, de La Coste A, Pollicino T, Neuveut C, Sitterlin D, Lecoeur H, Gougeon ML, Kahn A, Buendia MA. The hepatitis B virus X protein abrogates Bcl-2-mediated protection against Fas apoptosis in the liver. Oncogene 2002;21:377-86.
277. Lee YI, Hwang JM, Im JH, Lee YI, Kim NS, Kim DG, Yu DY, Moon HB, Park SK. Human hepatitis B virus-X protein alters mitochondrial function and physiology in human liver cells. J Biol Chem 2004;279:15460-71.
278. Su F, Schneider RJ. Hepatitis B virus HBx protein sensitizes cells to apoptotic killing by tumor necrosis factor alpha. Proc Natl Acad Sci U S A 1997;94:8744-9.
279. Wang WH, Gregori G, Hullinger RL, Andrisani OM. Sustained activation of p38 mitogen-activated protein kinase and c-Jun N-terminal kinase pathways by hepatitis B virus X protein mediates apoptosis via induction of Fas/FasL and tumor necrosis factor (TNF) receptor 1/TNF-alpha expression. Mol Cell Biol 2004;24:10352-65.
280. Benhenda S, Cougot D, Buendia MA, Neuveut C. Hepatitis B virus X protein molecular functions and its role in virus life cycle and pathogenesis. Adv Cancer Res 2009;103:75-109.
281. Ng SA, Lee C. Hepatitis B virus X gene and hepatocarcinogenesis. J Gastroenterol 2011;46:974-90.
282. Arzberger S, Hösel M, Protzer U. Apoptosis of hepatitis B virus-infected hepatocytes prevents release of infectious virus. J Virol 2010;84:11994-2001.
283. Brenner DA. Signal transduction during liver regeneration. J Gastroenterol Hepatol 1998;13:S93-5.
284. FitzGerald MJ, Webber EM, Donovan JR, Fausto N. Rapid DNA binding by nuclear factor kappa B in hepatocytes at the start of liver regeneration. Cell Growth Differ 1995;6:417-27.
285. Koike K, Moriya K, Yotsuyanagi H, Shintani Y, Fujie H, Tsutsumi T, Kimura S. Compensatory apoptosis in preneoplastic liver of a transgenic mouse model for viral hepatocarcinogenesis. Cancer Lett 1998;134:181-6.
286. Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005;309:1577-81.
287. Henke JI, Goergen D, Zheng J, Song Y, Schuttler CG, Fehr C, Junemann C, Niepmann M. MicroRNA-122 stimulates translation of hepatitis C virus RNA. EMBO J 2008;27:3300-10.
288. Kitab B, Alj HS, Ezzikouri S, Benjelloun S. MicroRNAs as important players in host-hepatitis b virus interactions. J Clin Transl Hepatol 2015;3:149-61.
289. Hou J, Lin L, Zhou W, Wang Z, Ding G, Dong Q, Qin L, Wu X, Zheng Y, Yang Y, Tian W, Zhang Q, Wang C, Zhang Q, Zhuang SM, Zheng L, Liang A, Tao W, Cao X. Identification of miRNomes in human liver and hepatocellular carcinoma reveals miR-199a/b-3p as therapeutic target for hepatocellular carcinoma. Cancer cell 2011;19:232-43.
290. Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol 2002;12:735-9.
291. Chen Y, Shen A, Rider PJ, Yu Y, Wu K, Mu Y, Hao Q, Liu Y, Gong H, Zhu Y, Liu F, Wu J. A liver-specific microRNA binds to a highly conserved RNA sequence of hepatitis B virus and negatively regulates viral gene expression and replication. FASEB J 2011;25:4511-21.
292. Li C, Wang Y, Wang S, Wu B, Hao J, Fan H, Ju Y, Ding Y, Chen L, Chu X, Liu W, Ye X, Meng S. Hepatitis B virus mRNA-mediated miR-122 inhibition upregulates PTTG1-binding protein, which promotes hepatocellular carcinoma tumor growth and cell invasion. J Virol 2013;87:2193-205.
293. Qiu L, Fan H, Jin W, Zhao B, Wang Y, Ju Y, Chen L, Chen Y, Duan Z, Meng S. MiR-122-induced down-regulation of HO-1 negatively affects miR-122-mediated suppression of HBV. Biochem Biophys Res Commun 2010;398:771-7.
294. Fornari F, Gramantieri L, Giovannini C, Veronese A, Ferracin M, Sabbioni S, Calin GA, Grazi GL, Croce CM, Tavolari S, Chieco P, Negrini M, Bolondi L. MiR-122/cyclin G1 interaction modulates p53 activity and affects doxorubicin sensitivity of human hepatocarcinoma cells. Cancer Res 2009;69:5761-7.
295. Gramantieri L, Ferracin M, Fornari F, Veronese A, Sabbioni S, Liu CG, Calin GA, Giovannini C, Ferrazzi E, Grazi GL, Croce CM, Bolondi L, Negrini M. Cyclin G1 is a target of miR-122a, a microRNA frequently down-regulated in human hepatocellular carcinoma. Cancer Res 2007;67:6092-9.
296. Fan CG, Wang CM, Tian C, Wang Y, Li L, Sun WS, Li RF, Liu YG. MiR-122 inhibits viral replication and cell proliferation in hepatitis B virus-related hepatocellular carcinoma and targets NDRG3. Oncol Rep 2011;26:1281-6.
297. Peng F, Xiao X, Jiang Y, Luo K, Tian Y, Peng M, Zhang M, Xu Y, Gong G. HBx down-regulated Gld2 plays a critical role in hbv-related dysregulation of miR-122. PloS One 2014;9:e92998.
298. Song K, Han C, Zhang J, Lu D, Dash S, Feitelson M, Lim K, Wu T. Epigenetic regulation of microRNA-122 by peroxisome proliferator activated receptor-gamma and hepatitis b virus X protein in hepatocellular carcinoma cells. Hepatology 2013;58:1681-92.
299. Wang S, Qiu L, Yan X, Jin W, Wang Y, Chen L, Wu E, Ye X, Gao GF, Wang F, Chen Y, Duan Z, Meng S. Loss of microRNA 122 expression in patients with hepatitis B enhances hepatitis B virus replication through cyclin G(1) -modulated P53 activity. Hepatology 2012;55:730-41.
300. Denzler R, Agarwal V, Stefano J, Bartel DP, Stoffel M. Assessing the ceRNA hypothesis with quantitative measurements of mirna and target abundance. Mol Cell 2014;54:766-76.
301. Xu G, Gao Z, He W, Ma Y, Feng X, Cai T, Lu F, Liu L, Li W. MicroRNA expression in hepatitis B virus infected primary treeshrew hepatocytes and the independence of intracellular miR-122 level for de novo HBV infection in culture. Virology 2014;448:247-54.
302. Thornton JE, Gregory RI. How does Lin28 let-7 control development and disease? Trends Cell Biol 2012;22:474-82.
303. Guo H, Liu H, Mitchelson K, Rao H, Luo M, Xie L, Sun Y, Zhang L, Lu Y, Liu R, Ren A, Liu S, Zhou S, Zhu J, Zhou Y, Huang A, Wei L, Guo Y, Cheng J. MicroRNAs-372/373 promote the expression of hepatitis B virus through the targeting of nuclear factor I/B. Hepatology 2011;54:808-19.
304. Jin J, Tang S, Xia L, Du R, Xie H, Song J, Fan R, Bi Q, Chen Z, Yang G, Liu J, Shi Y, Fan D. MicroRNA-501 promotes HBV replication by targeting HBXIP. Biochem Biophys Res Commun 2013;430:1228-33.
305. Wang Y, Jiang L, Ji X, Yang B, Zhang Y, Fu XD. Hepatitis B viral RNA directly mediates down-regulation of the tumor suppressor microRNA miR-15a/miR-16-1 in hepatocytes. J Biol Chem 2013;288:18484-93.
306. Wang Y, Lu Y, Toh ST, Sung WK, Tan P, Chow P, Chung AY, Jooi LL, Lee CG. Lethal-7 is down-regulated by the hepatitis B virus x protein and targets signal transducer and activator of transcription 3. J Hepatol 2010;53:57-66.
307. Zhang ZZ, Liu X, Wang DQ, Teng MK, Niu LW, Huang AL, Liang Z. Hepatitis B virus and hepatocellular carcinoma at the miRNA level. World J Gastroenterol 2011;17:3353-8.
308. Dai X, Zhang W, Zhang H, Sun S, Yu H, Guo Y, Kou Z, Zhao G, Du L, Jiang S, Zhang J, Li J, Zhou Y. Modulation of HBV replication by microRNA-15b through targeting hepatocyte nuclear factor 1α. Nucleic Acids Res 2014;42:6578-90.
309. Gramantieri L, Fornari F, Callegari E, Sabbioni S, Lanza G, Croce CM, Bolondi L, Negrini M. MicroRNA involvement in hepatocellular carcinoma. J Cell Mol Med 2008;12:2189-204.
310. Liu Y, Zhao JJ, Wang CM, Li MY, Han P, Wang L, Cheng YQ, Zoulim F, Ma X, Xu DP. Altered expression profiles of microRNAs in a stable hepatitis B virus-expressing cell line. Chin Med J 2009;122:10-4.
311. Wu CS, Yen CJ, Chou RH, Chen JN, Huang WC, Wu CY, Yu YL. Downregulation of microRNA-15b by hepatitis B virus X enhances hepatocellular carcinoma proliferation via fucosyltransferase 2-induced Globo H expression. Int J Cancer 2014;134:1638-47.
312. Wu G, Yu F, Xiao Z, Xu K, Xu J, Tang W, Wang J, Song E. Hepatitis B virus X protein downregulates expression of the miR-16 family in malignant hepatocytes in vitro. Br J Cancer 2011;105:146-53.
313. Coppola N, Potenza N, Pisaturo M, Mosca N, Tonziello G, Signoriello G, Messina V, Sagnelli C, Russo A, Sagnelli E. Liver microRNA hsa-miR-125a-5p in HBV chronic infection: correlation with HBV replication and disease progression. PloS One 2013;8:e65336.
314. Mosca N, Castiello F, Coppola N, Trotta MC, Sagnelli C, Pisaturo M, Sagnelli E, Russo A, Potenza N. Functional interplay between hepatitis B virus X protein and human miR-125a in HBV infection. Biochem Biophys Res Commun 2014;449:141-5.
315. Jung YJ, Kim JW, Park SJ, Min BY, Jang ES, Kim NY, Jeong SH, Shin CM, Lee SH, Park YS, Hwang JH, Kim N, Lee DH. C-Myc-mediated overexpression of miR-17-92 suppresses replication of hepatitis B virus in human hepatoma cells. J Med Virol 2013;85:969-78.
316. Cheng PN, Liu WC, Tsai HW, Wu IC, Chang TT, Young KC. Association of intrahepatic cccDNA reduction with the improvement of liver histology in chronic hepatitis B patients receiving oral antiviral agents. J Med Virol 2011;83:602-7.
317. Ghany MG, Doo EC. Antiviral resistance and hepatitis B therapy. Hepatology 2009;49:S174-84.
318. Yang HC, Kao JH. Viral hepatitis. HBV cure--can we pin our hopes on immunotherapy? Nat Rev Gastroenterol Hepatol 2015;12:129-31.
319. Zoulim F. Antiviral therapy of chronic hepatitis B: can we clear the virus and prevent drug resistance? Antivir Chem Chemother 2004;15:299-305.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Lamontagne RJ, Bagga S, Bouchard MJ. Hepatitis B virus molecular biology and pathogenesis. Hepatoma Res 2016;2:163-86. http://dx.doi.org/10.20517/2394-5079.2016.05
AMA Style
Lamontagne RJ, Bagga S, Bouchard MJ. Hepatitis B virus molecular biology and pathogenesis. Hepatoma Research. 2016; 2: 163-86. http://dx.doi.org/10.20517/2394-5079.2016.05
Chicago/Turabian Style
Lamontagne, R. Jason, Sumedha Bagga, Michael J. Bouchard. 2016. "Hepatitis B virus molecular biology and pathogenesis" Hepatoma Research. 2: 163-86. http://dx.doi.org/10.20517/2394-5079.2016.05
ACS Style
Lamontagne, RJ.; Bagga S.; Bouchard MJ. Hepatitis B virus molecular biology and pathogenesis. Hepatoma. Res. 2016, 2, 163-86. http://dx.doi.org/10.20517/2394-5079.2016.05
About This Article
Copyright

Data & Comments
Data


 Cite This Article 179 clicks
Cite This Article 179 clicks

 Like This Article 28
likes
Like This Article 28
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.