
Physiological potential of cytokines and liver damages


Abstract
Cytokines are soluble extracellular small molecular weight protein or peptide. They are produced by virtually every nucleated cell type in response to injurious stimuli to control body metabolism, infection, inflammation and tissue or neuronal damage; therefore acting as messengers between tissues and the immune system; and participating in many physiological processes through their either anti-inflammatory or pro-inflammatory characteristics. Many cytokines have multiple cellular sources and targets, as well as many natural inducers and inhibitors. In pathophysiological conditions and during the early phase of chronic liver diseases, agent like virus, bacteria, parasites, ethanol, or toxins, induce secretion of cytokines at high levels. The presence of cytokine antagonists and soluble cytokine receptors, often released in concert with their respective cytokine agonist, presents additional complexity to interpretation.
Keywords
Introduction
Cytokines are small molecular weight proteins or peptides messengers between tissues and the immune system[1] and participate in many physiological processes.[2] They are either poor anti-inflammatory, suppress the activity and production of pro-inflammatory signals limiting inflammation and host damage; or pro-inflammatory, induce inflammation as a result of infection or injury.[3] Different cytokine combinations give rise to distinct consequences, such as inflammation, proliferation, and angiogenesis.[4]
Many cytokines have multiple cellular sources and targets, as well as many natural inducers and inhibitors.[5] They are produced to control infection, inflammation and tissue or neuronal damage. The inflammatory ones are fundamental regulators of body metabolism.[6]
On several bases, cytokines may be classified depending on their (1) cell of origin; (2) spectrum of activity; (3) the category of activity they influence; (4) the cells that are their targets; or (5) on specific features of their ligand-receptor interaction,[7] although the nomenclature is somewhat arbitrary, having arisen in different branches of biology [Table 1].[8] Extensive genetic polymorphisms have also been described, which in many cases appear to play an important part in their level of expression and have been linked to a variety of diseases,[6] as a variety of experiments has shown that either excessive or insufficient production of cytokines may contribute significantly to the pathophysiology of a range of diseases[2] including hepatic diseases.[9]
Important classes of cytokines (Ikram et al.[8])
| Cytokines classes |
|---|
| Growth factors |
| Haemopoietic growth factors; granulocyte-colony stimulating factor; granulocyte macrophage-colony stimulating factor; erythropoietin; thrombopoietin; stem cell factor or c-kit ligand |
| Epidermal growth factor |
| Platelet derived growth factor |
| Transforming growth factor β |
| Fibroblast growth factor |
| Insulin like growth factor |
| Nerve growth factor |
| ILs |
| IL-1 to IL-18, etc. |
| IFNs |
| IFN-α |
| IFN-β |
| IFN-γ |
| Miscellaneous |
| Tumour necrosis factor, etc. |
General properties of cytokines
Cytokines can be produced by virtually every nucleated cell type in response to injurious stimuli [Table 2].[10] Mostly, cytokines are produced and act locally. A minority enter the systemic circulation in biologically relevant amounts and a few have an important physiological role there. However, their “endocrine” role is subtly different from that of classical endocrine hormones. Whereas the purpose of endocrine hormones is to ensure the efficient function of normal tissues and the whole organism, cytokines with a physiological role in the circulation are concerned with restoring normal function to the tissue in which they were produced. Indeed, when tissues are severely challenged, and larger amounts of cytokines do enter the circulation, they may be responsible for upsetting systemic homeostasis, inducing fever, sickness behavior, cachexia and a variety of endocrine hormone imbalances.[11] Individual cytokine either in tissues or in the circulation may exhibit a range of activities and many of these overlap with activities of other cytokines.[12]
Cytokine properties (Oppenheim[10])
| Cytokine properties |
|---|
| Low molecular weight protein/glycoproteins |
| Almost all cells produce some cytokines |
| A single cytokine may be produced by many cell types |
| Cytokine expression is usually induced, not constitutive |
| Have a pleiotropy: one cytokine may exhibit many biological activities |
| Have redundancy: several cytokines may share the same/similar activities |
Structural organization of the liver and cytokines potential in its damage
The liver consists of several cell types that under normal circumstances produce only minimal levels of cytokines. When liver cells, particularly immune cells called Kupffer cells (KC), become activated cytokine production increases dramatically; therefore, if the liver has been damaged, cytokines mediate the regeneration of liver tissue. Also, KC can be activated by diseases caused by microorganisms or substances (i.e., pathogens). In this case, cytokines produced and released by the KC induce an inflammatory response in the liver (hepatitis), which is required to start the healing process. However, if the inflammation does not subside after a short time, persistent production of these same cytokines may lead to formation of fibrosis and cirrhosis. Thus, cytokine production can have both beneficial and harmful effects, depending on the amount and duration of cytokine release. The architecture and cellular composition of the healthy liver in numbers indicating the estimated frequency of each population relative to the total number of parenchymal and nonparenchymal cells in the liver is shown in Figures 1 and 2. This discontinuous structure allows contact between hepatocytes and lymphocytes. The contact can either be produced through hepatocyte microvilli protruding into the lumen or by lymphocyte pseudopod extensions penetrating into the space of Disse. The space of Disse contains hepatic stellate cells (HSCs, fat storing). KC reside within the liver sinusoidal vascular space, predominantly in the periportal area. Together sinusoidal endothelial cells and resident dendritic cells represent the liver antigen presenting cells. Lymphocytes are scattered throughout the parenchyma and portal tracts, and include conventional and unconventional T cells. A low frequency of B cells and abundance of natural killer (NK) are also characteristic of the liver immune microenvironment.[13]
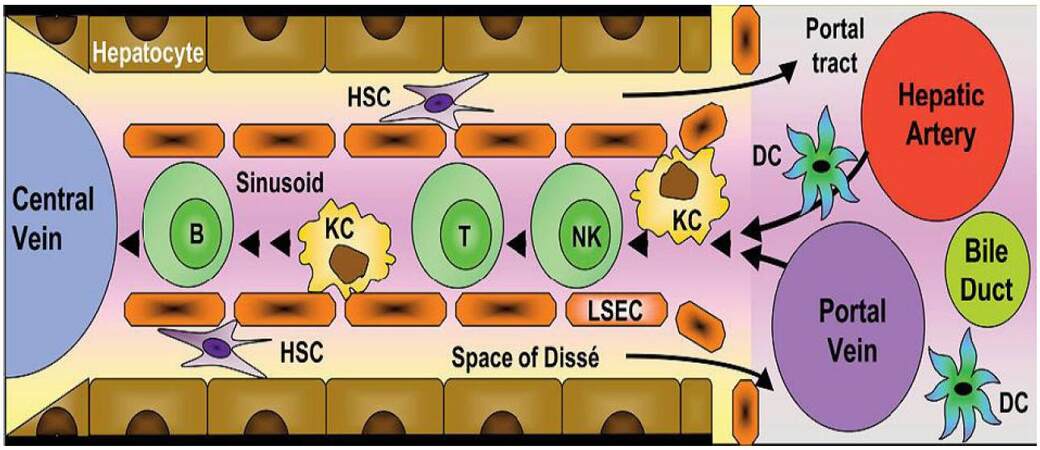
Figure 1. Architecture of the liver: sinusoids, hepatocytes and immune cells. LSEC form a fenestrated monolayer within the sinusoidal endothelium. HSC: hepatic stellate cells; NK: natural killer; KC: Kupffer cells; LSEC: liver sinusoidal endothelial cells; DC: dendritic cells
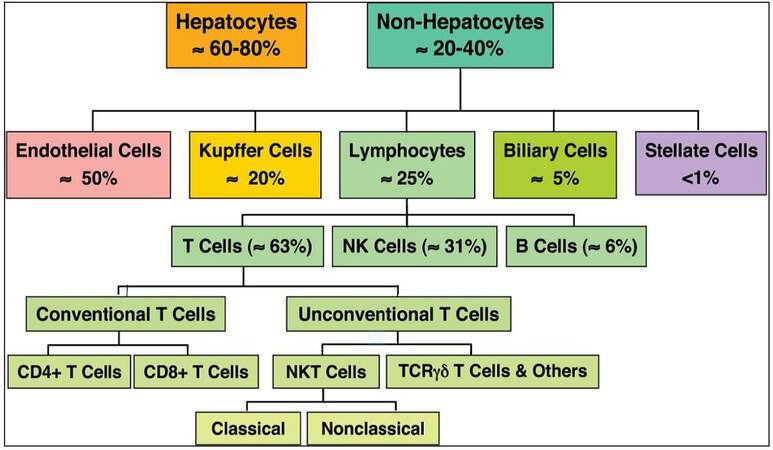
Figure 2. The estimated frequency of each population relative to the total number of parenchymal and nonparenchymal cells in the liver. NK: natural killer; TCR: T-cell receptor
Cytokine inhibitors, regardless of the mechanism by which they block the action of cytokines, appear to be the host’s own defense against the cytokines. The finding of elevated plasma concentrations of cytokines antagonist in patients with various diseases suggests that antagonism to cytokines is part of the host’s natural response to illness. One might therefore ask what the balance is between the amount of cytokines and these natural cytokine inhibitors in disease and whether disease can result, at least in part, from the failure to produce sufficient amounts of these inhibitors. For instance, the principle agonist forms of the interleukin (IL)-1 family are IL-1α and IL-1β. A third member of the IL-1 family, IL-1 receptor antagonist (IL-1ra), is also induced during inflammatory responses [Figure 3][14] but has preventive effect against the binding of IL-1α and β to their receptors IL-1ra is required to be in approximately 100-fold excess to inhibit IL-1α or IL-1β effectively; that is why IL-1ra is produced in greater amounts and is present at greater concentration matched with rare occurrence of α and β in the circulation. Additionally, cellular receptors for both IL-1 and tumor necrosis factor (TNF) exist in soluble forms, after being cleaved from the cell surface, and are able to bind and neutralize the cytokine [Figure 4].[15]
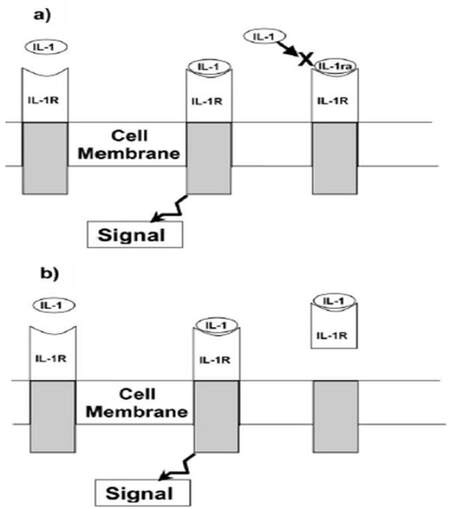
Figure 3. Interleukin 1 (IL-1) antagonism: IL-1 activity at the receptor may be blocked either by (a) competitive inhibition by IL-1 receptor antagonist (IL-1ra) or by (b) soluble forms of the type II receptor that bind to free IL-1
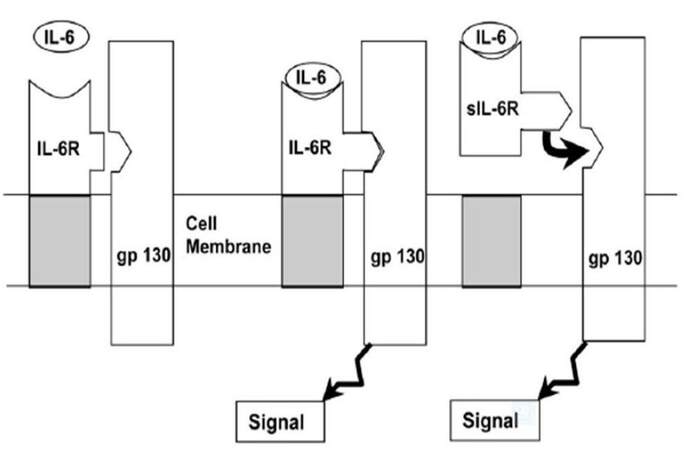
Figure 4. Maintained activity by soluble interleukin 6 receptor (sIL-6R); ligation of IL-6 with its membrane binding protein (IL-6R) results in association of the complex with a 130 kDa signal transduction glycoprotein (gp130). When cleaved from the cell surface, the IL-6/sIL-6R complex remains able to bind and activate gp130
The goal of this review is to highlight, in brief account, the major cytokines involved in different liver damage and discuss their basic biology and clinical applications.
Cytokines and alcoholic liver disease
Alcohol-related liver disease (ALD) is a major cause of morbidity and mortality worldwide. Chronic alcohol consumption leads to hepatocellular injury, fat accumulation, and liver inflammation and sometimes leads to liver cirrhosis or hepatocellular carcinoma (HCC) [Figure 5].[16] In the liver, TNF-α is mainly produced by KC.[17] The role of TNF-α as a critical inflammatory cytokine in the progression of ALD is well known.[18] KC secrete inflammatory cytokines[19] and reactive oxygen species (ROS)[20] which activate cells such as hepatocytes, HSCs, and endothelial cells.[21] After chronic alcohol consumption, KC exhibit enhanced sensitivity to lipopolysaccharide (LPS) -stimulated TNF-α production.[22] Elevated serum levels of TNF-α inducible cytokines or chemokines, including IL-6, IL-8, and IL-18, have also been reported in patients with alcoholic hepatitis.[23] Serum TNF-α is increased in patients with ALD and correlates with mortality. Treatment with pentoxifylline (an inhibitor of TNF-α synthesis) improved the survival of patients with severe alcoholic hepatitis (AH).[24] Anti-TNF-α antibody, infliximab, is also effective in severe AH patients.[25] These results suggest that TNF-α plays an important role in the progression of ALD.
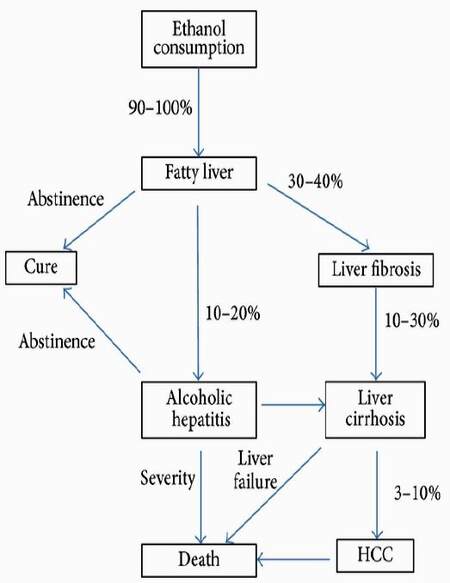
Figure 5. The natural history of alcoholic liver disease. Chronic ethanol consumption leads to fatty liver for more than 90%. But only up to 40% of this population develops more severe forms of alcoholic liver disease, including fibrosis and alcoholic hepatitis. Continuous ethanol consumption finally leads to liver cirrhosis or HCC and leads to death. HCC: hepatocellular carcinoma
IL-6 appears to have some beneficial effects on the liver. IL-6 may protect against hepatocyte apoptosis and participates in mitochondrial DNA repair after alcoholic liver injury.[26] IL-6 may promote human thymus derived monocytes helper 17 (Th17) differentiation and IL-17 production, therefore contributing to ethanol induced liver inflammation. IL-6 is also released along with IL-10, TNF-α and other cytokines by KC after alcohol consumption. IL-6 and IL-10 are two cytokines that play roles in reducing alcoholic liver injury and inflammation.[27] Elevated IL-6 is found in patients with ALD.[28] On the other hand, IL-6 knockout mice fed chronic alcohol showed increased liver fat accumulation, lipid peroxidation, mitochondrial DNA damage, and sensitization of hepatocytes to TNF-α induced apoptosis, which was prevented by the administration of recombinant IL-6.[29] These findings suggest that IL-6 has a protective effect at the early phase of ALD. Furthermore, IL-17 can act with other cytokines to activate NF-kB which plays a central role in regulating genetic transcription and encoding of inflammatory cytokines, and induce IL-8. Recently it was shown that patients with ALD had higher IL-17 plasma levels compared with healthy subjects.[30] IL-1α is also a potent proinflammatory cytokine. In both animal model and patient with ALD, the levels of pro-IL-1β are significantly increased in the liver and serum.[31]
Cytokines and fatty liver disease
Non-alcoholic fatty liver disease (NAFLD) is now the most frequent chronic liver disease that occurs across all age groups and is recognized to occur in 14-30% of the general population, representing a serious and growing clinical problem due to the growing prevalence of obesity and overweight.[32] The first manifestation of hepatic injury is the accumulation of fat within hepatocytes (steatosis), this is followed by the development of necroinflammatory (steatohepatitis) activity that leads to cirrhosis.[33]
The importance of cytokines as molecular effectors in liver damage has been particularly well demonstrated in patients and animals ranging from steatosis to cirrhosis. TNF-α is involved in the progression from steatohepatitis to cirrhosis, since it promotes activation of stellate cells, matrix-gene expression, and matrix remodeling.[34] Recent studies have indicated that deficiency of IL-1α in KC reduces liver inflammation and expression of inflammatory cytokines, which may implicate KC-derived IL-1α in steatohepatitis development.[35]
Obesity, especially visceral adiposity, is a major risk factor for NASH in humans.[36] Adipose tissue is a source of free fatty acids (FFA) that are delivered to the liver and a depot for triglycerides that are synthesized by hepatocytes and released into the blood. As producers of TNF-α and IL-6, adipocytes are considered a component of the immune system.[37] Visceral fat, which appears to be less “mature” than subcutaneous fat, produces more TNF-α and free fatty acids but less adiponectin than subscutaneous fat. Adiponectin antagonizes both the production and activity of TNF-α; thus the effect of this cytokine is potentiated when adiponectin is scarce. In addition, TNF-α inhibits adiponectin. Adiponectin also inhibits synthesis and uptake of FFA by hepatocytes, while stimulating FA oxidation enhancing their sensitivity to insulin. The combination of low adiponectin and high TNF-α levels in the context of increased hepatic exposure to FFA results in hepatic steatosis and severe hepatic insulin resistance.[38]
Leptin, as one of adipocyte secretions, together with its receptor share structural and functional similarities with the IL-6 family of cytokines, and leptin appears to play a critical role in the inflammatory response by stimulating leukocyte proliferation and the resulting increased plasma levels of the proinflammatory cytokines such as IL-6 and TNF-α.[39] These cytokines influence nitric oxide[40] that induces free radicals production and lipid peroxidation. Elevation of the inflammatory markers above normal levels is an independent predictor of several chronic diseases, including coronary heart disease, stroke, diabetes, atherosclerosis and insulin resistance.[41]
Cytokines and hepatic cholestasis
Cholestasis is defined as a decrease in canalicular bile flow that results in accumulation of bile in hepatocytes and canaliculi.[42] Hepatocellular cholestasis may be due to functional or structural alterations in the biliary tree. The clinical consequences of prolonged cholestasis are due to the failure of bile acids to reach the duodenum with subsequent malabsorption of fat and fat-soluble vitamins A, D, E and K as well as the accumulation of biliary constituents such as bile acids, bilirubin and cholesterol in the liver. Bile acids retention causes liver cell damage and pruritus.[43]
TNF-α plays a critical role in epithelial cell injury as well as in immune-mediated cholangiocyte injury.[44] Systemic levels of TNF-α are increased following biliary obstruction in experimental cholestasis produced by ethinylestradiol in rats.[45] Furthermore, TNF-α (in combination with other inflammatory cytokines) inhibits cholangiocyte secretory function in vitro.[44]
In cholestatic diseases, the intrahepatic bile acids induce hepatocellular apoptosis by stimulating Fas (a surface receptor that mediates apoptosis upon oligomerization by its ligands) translocation from the cytoplasm to the plasma membrane where self-aggregation occurs to trigger apoptosis.[46] Apoptosis is known to be the mechanism leading to progressive inflammation and destruction of bile ducts.[47] Also, bile acids can induce hepatic inflammatory response via the activation of hepatic macrophages[46] that follows the activation of the transcription factor NF-kB, since NF-kB activation has been shown to have a key role in the inflammatory process.[48] It is well known that NF-kB is activated by a wide range of agents and cytokines including TNF-α and IL-1α secreted from the injured hepatic macrophages.[48]
Proinflammatory cytokines were reported to stimulate the billiary epithelium to generate nitric oxide (NO), via nitric oxide synthase induction. NO causes ductular cholestasis by a reactive nitrogen oxide species mediated inhibition of adenyle cyclase and cAMP-dependent HCO3- and Cl- secretory mechanisms. This pathogenetic sequence may contribute to ductal cholestasis in inflammatory cholangiopathie.[44]
Cytokines and hepatic HCV infection
In HCV infection, the production of abnormal cytokine levels appears to contribute in the progression of the disease, viral persistence, and affects response to therapy. Cytokine genes polymorphisms located within the coding/regulatory regions have been shown to affect the overall expression and secretion of cytokines.[6]
The pathogenesis of liver cell damage in HCV infection may be related to several immunologic mechanisms and the subsequent T-cell responses.[49] Patients with chronic HCV infection, viral persistence which is a characteristic feature of chronic hepatitis C may be due to selective immune responses deficiencies and the production of inappropriate cytokine patterns.[50]
The involvement of macrophage derived cytokines such as TNF-α and IL1β in the production of inflammation has been described.[51] TNF-α acts as important mediator in liver injury and generally associated with several known cirrhosis-related complications. Moreover, TNF-α is positively related with the extent of liver necrosis.[52]
Adhesion molecules are necessary for leucocytes to adhere tightly to endothelial cells and have been reported to be cytokine-induced. Intercellular adhesionmolecule-1 (ICAM-1) is one of the principal adhesion molecules expressed on sinusoidal and venular endothelial cells and involved in firm adhesion and trans endothelial cell migration.[53] Consequently, The predominant features of HCV-C are more related with those that allow viral evasion of the immune defenses, especially although not exclusively, inhibition of interferons secretion, natural killer cells activation and T cell-mediated cytotoxicity.[54]
Several researchers have suggested that an adequate T-helper 1 (Th1) response [i.e., high interferon (IFN)-γ secretion by peripheral blood mononuclear cells] may be associated with a protective antiviral immune response,[55] while insufficient systemic Th1 cytokine secretion may be associated with increased viral load and disease progression.[56] Indeed, serum samples from HCV patients contain significantly lower level of soluble IFN-γ compared with controls.[57] In this sense, it has been reported that the IL-18 and IFN-γ mRNA expression in the liver were significantly correlated with each other and both upregulated in chronic HCV patients.[58] It was suggested that inheritance of IL-28B CT and TT, transforming growth factor (TGF)-β1 CT and TT and TNF-α AG and AA genotypes which appear to affect the cytokine production may be associated with susceptibility to HCV infection and resistance to combined antiviral therapy.[6]
Mitochondria are a major source of ROS under physiologic conditions, because 2% to 3% of the O2− consumed is converted to O2•− mainly by auto oxidation of ubisemiquinone which transfer electrons from complexes I and II to complex III. Hepatocyte ischemia described in chronic liver pathology, enhances O2•− production by impairing function of complex III.[59] TNF-α as one of the cytokines released from endotoxin-stimulated KC, through intracellular signaling, leads to decreased function of complex III.[60] Endotoxemia has been described in chronic hepatitis. Furthermore, activation of sinusoidal inflammatory cells such as lymphocytes and KC has been described in chronic hepatitis C virus infection.[61] Therefore, ROS serve as signaling molecules for the initiation and perpetuation of the inflammatory process that occurs with conditions of oxidative stress. This involves genetic regulation. Transcription factors that are directly influenced by reactive species and proinflammmatory signaling include NF-kB. NF-kB plays a central role in regulating genetic transcription and encoding of inflammatory cytokines, growth factors, acute phase proteins, adhesion molecules, other transcription factors, and cell death regulators. These NF-kB regulated genes are important in regulating genetic activity during critical illness, inflammatory diseases, and cancer.[62]
Cytokines and hepatic hepatitis B virus infection
Hepatitis B, which is caused by hepatitis B virus (HBV) infection, remains a major health threat worldwide. Hepatic injury and regeneration from chronic inflammation are the main driving factors of liver fibrosis and cirrhosis in chronic hepatitis B.[63]
During HBV infection, intrahepatic production of Th1 inflammatory cytokines and type-I IFNs activates two functionally independent pathways: an early elimination of HBV nucleocapsid particles from the hepatocytes; and a later post-transcriptional downregulation of viral RNA. Most of these effects are mediated direct or indirectly by IFN-α, β and γ.[64] Additionally, chronic HBV patients who clear the virus have higher levels of IL-12 than patients who remain HBV positive.[65] IL-12 can inhibit the replication of HBV through the induction of IFN-γ.[66]
Cytokines and hepatitis E virus
Hepatitis E virus (HEV) is a small non enveloped single-stranded positive-sense RNA virus and is one of the major causes for acute hepatitis worldwide. C-X-C motif ligand 8 (CXCL-8) is a small multifunctional proinflammatory chemokine. It was reported recently that HEV infection significantly upregulates CXCL-8 gene expression.[67]
The severity of HEV infection and associated adverse outcome might be mediated by cytokine1. In a pregnant and non-pregnant HEV infected women study, HEV viral load in acute viral hepatitis and fulminant hepatic failure were comparatively higher levels of TNF-α, IL-6, IFN-γ and TGF-β1 than those in controls; moreover TNF-α, IL-6 and IFN-γ had significant positive correlation with viral load, serum bilirubin and prothrombin time within infected women.[68]
Cytokines and hepatic schistosoma infection (Schistosomiasis)
Schistosomiasis is a chronic and debilitating disease that affects over 200 million people worldwide.[69] The pathology, resulting from infection with the helminth parasite Schistosoma mansoni or Schistosoma japonicum, is predominantly caused by the host immune response to parasite eggs that are laid in the portal venous system and then become trapped in hepatic sinusoids and sequestered within granulomatous lesions.[70] Cytokines, which communicate between the fibrotic areas and the immune system, form a network of host-parasite responses. Nevertheless, the mechanisms involved in the pathogenesis and progression of hepatic fibrosis in patients with schistosomiasis have not yet been fully elucidated.[71]
Studies on certain-cytokines knockout mice which had been infected with Schistosoma mansoni showed that egg granulomas and the hepatic fibrosis are dependent on the regulation of cytokines.[72] Higher levels of eosinophil-derived cytokines were observed in periportal fibrosis. A mixed cytokine pattern, characterized by positive correlation between TNF-α, IL-4 and IL-5 was observed in periportal fibrosis. Also, the positive association between lymphocyte-derived IL-10 and the eosinophils cytokine profile was observed exclusively in intestine further emphasize the hypothesis that immunoregulatory events take place controlling disease morbidity in human schistosomiasis[73] or in experimental models.[74] However, in human and animal schistosomiasis, studies have shown that high levels of TNF-α produced by peripheral blood mononuclear cells stimulated with schistosome antigen (Ag) are significantly associated with the presence of hepatosplenomegaly.[75,76] As hepatosplenic disease is a long-term complication of schistosomiasis and is considered to be indicative of severe hepatic and periportal fibrosis, it is conceivable that the immune mechanisms responsible for this lesion occur much earlier during infection and precede the downstream development of hepatosplenomegaly.[75] In addition Schistosoma japonicum significantly activates collagen deposition and hepatic stellate cell in the liver, however, fibrosis was accompanied by increased IFN-α, IFN-β, IFN-γ, IL-12, TNF-α, and IL-10 mRNA expression as well as decreased the expression of IL-4, IL-5 mRNA, natural killer group 2 member D (NKG2D) mRNA and tumor necrosis factor related apoptosis-inducing ligand (TRAIL).[77]
Cytokines and autoimmune hepatitis
Autoimmune hepatitis (AIH) is an inflammatory liver disorder, characterized by female preponderance, hypergammaglobulinaemia and interface hepatitis on histology. AIH is associated with impairment of regulatory T-cells,[78] a lymphocyte subset key in maintaining immune-tolerance to autoantigens.[79]
Limited data are available for the participation of cytokines in the development of AIH. In a previous study, Chernavsky et al.[80] analyzed the expression of cytokines in liver biopsies from pediatric autoimmune hepatitis (PAIH) patients in comparison with liver control samples obtained from cadaveric liver donors. While the expression of IFN-γ and IL-12p40 was not detectable in control livers, it was clearly unregulated,[80] and showed an increased expression of IL-18, IL-4 and the 1L-12 β2 chain receptor in PAIH patients. The unexpected increase of mRNA for IL-4, a typical Th2 cytokine, was found in conjunction with a severe histological inflammation in AIH. The up regulation of IL-4 in PAIH but not in another disease clearly suggests a more complex immunopathologic mechanism.
Th2 cytokines activate B cells and induce their differentiation into antibody-producing cells. Liver-infiltrating autoreactive B cells, in addition to their role in producing autoantibodies, also play a critical role in the development of fibrosis. The mechanism of suppressing fibrosis by B-cell depletion is independent of antibodies or T cells, raising the possibility that cytokines, produced or induced by autoimmune B cell, are responsible for fibrosis in autoimmune diseases targeting the liver.[81]
Human liver contains an uncommonly high number of NKT cells that participate in the early regulation of Thl/Th2 cell differentiation through the release of IFN-γ and IL-4. Moritoki et al.[81] and Solari et al.[82] found an increased number of Vα24 positive cells and transcripts coding for this invariant Vα24 chain in the liver of PAIH patients, pointing to a probable involvement of these regulatory cells as mediators of the hepatocellular injury in PAIH.
Cytokines and hepatic fibrosis, cirrhosis and cellular carcinoma
Chronic hepatic injury is associated with both liver cirrhosis and liver cancer.[71] Several cytokines and ROS, produced in the injured liver by resident macrophages and infiltrating leukocytes during inflammatory conditions, cause transformation of the quiescent HSCs into the activated phenotype, which is responsible for fibrosis, cirrhosis and cancer.[71,83]
The perisinusoidal retinoid- storing quiescent HSCs physiologically regulate liver architecture and blood flow by producing components of extracellular matrix and contractility respectively. During hepatic injury, HSCs transform into retinoid-free proliferating myofibroblast-like cells (activated HSCs, aHSCs), which express α-smooth muscle actin (α-SMA). aHSCs are highly fibrogenic and contractile, and play major role in causing architectural damage and portal hypertension.[83]
A phenomenon of aHSCs rapid apoptosis was observed among the proliferating cells during CCl4-induced active fibrosis.[84] During inflammatory liver injury, ROS are produced by resident macrophages and infiltrating blood cells, particularly neutrophils.[85] ROS-induced increased expressions of α-SMA, collagen I and collagen III in rat and human HSCs[86] indicate their role in HSC activation and fibrosis. While investigating the actions of ROS on aHSCs it is noted that superoxide (SO) reduced their viability revealing that SO causes apoptosis of aHSCs that involves mitochondrial release of cytochrome-C, activation of caspase-3 and increased expression of Bax.[87] During inflammation, the activated inflammatory cells produce fibrogenic cytokines and growth factors that activate HSCs.[88] The role of cytokine gene polymorphism in the progression of liver fibrosis or development of cirrhosis in patients with hepatic diseases has been investigated extensively. Yee et al.[89] indicated that TNF2 (-238A) and TNF3 (-308A) alleles are frequently found in patients with cirrhosis in chronic HCV infection. Polymorphisms of TGF-β gene are thought to be one of the determinants of fibrosis progression in viral hepatitis.[90] HSCs play an important role in hepatic fibrogenesis and that IL-1 is a potent cytokine that induces the myofibroblastic activation of HSCs. IL-1 is also implicated in the proliferation of HSCs and the regulation of the expression of various matrix metalloproteinases, which play a key role in the turnover and the deposition of extracellular matrix (ECM). Therefore, it is possible that genetic polymorphism of IL-1B gene may influence the progression of hepatic fibrosis by affecting the hepatic expression of IL-1 during the process of liver injury.[91] TGF-β has been implicated in hepatic fibrogensis; as it stimulates the production of extracellular matrix proteins and their receptors, and inhibits the synthesis of matrix-regrading proteolytic enzymes in chronic HCV; moreover, its serum or liver level has a positively correlation with the fibrosis score in both untreated patients or those respond to IFN-α treatment.[92]
IL-10 has a protective role in hepatic fibrogenesis, as it showed a decreased hepatic inflammation and increased serum levels of HCV-RNA matched with reduced liver fibrosis score either in chronic HCV-infected patients who received a short or after 12 months therapy with recombinant IL-10.[93]
A characteristic feature of HCV infection is a high frequency of persistence and progression to chronic liver disease (CLD). Persistent infection upsets the balance between immunostimulatory and inhibitory cytokines, which can prolong inflammation, and lead to necrosis, fibrosis, and CLD.[94] Elevated concentrations of cytokines also represent a characteristic feature of CLD, regardless of underlying etiology, which may represent a consequence of liver dysfunction instead of inflammatory disorder.[95]
T lymphocytes and immunoregulatory cytokines are of critical importance in the host defense against HCV infection. T-helper type 1 (Thl) cytokines (IL-2, IFN-γ) are required for host anti-viral responses, while T-helper type 2 (Th2) cytokines (IL-4, IL-10) can inhibit the development of these effectors.[96] Significant elevations in circulating Th22 cells, Th17 cells, Th1 cells, IL-22, IL-17A, and IFN-γ were observed in the hepatic fibrosis groups compared with the control group.[97]
It has been demonstrated that the proinflammatary IL-6 and IL-10 have been implicated to associate with certain human cancers and HCC. Previous study indicated that both IL-6 and IL-10 levels were elevated in HCC patients compared to normal controls, and the high levels would invariably decrease after surgical resection.[98] In addition, a high IL-10 level predicted a poor disease-free survival in patients undergoing curative surgery.[99] Hsia et al.[98] found that both IL-6 and IL-10 expression were more often higher in HCC patients compared to patients in other disease categories.
It has been postulated that an imbalance between Th1 and Th2 cytokine production is implicated in disease progression or inability to clear infections. It was reported that HCV-infected patients who develop chronicity have a predominant Th2 response, but a weak Th1 response, suggesting that this immune response imbalance can result from HCV interaction with dendritic cell functions.[100] These results support the notion that Th-lymphocyte polarization may play an important pathophysiologic role in influencing the outcome of HCV infection. All these immunological findings are mostly due to HCV infection rather than schistosomal infection, because patients with no schistosomal antibody had the same elevation of the same cytokines, late Schistosoma mansoni cases showed a suppressed cell-mediated immunity and a significant depletion of T-helper/inducer subset.[101]
In the majority of cases, HCC is found in conjunction with cirrhosis of the liver. Chronic inflammation and cirrhosis, accompanied by regenerative process, function as a tumor promoter, providing a common pathway from chronic HBV or HCV-infection to HCC. The direct etiologic role of HBV and HCV for HCC is obscure. Tumor progression may be brought about in HCC by mutation of p53 tumor suppressor gene. The prevalence of p53 mutations is similar in HBV-associated and HCV-associated HCCs. Other mechanisms of host defense are the production of TGF-β1, and the induction of cytotoxic T lymphocytes; the failure of these mechanisms permits the process of hepatocarcinogenesis. Treatment with alpha interferon of chronic hepatitis is necessary to delay or prevent the progression to liver cirrhosis and development of HCC.[102]
To date, two cytokines have achieved Food and Drug Administration approval as single agents for cancer treatment: high-dose, bolus IL-2 for metastatic melanoma and renal cell carcinoma and IFN-α for the adjuvant therapy of Stage III melanoma.[103]
The classical and current view of the cytokines role and mechanisms in both healthy and diseased liver is presented in Figure 6.[104]
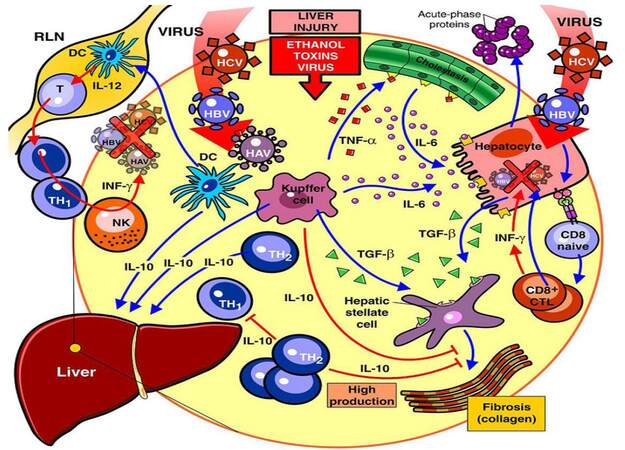
Figure 6. Overview of immune and parenchymal cells during liver injury. A steady-state migration of immature DC to the RLN and the production of IL-10 by KC and resident DC are involved in the phenomenon of tolerance to self-antigens within a healthy liver. After a virus infection, viral particles are incorporated into DC either because they become infected or through cross-priming and then migrate to the RLN, where they differentiate and activate naive T cells. Effector CD4+ T cells return to the liver and through secretion of Th1 cytokines and collaboration with activated NK cells, might contribute to the virus clearance. In an alternative view, exogenous antigen (Ag) expressed in hepatocytes can be presented to naive CD8+ T cells which after clonal expansion become efficient CTLs and secrete Th1 cytokines Under conditions of liver injury, KC play a critical role through secretion of TNF-a, TGF-b and IL-6. The latter acting on hepatocytes induces the production of the acute phase proteins. TGF-b activates the induction of fibrosis through the action of stellate cells and TNF-a plays a critical role in the induction of cholestasis. A high production of IL-10 is able to modulate the development of fibrosis. IL: interleukin; DC: dendritic cell; RLN: regional lymph node; NK: natural killer; CTL: cytotoxic T lymphocytes; KC: Kupffer cell; TNF-a: tumor necrosis factor-a; TGF-b: transforming growth factor-b; HAV: hepatitis A virus; HBV: hepatitis B virus; HCV: hepatitis C virus (Fainboim et al.[104])
Summary and conclusion
Cytokines are a large family of small proteins secreted by leukocytes and having an essential role in mediating the immune function. Many cytokines have multiple cellular sources and targets, as well as many natural inducers and inhibitors. Cytokines are produced to control body metabolism, infection, inflammation and tissue or neuronal damage. The pharmacological agents that can either suppress the production of the cytokines or block its biological actions may have potential therapeutic value against a wide variety of liver diseases. However, a stress is needed for a better knowledge about the adverse side effects for the anti-cytokine agents on the autoimmune responses; therefore future studies, leading to a combination of drugs that modulate the cellular immunity system but selectively block cytokines action, may be more useful for use to overcome the side effects of anti-cytokine therapy in the long-term. Again, proinflammation and prooxidation is the main cause of the complications of various inflammatory diseases. Since the high levels of cytokines directly induces the oxidative stress of the cells by depleting the vital antioxidant substances (such as glutathione) of the body and therefore elevate the ROS levels of the cells, it would be interesting to check the effectiveness of combination of drugs including antioxidant enhancer to effectively combat the side effects of anti-cytokine therapy.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
1. Guven-Maiorov E, Acuner-Ozbabacan SE, Keskin O, Gursoy A, Nussinov R. Structural pathways of cytokines may iluminate their roles in regulation of cancer development and immunotherapy. Cancers (Basel) 2014;6:663-83.
2. Ai W, Li H, Song N, Li L, Chen H. Optimal method to stimulate cytokine production and its use in immunotoxicity assessment. Int J Environ Res Public Health 2013;10:3834-42.
3. Sultani M, Stringer AM, Bowen JM, Gibson RJ. Anti-inflammatory cytokines: important immunoregulatory factors contributing to chemotherapy induced gastrointestinal mucositis. Chemother Res Pract 2012;2012:490804.
4. Allavena P, Germano G. Marchesi F, Mantovani, A. Chemokines in cancer related inflammation. Exp Cell Res 2011;317:664-73.
5. Matei I, Matei L. Cytokine patterns and pathogenicity in autoimmune disease. Rom J Intern Med 2002;40:27-41.
6. Pasha HF, Radwan MI, Hagrass HA, Tantawy EA, Emara MH. Cytokines genes polymorphisms in chronic hepatitis C: impact on susceptibility to infection and response to therapy. Cytokine 2013;61:478-84.
7. Cohen MC, Cohen S. Cytokine function: a study in biologic diversity. Am J Clin Pathol 1996;105:589-98.
8. Ikram N, Hassan K, Tufail S. Cytokines. Int J Pathol 2004;2:47-58.
9. SekIyama KD, Yoshiba M, Thomson AW. Circulating proinflammatory cytokines (IL-ll3, TNF-a, and IL-6) and IL-1 receptor antagonist (IL-lRa) in fulminant hepatic failure and acute hepatitis. Clin Exp Immunol 1994;98:71-7.
11. Slifka MK, Whitton JL. Clinical implication of dysregulated cytokine production. J Mol Med (Berl) 2000;78:74-80.
12. Angkasekwinai P, Dong C. TH17 Cytokines: Characteristics, Regulation, and Biological Function. In: Jiang S, editor. TH17 Cells in Health and Disease. Berlin: Springer Science+Business Media LLC; 2011 .
13. Racanelli V, Rehermann B. The liver as an immunological organ. Hepatology 2006;43:S54-62.
14. Arend WP, Malyak M, Guthridge CJ, Gabay C. Interleukin-1 receptor antagonist: role in biology. Annu Rev Immunol 1998;16:27-55.
15. Symons JA, Young PR, Duff GW. Soluble type II interleukin I (IL-1) receptor binds and blocks processing of IL-1β precursor and loses affinity for IL-1 receptor antagonist. Proc Natl Acad Sci U S A 1995;92:1714-8.
16. Ceni E, Mello T, Galli A. Pathogenesis of alcoholic liver disease: Role of oxidative metabolism. World J Gastroenterol 2014;20:17756-72.
17. McClain CJ, Song Z, Barve SS, Hill DB, Deaciuc I. Recent advances in alcoholic liver disease: IV- Dysregulated cytokine metabolism in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol 2004;287:G497-502.
18. Kitazawa T, Nakatani Y, Fujimoto M, Tamura N, Uemura M, Fukui H. The production of tumor necrosis factor-a by macrophages in rats with acute alcohol loading. Alcohol Clin Exp Res 2003;27:S72-5.
19. Tsujimoto T, Kuriyama S, Yamazaki M, Nakatani Y, Okuda H, Yoshiji H, Fukui H. Augmented hepatocellular carcinoma progression and depressed Kupffer cell activity in rat cirrhotic livers. Int J Oncol 2001;18:41-7.
20. Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver Int 2006;26:1175-86.
21. Diehl AM. Recent events in alcoholic liver disease: V. Effects of ethanol on liver regeneration. Am J Physiol Gastrointest Liver Physiol 2005;288:G1-6.
22. Aldred ND, Nagy LE. Ethanol dissociates hormone stimulated cAMP production from inhibition of TNF-a production in rat Kupffer cells. Am J Physiol 1999;276:G98-106.
23. Hill DB, Marsano L, Cohen D, Allen J, Shedlofsky S, McClain CJ. Increased plasma interleukin-6 concentrations in alcoholic hepatitis. J Lab Clin Med 1992;119:547-52.
24. Akriviadis E, Botla R, Briggs W, Han S, Reynolds T, Shakil O. Pentoxifylline improves short-term survival in severe acute alcoholic hepatitis: a double-blind, placebo-controlled trial. Gastroenterology 2000;119:1637-48.
25. Tilg H, Jalan R, Kaser A, Davies NA, Offner FA, Hodges SJ, Ludwiczek O, Shawcross D, Zoller H, Alisa A, Mookerjee RP, Graziadei I, Datz C, Trauner M, Schuppan D, Obrist P, Vogel W, Williams R. Anti-tumor necrosis factor alpha monoclonal antibody therapy in severe alcoholic hepatitis. J Hepatol 2003;38:419-25.
26. Hong F, Kim WH, Tian Z, Jaruga B, Ishac E, Shen X, Gao B. Elevated interleukin-6 during ethanol consumption acts as a potential endogenous protective cytokine against ethanol-induced apoptosis in the liver: involvement of induction of Bcl-2 and Bcl-xL proteins. Oncogene 2002;21:32-43.
27. Gao B. Cytokines, STATs and liver disease. Cell Mol Immunol 2005;2:92-100.
28. Kasztelan-Szczerbińska B, Surdacka A, Celiński K, Roliński J, Zwolak A, Miącz S, and Szczerbiński M. Prognostic significance of the systemic inflammatory and immune balance in alcoholic liver disease with a focus on gender-related differences. PLoS One 2015;10:e0128347.
29. Zhang X, Tachibana S, Wang H, Hisada M, Williams GM, Gao B, Sun Z. Interleukin-6 is an important mediator for mitochondrial DNA repair after alcoholic liver injury in mice. Hepatology 2010;52:2137-47.
30. Lemmers A, Moreno C, Gustot T, Maréchal R, Degré D, Demetter P, de Nadai P, Geerts A, Quertinmont E, Vercruysse V, Le Moine O, Devière J. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology 2009;49:646-57.
31. Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, Barrieau M, Min SY, Kurt-Jones EA, Szabo G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest 2012;122:3476-89.
32. Abd El-Kader SM, El-Den Ashmawy EM. Non-alcoholic fatty liver disease: the diagnosis and management. World J Hepatol 2015;7:846-58.
33. Lalor PF, Faint J, Aarbodem Y, Hubscher SG, Adams DH. The role of cytokines and chemokines in the development of steatohepatitis. Semin Liver Dis 2007;27:173-93.
34. Brenner DA, O'Hara M, Angel P, Chojkier M, Karin M. Prolonged activation of jun and collagenase genes by tumor necrosis factor-alpha. Nature 1989;337:661-3.
35. Olteanu S, Kandel-Kfir M, Shaish A, Almog T, Shemesh S, Barshack I, Apte RN, Harats D, Kamari Y. Lack of interleukin-1α in Kupffer cells attenuates liver inflammation and expression of inflammatory cytokines in hypercholesterolaemic mice. Dig Liver Dis 2014;46:433-9.
36. Ikejima K, Okumura K, Kon K, Takei Y, Sato N. Role of adipocytokines in hepatic fibrogenesis. J Gastroenterol Hepatol 2007:S87-92.
37. Rajala MW, Scherer PE. Minireview: The adipocyte-at the cross roads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology 2003;144:3765-73.
38. Bruun JM, Lihn AS, Verdich C, Pedersen SB, Toubro S, Astrup A, Richelsen B. Regulation of adiponectin by adipose tissue-derived cytokines: in vivo and in vitro investigations in humans. Am J Physiol Endocrinol Metab 2003;285:E527-33.
39. Hukshorn CJ, Lindeman JH, Toet KH, Saris WH, Eilers PH, Westerterp-Plantenga MS, Kooistra T. Leptin and the proinflammatory state associated with human obesity. J Clin Endocrinol Metab 2004;89:1773-8.
40. You T, Berman DM, Rayan S, Nicklas BJ. Effect of hypocaloric diet and exercise training on inflammation and adipocyte lipolysis in obese post-menopausal women. J Clin Endocrinol Metab 2004;89:1739-46.
41. Pravo PE, Morse S, Borne DM, Aguilar EA, Reisin E. Leptin and hypertension. Vasc Health Risk Manag 2006;2:163-9.
43. Keith G, Tolman MD, Rej R. Liver function. In: Burtis CA, Ashwood ER, Tietz NW, editors. Tietz Textobook of Clinical Chemistry, Volume 3, Chapter 33. Philadelphia, PA: W.B. Saunders; 1999.
44. Spirlì C, Fabris L, Duner E, Fiorotto R, Ballardini G, Roskams T, Larusso NF, Sonzogni A, Okolicsanyi L, Strazzabosco M. Cytokine-stimulated nitric oxide production inhibits adenyl cyclase and cAMP-dependent secretion in cholangiocytes. Gastroenterology 2003;124:737-53.
45. Ahmed HH, Mannaa F. Curcumin as an effective agent against ethinylestradiol-induced hepatocellular cholestaris. Egypt J Med Lab Sci 2004;13:1-16.
46. Jaeschke H, Gores GJ, Cederbaum AI, Hinson JA, Pessayre D, Lemasters JJ. Mechanism of hepatotoxicity. Toxicol Sci 2002;65:166-76.
47. Sträter J, Möller P. Pathogenesis of primary biliary cirrhosis: CD 95-induced apoptosis at last? Eur J Gastroenterol Hepatol 1998;10:539-41.
48. Fox ES, Kim JC, Tracy TF. NF-Kappa β activation and modulation in hepatic macrophages during cholestatic injury. J Surg Res 1997;72:129-34.
49. Ferrari C, Valli A, Galati L, Penna A, Scaccaglia P, Giuberti T, Schianchi C, Missale G, Marin MG, Fiaccadori F. T-cell response to structural and non-structural hepatitis C virus antigen in persistent and self limited hepatitis C virus infection. Hepatology 1994;19:286-95.
50. Abd-Elghaffar Y, Fouad HH, Eid A. Effect of interferon-α therapy on oxidative stress in hepatitis C virus infection. Arab J Lab Med 1999;25:345-54.
51. Lee J, Tian Y, Chan ST, Kim JY, Cho C, Ou JH. TNF-α induced by hepatitis C virus via TLR7 and TLR8 in hepatocytes supports interferon signaling via an autocrine mechanism. PLoS Pathog 2015;11:e1004937.
52. Zalata AA, Morsy HK, Atwa AA, El-bendary MM. Relationship between S-Adenosylmethionine, S-Adenosylhomocysteine, Tumor necrosis factor-α, reduced glutathione and nitrite in liver cirrhosis. Egypt J Biochem Molecular boil 2007;25:362-82.
53. Nessim I, Abu-Zikri N, Moussa M, Hafez T, Hazem A, Reda M, Abdel-Hadi A, Edward N, El-Ghafir W. Implication of tumor-necrosis factor alpha (TNF-α): Interleukin-1 Beta (IL1-β) and intercellular adhesion molecule-1 (ICAM-1) in warm hepatic ischaemia-reperfusion injury: Effect of intraportal infusion of prostaglandin E-1. Egypt J Lab Med 2002;14:243-74.
54. Martínez-Esparza M, Tristán-Manzano M, Ruiz-Alcaraz AJ, García-Pe-arrubia P. Inflammatory status in human hepatic cirrhosis. World J Gastroenterol 2015;21:11522-41.
55. Koziel MJ, Dudley D, Afdhal N, Grakoui A, Rice CM, Choo QL, Houghton M, Walker BD. HLA class I-restricted cytotoxic T lymphocytes specific for hepatitis C virus. Identification of multiple epitopes and characterization of patterns of cytokine release. J Clin Invest 1995;96:2311-21.
56. Lechmann M, Woitas RP, Langhans B, Kaiser R, Ihlenfeldt HG, Jung G, Sauerbruch T, Spengler U. Decreased frequency of HCV core-specific peripheral blood mononuclear cells with type 1 cytokine secretion in chronic hepatitis C. J Hepatol 1999;31:971-8.
57. Cribier B, Schmitt C, Rey D, Landg JM, Kirn A, Stoll-Keller F. Production of cytokines in patients infected by hepatitis C virus. J Med Virol 1998;55:89-91.
58. McGuinness PH, Painter D, Davies S, McCaughan GW. Increases in intrahepatic CD68 positive cells, MAC387 positive cells, and proinflammatory cytokines (particularly interleukin 18) in chronic hepatitis C infection. Gut 2000;46:260-9.
59. Gonzalez-Flecha B, Reides C, Cutrin JC, Llesuy SF, Boveris A. Oxidative stress produced by suprahepatic occlusion and reperfusion. Hepatology 1993;18:881-9.
60. Goossens V, Grooten J, Devos K, Fiers W. Direct evidence for tumor necrosis factor induced mitochondrial reactive oxygen intermediates and their involvement in cytotoxicity. Proc Nat Acad USA 1995;92:8115-9.
61. Lefkowitch JH, Schiff ER, Davis GL, Perrillo RP, Lindsay K, Bodenheimer HC Jr, Balart LA, Ortego TJ, Payne J, Dienstag JL, Gibas A, Jacobson IM, Tamburro CH, Carey W, O'Brien C, Sampliner R, Van Thiel DH, Feit D, Albrecht J, Meschievitz C, Sanghvi B, Vaughan RD; The Hepatitis Interventional Therapy Group. Pathological diagnosis of chronic hepatitis C: a multicenter comparative study in chronic hepatitis B. Gastroenterology 1993;104:595-603.
62. Mandelker L. Oxidative stress, free radicals, and cellular damage In: Mandelker L, Vajdovich P, editors. Studied on Veterinary Medicine. Berlin: Springer Science+Business Media LLC; 2011. pp. 1-18.
63. Jia B, Guo M, Li G, Yu D, Zhang X, Lan K, Deng Q. Hepatitis B virus core protein sensitizes hepatocytes to tumor necrosis factor-induced apoptosis by suppression of the phosphorylation of mitogen-activated protein kinase Kinase 7. J Virol 2015;89:2041-51.
64. Wieland SF, Guidotti LG, Chisari FV. Intrahepatic induction of alpha/beta interferon eliminates viral RNA-containing capsids in hepatitis B virus transgenic mice. J Virol 2000;74:4165-73.
66. Cavanaugh VJ, Guidotti LG, Chisari FV. Interleukin-12 inhibits hepatitis B virus replication in transgenic mice. J Virol 1997;71:3236-43.
67. Li Z, Chen L, Liu Q. Activation of CXCL-8 Transcription by hepatitis E virus ORF-1 via AP-1. Mediators Inflamm 2015;2015:495370.
68. Kumar A, Devi SG, Kar P, Agarwal S, Husain SA, Gupta RK, Sharma S. Association of cytokines in hepatitis E with pregnancy outcome. Cytokine 2014;65:95-104.
69. Iarotski LS, Davis A. The schistosomiasis problem in the world: results of a WHO questionnaire survey. Bull World Health Organ 1981;59:115-27.
70. Duan Y, Gu X, Zhu D, Sun W, Chen J, Feng J, Song K, Xu F, He X, He X. Schistosoma japonicum soluble egg antigens induce apoptosis and inhibit activation of hepatic stellate cells: a possible molecular mechanism. Int J Parasitol 2014;44:217-24.
71. Huang J, Tao R, Li L, Ma K, Xu L, Ai G, Fan X, Jiao Y, Ning Q. Involvement of heat shock protein 47 in Schistosoma japonicum-induced hepatic fibrosis in mice. Int J Parasitol 2014;44:23-35.
73. Silveira-Lemos D, Teixeira-Carvalho A, Martins-Filho OA, Oliveira LFA, Costa-Silva MF, Matoso LF, de Souza LJ, Gazzinelli A, Correˆa-Oliveira R. Eosinophil activation status, cytokines and liver fibrosis in Schistosoma mansoni infected patients. Acta Trop 2008;108:150-9.
74. Wilson MS, Mentink-Kane MM, Pesce JT, Ramalingam TR, Thompson R, Wynn TA. Immunopathology of schistosomiasis. Immunol Cell Biol 2007;85:148-54.
75. Mwatha JK, Kimani G, Kamau T, Mbugua GG, Ouma JH, Mumo J, Fulford AJ, Jones FM, Butterworth AE, Roberts MB, Dunne DW. High levels of TNF, soluble TNF receptors, soluble ICAM-1, and IFN-gamma, but low levels of IL-5, are associated with hepatosplenic disease in human Schistosomiasis mansoni. J Immunol 1998;160:1992-9.
76. Sombetzki M, Fuchs CD, Fickert P, ضsterreicher CH, Mueller M, Claudel T, Loebermann M, Engelmann R, Langner C, Sahin E, Schwinge D, Guenther ND, Schramm C, Mueller-Hilke B, Reisinger EC, Trauner M. 24-nor-ursodeoxycholic acid ameliorates inflammatory response and liver fibrosis in a murine model of hepatic schistosomiasis. J Hepatol 2015;62:871-8.
77. Hou X, Yu F, Man S, Huang D, Zhang Y, Liu M, Ren C, Shen J. Polyinosinic-polycytidylic acid attenuates hepatic fibrosis in C57BL/6 mice with Schistosoma japonicum infection. Acta Trop 2012;121:99-104.
78. Ferri S, Longhi MS, De Molo C, Lalanne C, Muratori P, Granito A, Hussain MJ, Ma Y, Lenzi M, Mieli-Vergani G, Bianchi FB, Vergani D, Muratori L. A multifaceted imbalance of T cells with regulatory function characterizes type 1 autoimmune hepatitis. Hepatology 2010;52:999-1007.
79. Holder BS, Grant CR, Liberal R, Maa Y, Heneghan MA, Mieli-Vergani G, Vergani D, Longhi MS. Retinoic acid stabilizes antigen-specific regulatory T-cell function in autoimmune hepatitis type 2. J Autoimmun 2014;53:26-32.
80. Cherٌavsky AC, Paladino N, Rubio AE, De Biasio MB, Periolo N, Cuarterolo M, Goٌi J, Galoppo C, Caٌero-Velasco MC, Muٌoz AE, Fainboim H, Fainboim L. Simultaneous expression of Thl cytokines and IL-4 confers severe characteristics to type I autoimmune hepatitis in children. Hum Immunol 2004;65:683-91.
81. Moritoki Y, Lian ZX, Ohsugi Y, Ueno Y, Gershwin ME. B cells and autoimmune liver diseases. Autoimmun Rev 2006;5:449-57.
82. Solari NEF, Galoppo C, Cuarterolo M, Goٌi J, Fernلndez-Salazar L, Arranz LE, Garrote J, Cherٌavsky AC. The simultaneous high expression of Vα24, IFN-γ and FoxP3 characterizes the liver of children with type I autoimmune hepatitis. Clin Immunol 2010;137:396-405.
83. Gresner AM. The up-and-down of hepatic stellate cells in tissue injury: apoptosis restores cellular homeostasis. Gastroenterology 2001;120:1285-8.
84. Iredale JP, Benyon RC, Pickering J, McCullen M, Northrop M, Pawley S, Hovell C, Arthur MJ. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest 1998;102:538-49.
85. Gressner AM. Liver fibrosis: perspectives in pathobiochemical research and clinical outlook. Eur J Clin Chem Clin Biochem 1991;29:293-311.
86. Svegliati Baroni G, D'Ambrosio L, Ferretti G, Casini A, Di Sario A, Salzano R, Ridolfi F, Saccomanno S, Jezequel AM, Benedetti A. Fibrogenic effect of oxidative stress on rat hepatic stellate cells. Hepatology 1998;27:720-6.
87. Thirunavukkarasu C, Watkins S, Harvey SAK, Gandhi CR. Superoxide-induced apoptosis of activated rat hepatic stellate cells. J Hepatol 2004;41:567-75.
89. Yee LJ, Tang J, Herrera J, Kaslow RA, van Leeuwen DJ. Tumor necrosis factor gene polymorphisms in patients with cirrhosis from chronic hepatitis C virus infection. Genes Immun 2000;1:386-90.
90. Bataller R, North KE, Brenner DA. Genetic polymorphisms and the progression of liver fibrosis: a critical appraisal. Hepatology 2003;37:493-503.
91. Han YP, Zhou L, Wang J, Xiong S, Garner WL, French SW, Tsukamoto H. Essential role of matrix metalloproteinases in interleukin-1-induced myofibroblastic activation of hepatic stellate cell in collagen. J Biol Chem 2004;279:4820-8.
92. Marek B, Kajdaniuk D, Mazurek U, Janczewska-Kazek E, Kos-Kudla B, Strzalka B, Fila A, Niedziolka D, Beniowski M, Ostrowska Z, Borgiel-Marek H, Kajdaniuk J, Sieminska L, Nowak M, Wilczok T, Pakula D, Filipczyk P. TGF-beta l mRNA expression in liver biopsy specimens and TGF-beta l serum levels in patients with chronic hepatitis C before and after antiviral therapy. J Clin Pharm Ther 2005;30:271-7.
93. Nelson DR, Lauwers GY, Lau Y J, Davis GL. Interleukin 10 treatment reduces fibrosis in patients with chronic hepatitis C: a pilot trial of interferon nonresponders. Gastroenterology 2000;118:655-60.
94. Jacobson Brown PM, Neuman MG. Immunopathogenesis of hepatitis C viral infection: Thl/Th2 responses and the role of cytokines. Clin Biochem 2001;34:167-71.
95. Goyal A, Kazim SN, Sakhuja P, Malhotra V, Arora N, Sarin SK. Association of TNF-beta polymorphism with disease severity among patients infected with hepatitis C virus. J Med Virol 2004;72:60-5.
96. Cacciarelli TV, Martintez OM, Gish RG, Villarnueva JC, Krams SM. Immunoregulatory cytokines in chronic hepatitis C virus infection: pre-and posttreatment with interferon alfa. Hepatology 1996;24:6-9.
97. Lu DH, Guo XY, Qin SY, Luo W, Huang XL, Chen M, Wang JX, Ma SJ, Yang XW, Jiang HX. Interleukin-22 ameliorates liver fibrogenesis by attenuating hepatic stellate cell activation and downregulating the levels of inflammatory cytokines. World J Gastroenterol 2015;21:1531-45.
98. Hsia CY, Huo TI, Chiang SY, Lu MF, Sun CL, Wu JC, Lee PC, Chi CW, Lui WY, Lee SD. Evaluation of interleukin-6, interleukin-10 and human hepatocyte growth factor as tumor markers of hepatocellular carcinoma. Eur J Surg Oncol 2007;33:208-12.
99. El-Medany OM, Abdel Wahab KS, Abu Shady EA, Gad El-Hak N. Chronic liver disease and hepatitis C virus in Egyptian patients. Hepatogastroenterology 1999;46:1895-903.
100. Stoll-Keller F, Schvoerer E, Thumann C, Navas MC, Aubertin AM. Immunomodulating effect of HCV during the development of chronic hepatitis C: toward new therapeutic approaches. Bull Acad Natl Med 2003;187:1147-60; discussion 1160-1. (in French)
101. Elrefaei M, El-Sheikh N, Kamal K, Cao H. HCV-specific CD27- CD28- memory T cells are depleted in hepatitis C virus and Schistosoma mansoni co-infection. Immunology 2003;110:513-8.
102. Marotta F, Vangieri B, Cecere A, Gattoni A. The pathogenesis of hepatocellular carcinoma is multifactorial event. Novel immunological treatment in prospect. Clin Ter 2004;155:187-99.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Mannaa FA, Abdel-Wahhab KG. Physiological potential of cytokines and liver damages. Hepatoma Res 2016;2:131-43. http://dx.doi.org/10.20517/2394-5079.2015.58
AMA Style
Mannaa FA, Abdel-Wahhab KG. Physiological potential of cytokines and liver damages. Hepatoma Research. 2016; 2: 131-43. http://dx.doi.org/10.20517/2394-5079.2015.58
Chicago/Turabian Style
Mannaa, Fathia A., Khaled G. Abdel-Wahhab. 2016. "Physiological potential of cytokines and liver damages" Hepatoma Research. 2: 131-43. http://dx.doi.org/10.20517/2394-5079.2015.58
ACS Style
Mannaa, FA.; Abdel-Wahhab KG. Physiological potential of cytokines and liver damages. Hepatoma. Res. 2016, 2, 131-43. http://dx.doi.org/10.20517/2394-5079.2015.58
About This Article
Copyright

Author Biographies

Data & Comments
Data


 Cite This Article 51 clicks
Cite This Article 51 clicks

 Like This Article 32
likes
Like This Article 32
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.